
Abstract
The pacific oyster Crassostrea gigas and the Mediterranean mussel Mytilus galloprovincialis are two widely farmed bivalve species which show contrasting behaviour in relation to microbial diseases, with C. gigas being more susceptible and M. galloprovincialis being generally resistant. In a recent study, we showed that different susceptibility to infection exhibited by these two bivalve species may depend on their different capability to kill invading pathogens (e.g., Vibrio spp.) through the action of haemolymph components. Specific microbial-host interactions may also impact bivalve microbiome structure and further influence susceptibility/resistance to microbial diseases. To further investigate this concept, a comparative study of haemolymph and digestive gland 16SrDNA gene-based bacterial microbiota profiles in C. gigas and M. galloprovincialis co-cultivated at the same aquaculture site was carried out using pyrosequencing. Bacterial communities associated with bivalve tissues (hemolymph and digestive gland) were significantly different from those of seawater, and were dominated by relatively few genera such as Vibrio and Pseudoalteromonas. In general, Vibrio accounted for a larger fraction of the microbiota in C. gigas (on average 1.7-fold in the haemolymph) compared to M. galloprovincialis, suggesting that C. gigas may provide better conditions for survival for these bacteria, including potential pathogenic species such as V. aestuarianus. Vibrios appeared to be important members of C. gigas and M. galloprovincialis microbiota and might play a contrasting role in health and disease of bivalve species. Accordingly, microbiome analyses performed on bivalve specimens subjected to commercial depuration highlighted the ineffectiveness of such practice in removing Vibrio species from bivalve tissues.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Recent advance in DNA sequencing technology is enabling new insights into microbial community diversity associated with human and animal tissues [1,2,3]. It is now recognized that host associated microbial communities (“microbiota”) are playing an important role in animal health by providing multiple roles, ranging from nutrient processing to protection from diseases [4]. Marine invertebrates host high microbial abundance and diversity [5], and alteration of the microbiota due to stressful conditions and/or environmental changes was previously linked with a condition of a compromised health status and susceptibility to diseases (e.g., through the rise of opportunistic pathogens and/or colonization by non-resident microbial species) [6, 7].
Microbial communities associated with bivalve tissues were historically investigated by means of culture-dependent methods [8, 9], while only few studies have been conducted so far to estimate bivalve microbiota using next generation sequencing (NGS) technology [7, 10,11,12,13]. In Europe mass mortality episodes of the pacific oyster (Crassostrea gigas) in farming areas were reported at increasing frequency in recent years [14] and are attributed to complex interactions among oysters, microbial pathogens and environmental variables [15]. For example, stressful environmental conditions such as warm seawater temperatures were observed to favour shift of C. gigas bacterial communities toward pathogen-dominated communities, also promoting colonization by secondary opportunistic pathogens [7]. In contrast to C. gigas, other species of widely farmed bivalves such as the Mediterranean mussel Mytilus galloprovincialis are generally not affected by mortality events being less sensitive to changes in environmental conditions and microbial infections [16]. Unfortunately, molecular studies investigating M. galloprovincialis microbiota are lacking and, in contrast to oysters, no information are currently available on the role that microbiome might play in health and disease of farmed mussel [17].
We have recently shown that the different susceptibility to infection exhibited by C. gigas and M. galloprovincialis may depend, at least in part, on their different capability to kill invading microbial pathogens through the combined action of cellular (hemocyte) and soluble components of the haemolymph [18]. In particular the extrapallial protein (EP) present in serum of M. galloprovincialis (MgEP) but lacking in C. gigas has been recently shown to work as an opsonin promoting D-mannose sensitive (MS) interactions of the bivalve pathogen Vibrio aestuarianus 01/032 strain and other bacteria (e.g., Vibrio cholerae ElTor N16961 and Escherichia coli MG1655) carrying MS sensitive ligands with the hemocytes [19]. Presence/absence of this and/or similar pathways driving microbial-host interactions in the haemolymph and other bivalve tissues may significantly impact microbiome structure and, in turn, influence susceptibility/resistance to microbial diseases in these animals.
Based on these previous observations, the aim of this study was to investigate whether differences exist in the overall microbiota structure of the two bivalve species that might partially be linked with their different susceptibility to microbial diseases. To this end, a comparative study of haemolymph and digestive gland bacterial microbiota profiles in C. gigas and M. galloprovincialis co-cultivated at the same aquaculture site was carried out using pyrosequencing techniques. The composition of bivalve microbial communities was also compared to that of ambient water where bivalve are grown and cultivated. As an additional objective of the study, microbiota analyses were performed in bivalve specimens that were subjected to commercial depuration, in order to investigate the impact of this practice on bivalve microbiota and its effectiveness in the removal of indigenous microbial pathogens.
Material and Methods
Bivalve Collection and Preparation of Haemolymph and Digestive Gland Samples for Molecular Microbiological Analysis
Adult mussels (M. galloprovincialis Lam) (n = 15, 5–7 cm long) and oysters (C. gigas) (n = 15, 8–10 cm long), were collected from the same batch in August 2015 from a shellfish farm located in the Gulf of La Spezia (44° 04′ 33″ N; 9° 51′ 20″ E, Ligurian Sea, Italy). The site is the main shellfish production area in northwestern Italy. Mussels and oysters are farmed in co-cultivation by the suspended culture technique (rack culture method), with an estimated annual production of about 6.000 tons. Spat origin differs for the two bivalve species with C. gigas spat being mainly imported from Brittany (France) and M. galloprovincialis spat being naturally re-collected from the local cultivation area. Abnormal mortality of both species did not occur during the study period.
Additional mussels (n = 15, 5–7 cm long) and oysters (n = 15, 8–10 cm long) cultivated in the same area and period were obtained after depuration and treatment according to European and FAO regulations (bivalve are held in tanks of clean seawater for 24 h; tanks seawater is treated through various steps to remove contaminants including sand filtration, UV treatment, ozonozation, and biofiltration). 1 L of surface seawater (n = 3) was also collected in the farming area by means of sterile plastic bottles. At the time of collection seawater temperature (26.7 °C), salinity (39.8‰) and pH (8.2) were recorded.
In the laboratory all animals (depurated and non-depurated) were cleaned of epibionts and gently washed with ASW to remove part of the non-resident microbiota. Haemolymph was extracted from the posterior adductor muscle using a sterile 1 ml of syringe with an 18 G1/200 needle. With the needle removed, hemolymph was filtered through a sterile gauze and pooled in 50-ml Falcon tubes at 18 °C. All individual animals were dissected under sterile conditions and digestive glands were removed, weighted and frozen for further microbiological analysis.
Nucleic Acid Extraction
Nucleic acids were extracted from pooled whole haemolymph (n = 15) and digestive gland (n = 15) samples with the High Pure PCR Template Preparation Kit (Roche Diagnostics) according to the manufacturer’s instructions. For water samples, 1 L of seawater was filtered on 0.2 Nucleopore filters (45 mm) and nucleic acids were extracted with the PowerWater DNA Isolation Kit (Mo Bio laboratories, inc), according to the manufacturer’s instructions. The amount of DNA extracted was determined fluorimetrically with QuantiFluorTM dsDNA System using a QuantiFluorTMfluorometer (Promega Italia srl, Milano, Italy).
Real-Time PCR
Real-time PCR for detection and enumeration of faecal indicators (E. coli), bivalve (V. aestuarianus, V. splendidus-clade, V. coralliilyticus) and human (V. cholerae, V. parahaemolyticus, and V. vulnificus) pathogenic bacteria in seawater and bivalve tissues were performed with the LightCyler (Roche Diagnostics, Mannheim, Germany) using primers and protocols reported in ESM 1: Table S1.
For V. aestuarianus, V. splendidus-clade, V. cholerae, V. parahaemolyticus, and V. vulnificus a Taq-Man-based PCR protocol was used (ESM S1: Table S1) [20,21,22,23,24,25,26]. Amplification reaction mixtures (20 μL) contained the following: 1× TaqMan Master Mix (Roche Diagnostics), 200 nM primers, 25 nM probe, and DNA sample (0.2–2 ng/μL). Five microliters of DNA template was added to the reaction mixture. The PCR program used was as follows: initial denaturation at 95 °C for 10 min; subsequent 45 cycles of denaturation at 95 °C for 10 s; annealing at 60 °C [V. aestuarianus and V.splendidus-clade] for 15 s, 59 °C [V. cholerae], 60 °C [V. vulnificus] or 59 °C [V. parahaemolyticus], for 20 s; and elongation at 72 °C for 1 s, followed by a cooling step at 40 °C for 30 s.
For E. coli and V. coralliilyticus, a PCR protocol based on SYBR-Green I fluorescence was used (ESM S1: Table S1). Each reaction mixture contained 1 × SYBR Green I Master Mix (Roche Diagnostics), 5.0 mmol of MgCl2 and 0.25 mmol of each primer in a final volume of 20 uL. The PCR programme was optimized as follows: initial denaturation at 95 °C for 10 min, subsequent 40 cycles of denaturation at 95 °C for 15 s [E.coli] or for 5 s [V. coralliilyticus], annealing at 60 °C for 30 s [E.coli.] or 5 s [V.coralliilyticus] and elongation at 72 °C for 4 s, followed by a final elongation at 72 °C for 10 min. PCR runs were analysed directly in the LightCycler using melting analysis and the software provided with the instrument. For each single real-time PCR assay each DNA template was analysed in triplicate (coefficient of variation 5%). The standards were prepared from pure nucleic acid templates at known molar concentrations.
16SrDNA Pyrosequencing
A 16SrDNA PCR amplicon library was generated from genomic DNA extracted from bivalve samples using the broad-range bacterial primers, 967f-5′-CAACGCGAAGAACCTTACC-3′ and 1046r-5′-CGACAGCCATGCANCACCT-3′ amplifying positions 965–1063 (V6 hyper variable region) of the E. coli numbering of the 16SrDNA [27]. Fusion primers were custom designed to include the 16SrDNA complementary regions plus the Roche 454 A (5′-GCCTCCCTCGCGCCATCAG-3′) and B (5′-GCCTTGCCAGCCCGCTCAG-3′) pyrosequencing adapters and a specimen-specific barcode sequence. All primers were synthesized and HPSF or HPLC purified by Tib Molbiol Srl (Genoa, Italy).
PCR products were cleaned using a Agencourt AMPure XP kit (Beckman Coulter s.r.l.) and checked on an Agilent Bioanalyzer using a Agilent DNA 7500 kit (Agilent Technologies, Inc.). Equal amounts (1 × 109 molecules/μL) of each product were then pooled together and finally adjusted to 1 × 106 molecules/μL. Amplicon libraries were bound to beads under conditions that favour one fragment per bead and beads were emulsified in a PCR mixture in oil. After breaking the emulsion, the DNA strands were denatured, and the beads carrying single-stranded DNA clones were deposited into the wells on a PicoTiter- Plate (454 Life Sciences, Branford, CT, USA) for pyrosequencing on a 454 GS Junior System (Roche, Basel,Switzerland). Sequence reads data were archived at NCBI Sequence Read Archive (SRA) with accession number: SRP113304.
Bioinformatics
Bioinformatic analysis of NGS data was performed using the Microbial Genomics module (version 1.3) work-flow of the CLC Genomics workbench (version 9.5.1). After quality trimming based on quality scores (quality nucleotide limit 0.05), trim of ambiguous nucleotides (n = 2) and length trimming, reads were clustered at 97% level of similarity into Operational Taxonomical Units (OTUs). Chimera detection and removal was performed by the kmer searches pipeline of the Microbial Genomics module (version 1.3). Ribosomal RNA gene reads were classified against the non-redundant version of the SILVA SSU reference taxonomy (release 119; http://www.arb-silva.de). Alpha diversity was calculated by rarefaction analyses of diversity measures (number of total OTUs and bias-corrected form of Chao1 index) using the CLC software. Bray-Curtis dissimilarity distance between each pair of samples were also calculated and Beta diversity was estimated by applying principal coordinate analysis (PCoA) on the resulting distance matrix.
Results
General Sequencing Results
A total of 408.783 amplicons were sequenced spanning the V6 hypervariable region of bacterial rDNA gene from haemolpymph and digestive gland of C. gigas and M. galloprovincialis. To minimize bias associated with random sequencing errors, raw sequences were trimmed by eliminating reads that contained one or more ambiguous bases, had errors in the barcode or primer sequence, were atypically short (70 bp), and had an average quality score <30 [27]. This process reduced the size of the whole dataset by ca 5%. To assess most abundant members of the microbiota, phylogenetic identity of generated sequences from bivalve samples were analysed using the CLC Microbial Genomics Module (v.9.5.1). Trimmed reads were first clustered at 97% similarity resulting in a total of 3.476 operational taxonomical units (OTUs). Singletons (OTUs with a single read in the data set) were excluded from the analysis. OTUs sequences were BLASTed against SILVA reference database of nearly 227.000 high quality rDNA genes for the prokaryotic kingdoms as described in the method session. The results of BLASTN were used to estimate the taxonomic content of the data set, using SILVA taxonomy with the CLC software. Only reads occurring at least five times in the trimmed data set were assigned to bacterial taxa and included in the results. Following these steps classification against SILVA reference database assigned 233.998 reads to the domain Bacteria while 65.982 remained unassigned.
Comparison of C. gigas and M. galloprovincialis Microbiota
In general, the composition of the bacterial community in both hemolymph and digestive gland of C. gigas and M. galloprovincialis was dominated by few OTUs accounting for the majority of reads in the analyzed samples (e.g., on average, the three most abundant OTUs, excluding non-classified phylotypes, accounted for >40% of total bacterial diversity).
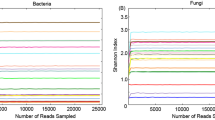
Alpha-diversity metrics (e.g., number of total OTUs and bias-corrected form of Chao1 index) indicated that bacterial diversity and richness were higher in the haemolymph compared to the digestive gland in both bivalve species (Fig. 1). Although rarefaction curves computed for total OTUs abundance failed to reach a plateau (indicating that more sequencing effort should be required to detect additional phylotypes), rarefaction curves calculated for Chao1 index and other comparable biodiversity indices (e.g., Simpson’s index and Shannon entropy, data not shown) reached a stable value suggesting that sequencing depth was good enough to measure and compare alpha-diversity metrics in all samples (Fig. 1).

Rarefaction curves for alpha-diversity metrics (number of total OTUs (a) and bias-corrected form of Chao1 index (b)) calculated from 16rDNA gene-based profiling analysis of the bacterial community in seawater and bivalve tissues (SW = seawater, HM = M. galloprovincialis haemolymph, HO = C. gigas haemolymph, GM = M. galloprovincialis digestive gland, GO = C. gigas digestive gland, DEP = depurated samples)
In the haemolymph, bacterial diversity was higher in C. gigas than in M. galloprovincialis and was dominated by the genera Pseudoalteromonas (~24 and ~25% relative abundance in C. gigas and M. galloprovincialis, respectively) and Vibrio (~24 and ~14% relative abundance in C. gigas and M. galloprovincialis, respectively) (Fig. 2). The Vibrio genus also dominated the composition of the bacterial community in the digestive gland, with higher relative abundance in C. gigas (~36%) than M. galloprovincialis (~28%) (Fig. 2). In contrast, bacteria belonging to the genus Pseudoalteromonas accounted for a smaller fraction of the bacterial community in the digestive gland in both bivalve species (on average less than 8% of total bacterial abundance). Interestingly, the bacterial community associated with the digestive gland of M. galloprovincialis was dominated by the genus Desulfovibrio (a bacterial genus not detected in C. gigas samples), that showed a relative abundance greater than 40% in these samples (Fig. 2). An additional large fraction of bacterial communities was represented by unclassified OTU sequences representing on average 20.1 ± 6.5% of the total bacteria diversity (Fig. 2).

Relative abundances of bacterial genera found in seawater and bivalve tissues by 16rDNA gene- based profiling analysis (SW = seawater, HM = M. galloprovincialis haemolymph, HO = C. gigas haemolymph, GM = M. galloprovincialis digestive gland, GO = C. gigas digestive gland, DEP = depurated samples)
Bacterial community composition of ambient seawater differed substantially from bivalve bacterial communities. In the water compartment, unclassified phylotypes made up a significant fraction of the microbial community composition (38%), followed by the phylum Cyanobacteria (28%) and bacteria belonging to the Rhodospirillaceae (14%) family (Fig. 2). In contrast, dominant phylotypes observed in bivalve tissues represent only a minor fraction of the water microbial community (e.g., 2.5 and 0.3% relative abundance for Vibrio and Pseudoalteromonas, in seawater, respectively) (Fig. 2).
Distinct bacterial communities in seawater and bivalve tissues was also evident from the analysis of beta-diversity (Fig. 3). Beta-diversity analysis also showed that the composition of the bacterial community was very similar in the haemolymph of C. gigas and M. galloprovincialis, whereas differences were observed in the bacterial community composition of digestive gland in the two bivalve species (Fig. 3).

3D PCoA plots of beta diversity for seawater and bivalve samples calculated using Principal Coordinate Analysis (PCoA) applied on Bray-Curtis distance matrix (SW = seawater, HM = M. galloprovincialis haemolymph, HO = C. gigas haemolymph, GM = M. galloprovincialis digestive gland, GO = C. gigas digestive gland, DEP = depurated samples)
Effect of Depuration on C. gigas and M. galloprovincialis Microbiota
The composition and diversity of the bacterial community were also evaluated in haemolymph and digestive gland of specimens of C. gigas and M. galloprovincialis collected after commercial depuration. In general, depurated bivalve samples showed lower diversity and richness of associated bacterial populations when compared with non-depurated samples, as clearly showed by alpha-diversity metrics (Fig. 1). In addition, both hemolymph and digestive gland samples from depurated bivalves clustered together in PCoA analysis, indicating a high level of similarity of bacterial diversity in these samples that differed significantly from beta-diversity metrics observed for non-depurated samples (Fig. 3).
A major shift in bivalve associated bacterial community structure following depuration practices regarded Vibrio populations that were found to significantly increase both in C. gigas and M. galloprovincialis tissues, with the only exception of C. gigas digestive gland (Fig. 2). In the hemolymph, the relative abundance of these bacteria increased from 24% to 42% in C. gigas and from 14 to 56% in M. galloprovincialis. A significant increase in the relative contribution of Vibrio to the whole bivalve bacterial community was also observed in the digestive gland of M. galloprovincialis, where this bacteria increased from 29 to 54% following depuration.
Pseudoalteromonas, the dominant bacterial genus in the hemolymph of the two bivalve species was found to significantly decrease in M. galloprovincialis following depuration (relative abundance in the haemolymph from 25 to 9% before and after depuration, respectively) while it remained fairly constant in C. gigas samples (relative abundance in the haemolymph from 24 to 29% before and after depuration, respectively) (Fig. 2). Other major effects of depuration observed on bivalve microbiota were linked to a significant increase in the relative abundance of the genus Stenotrophomonas in the digestive gland of both C. gigas (relative abundance from <1 to 23% before and after depuration, respectively) and M. galloprovincialis (relative abundance from <1 to 26% before and after depuration, respectively) and to the disappearance of Desulfovibrio (relative abundance from 43 to 0% before and after depuration, respectively) in M. galloprovincialis digestive gland (Fig. 2).
Indigenous Potential Pathogenic Vibrio Species in C. gigas and M. galloprovincialis Tissues
Considering that the Vibrio genus represents a dominant group in bivalve tissues and play a role in bivalve disease, the presence of potential pathogenic Vibrio species, was investigated by real-time PCR in all samples (Table 1). The presence of Vibrio species pathogenic for humans and the fecal indicator E. coli were also assessed (Table 1).
The bivalve pathogen V. aestuarianus was found in oyster haemolymph, but not in oyster digestive gland and in M. galloprovincialis samples. V. coralliilyticus, another potential bivalve pathogen, albeit found in seawater, was not detected in bivalve tissues. In contrast, bacteria belonging to the Vibrio splendidus clade, which includes a number of potential pathogenic species for bivalves (e.g., Vibrio splendidus and Vibrio crassostrea), were consistently found in C. gigas and M. galloprovincialis tissues at concentration ranging from 9.2 × 103 to 6.4 × 104 genomic unit (GU)/g (Table 1). Human pathogenic vibrio species V. cholerae, V. parahaemolyticus, and V. vulnificus were never detected in bivalve samples.
Depuration practices did not reduce the concentration of indigenous pathogenic vibrios (Table 1). In contrast, V. aestuarianus concentration increased from 2.4 × 102 cell/mL to 6.6 × 102 GU/g in the haemolymph of C. gigas following depuration. Likewise, concentration of bacteria belonging to the V. splendidus clade increased up to 3-fold in depurated samples as compared with non-depurated ones. The indicator bacteria E. coli used as control of the efficiency of depuration was only found in non-depurated bivalves (6.3 × 102 GU/g and 8.7 × 102 GU/g in digestive gland of C. gigas and M. galloprovincialis, respectively).
Discussion
Comparison of C. gigas and M. galloprovincialis Microbiota
In this work, for the first time to our knowledge, the microbiota structure in haemolymph and digestive gland of two contrasting bivalve species (C. gigas and M. galloprovincialis) that show different susceptibility to microbial diseases was evaluated by 16SrDNA gene amplification and pyrosequencing. Such comparison was made possible by taking advantage of co-cultivation of oysters and mussels in a Mediterranean fish farm where both species are grown under the same environmental conditions. The study was carried out during the warmest month of the year (August, SST 26.7 °C at the time of sampling), when susceptibility to diseases by bivalve species is generally highest.
Microbial communities associated with bivalves were significantly different from those of seawater, indicating the existence of a host-specific microbiota in bivalve tissues. Surprisingly, the microbiota associated to the haemolymph of the two bivalve species showed a high degree of similarity and it was dominated by the genera Vibrio and Pseudoalteromonas, both accounting on average for more than a third of the total bacterial community. These genera were reported in previous studies to represent dominant members of oyster microbiota [8, 28,29,30]. This not only indicates that these genera are able to persist in the haemolymph environment, and therefore to survive the antibacterial activity of hemolymph components, but also suggests that they might also contribute to bivalve antimicrobial defence for example through the production of antimicrobial compounds. Accordingly, Pseudoalteromonas species are generally found in association with marine eukaryotes and display anti-bacterial, bacteriolytic, agarolytic, and algicidal activities [31]. Vibrios are also commonly found in association with aquatic plants and animals, to which they may provide a chemical defence for the host [32]. However, the Vibrio genus also include bivalve pathogenic species, some of which are known to be associated with abnormal mortality outbreaks in farmed animals (e.g. V. aestuarianus and V. splendidus clade in farmed oysters). Interestingly, vibrios accounted for a larger fraction of the haemolymph microbiota in C. gigas compared to M. galloprovincialis (on average 1.7-fold), and the pathogen V. aestuarianus was detected by PCR in the hemolymph of C. gigas, but not in that of M. galloprovincialsis (Table 1). This is in line with the lower susceptibility of mussels, with respect to oysters, to pathogenic Vibrio species also in relation to the presence in mussels of specific hemolymph soluble components (MgEP) that mediate adhesion, internalization and killing of V. aestuarianus by the hemocytes [18, 19, 33]. Overall, these results indicate that Vibrio might play a contrasting role in health and disease of different bivalve species.
In contrast to hemolymph, microbial communities in digestive gland displayed a more diversified composition in the two bivalves (Fig. 2). Vibrio still represented a significant fraction of the microbiota, whilst bacteria belonging to the Pseudoalteromonas genus were far less abundant in this tissue. As found in the haemolymph, Vibrio contribution to the overall microbial community structure was greater in C. gigas than in M. galloprovincialis, further supporting the hypothesis that the oyster environment may provide better conditions for survival of these bacteria. From a functional perspective, the bivalve digestive gland plays a key role in the intracellular and extracellular digestion and storage of nutrients [34], and bacteria associated with this tissue may also contribute to this role. In this light, Desulfovibrio found in association with the mussel digestive gland is of particular interest, as bacteria belonging to this genus are capable to conduct sulphate respiration under strict anaerobic conditions [35]. Sulphate-reducing bacteria (SRB) are widespread in anaerobic environments, including the gastrointestinal tract of humans and other animals [35] (anaerobic conditions may establish in bivalve grow-out bags especially when bivalve are cultivated at high density as in the case of mussels). A potential role of these bacteria in anaerobic energy metabolism of M. galloprovincialis might thus be envisaged. In particular, they may contribute to the well-known capability of mussels to tolerate extended periods of hypoxia and anoxia, which may be induced by shell valve closure during emersion, depletion of oxygen in the surrounding water and other stressful conditions [36]. However, this remains a speculation and studies are in progress to confirm such observations and evaluate the role of SRB in mussel metabolism. It is finally worth noting that an additional large fraction of the hemolymph and digestive gland microbiota in the two bivalves is represented by unidentified microorganisms and rare taxa the role of which is yet to be assessed.
Effect of Depuration on C. gigas and M. galloprovincialis Microbiota
Depuration is a process by which bivalves are held in tanks of clean seawater under conditions which maximize the natural filtering activity enabling the removal of microbial and other contaminants before commercialization [37]. This process is used after collection and prior to commercialization of farmed bivalves resulting in a major decrease of coliforms and other transient bacteria in bivalve tissues. Albeit it is generally accepted that depuration practices are effective in removing allochthonous bacteria (e.g., enteric microorganisms) they are generally considered only partly effective, or ineffective, in removing other microbial contaminants such as naturally occurring marine vibrios [37].
Accordingly, the results of Real time-PCR indicate that E. coli, generally utilized as a marker of faecal contamination [38, 39], was completely removed from mussel and oyster tissues following depuration (Table 1). In contrast, indigenous Vibrio species (e.g., Vibrio aestuarianus and V. splendidus clade) were unaffected by the depuration treatment (Table 1). Although the existence of a host-specific microbiota in bivalves is still questioned, Vibrio species were found to represent a dominant fraction of both C. gigas and M. galloprovincialis microbiota (see previous paragraph). This observation suggests that these bacteria probably have a long co-evolutionary history with their invertebrate hosts, probably representing permanent residents of the microbiota, which in turn may lead to an increased resistance (persistence) to depuration. Interestingly, bacteria belonging to the Vibrio genus not only were unaffected by depuration practice but also tend to increase in abundance in depurated oysters and mussels when compared to non-depurated ones. Such findings might be better interpreted if we consider the more general influence that depuration had on the bivalve microbiota structure. In fact, the most prominent effect of depuration was a significant loss of microbial diversity in all samples from both bivalve species (Fig. 1). Phylotype loss was particularly evident for rare microbial taxa and, in turn, lead to a re-organization of dominant bacterial genera within the bivalve microbiota (Fig. 2). The increased dominance of bacteria belonging to the Vibrio genus within the overall bacterial community associated to both haemolymph and digestive gland of oysters and mussels may be probably related to new environmental niches that are made available through microbiota re-organization following depuration. Such an event is in line with the opportunistic nature of these bacteria, as also observed in other marine invertebrates for Vibrio pathogenic species during infection. For example, alteration of the coral microbiome under environmental stressful conditions, leading to decrease in dominant holobiont members such as the genus Endozoicomonas, has been linked to coral susceptibility to disease and infection by the coral pathogen Vibrio coralliilyticus [6, 40]. Similarly to vibrios, other bacterial groups such as the genus Stenotrophomonas, previously observed in association with oysters [41], significantly increased in dominance in the digestive gland of C. gigas and M. galloprovincialis following depuration. In contrast, bacterial group such as Desulfovibrio completely disappeared in M. galloprovincialis digestive gland probably in relation with the changing oxygen environment in depuration tanks.
Overall, these results show that depuration significantly affects bivalve microbiota. However, since this process may also favour opportunistic members of the bacterial community such as vibrios, this practice results ineffective in the removal of these indigenous pathogens from bivalve tissues. Alternative or additional protocols of depuration might thus be developed to challenge the threat posed by these pathogens in edible bivalves. For instance, settings of environmental conditions in depuration plants favoring hemolymph-mediated bacterial killing (e.g., increasing the production of serum opsonins) [19] and phage therapy [42] have been proposed. However, none of these practices has yet been tested and proved to be successful in real aquaculture depuration facilities.
Conclusions
The results obtained in this work provide some points of novelty. First, they represent to our knowledge the first description of 16SrDNA gene-based microbiota profiles of the bivalve M. galloprovincialis. They also provide the first data on the comparison of microbiota profiles in two aquacultured bivalves (C. gigas and M. galloprovincialis) showing different susceptibility to microbial diseases. They finally represent the first NGS data on the effect of depuration on the bivalve microbiota.
The results underline both similarities and differences in the composition of the microbiota in different tissues (hemolymph and digestive gland) of oyster and mussels. In general bivalve bacterial community appeared to be host-specific and may have contrasting role (e.g., Vibrio spp.) in health and disease of bivalve species. Depuration was found to significantly affect bivalve microbiota; however, it must be taken into account that this process may also favour opportunistic members of the bacterial community such as vibrios. Overall, understanding microbiota-host interactions in farmed bivalves remains an open challenge with the potential to bring new essential knowledge in the field of biology and control of bivalve infectious diseases.
References
Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. (2012) Human gut microbiome viewed across age and geography. Nature 486:222–227
Yarza P, Yilmaz P, Pruesse E, Glockner FO, Ludwig W, Schleifer K-H, et al. (2014) Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol 12:635–645
Lederberg J, McCray AT (2001) ‘Ome sweet’ omics – a genealogical treasury of words. The. Scientist 15:8
Sweet MJ, Bulling MT (2017) On the importance of the microbiome and Pathobiome in coral health and disease. Front Mar Sci. doi:10.3389/fmars.2017.00009
Olson JB, Kellogg CA (2010) Microbial ecology of corals, sponges, and algae in mesophotic coral environments. FEMS Microbiol Ecol 73(1):17–30
Vezzulli L, Pezzati E, Huete-Stauffer C, Pruzzo C, Cerrano C (2013) 16SrDNA pyrosequencing of the Mediterranean gorgonian Paramuricea Clavata reveals a link among alterations in bacterial Holobiont members, anthropogenic influence and disease outbreaks. PLoS One 8(6):e67745
Lokmer A, Wegner KM (2015) Hemolymph microbiome of Pacific oysters in response to temperature, temperature stress and infection. ISME J 9:670–682
Garnier M, Labreuche Y, Garcia C, Robert M, Nicolas JL (2007) Evidence for the involvement of pathogenic bacteria in summer mortalities of the Pacific oyster Crassostrea gigas. Microb. Ecol. 53:187–196
Wendling CC, Batista FM, Wegner MK (2014) Persistence, seasonal dynamics and pathogenic potential of Vibrio communities from Pacific oyster hemolymph. PLoS One 9:e94256
King GM, Judd C, Kuske CR, Smith C (2012) Analysis of stomach and gut microbiomes of the eastern oyster (Crassostrea virginica) from coastal Louisiana, USA. PLoS One 7:e51475
Trabal Fernandez N, Mazon-Suastegui JM, Vazquez-Juarez R, Ascencio-Valle F, Romero J (2013) Changes in the composition and diversity of the bacterial microbiota associated with oysters (Crassostrea corteziensis, Crassostrea gigas and Crassostrea sikamea) during commercial production. FEMS Microbiol Ecol 88:69–83
Wegner KM, Volkenborn N, Peter H, Eiler A (2013) Disturbance induced decoupling between host genetics and composition of the associated microbiome. BMC Microbiol 13:252
Lokmer A, Kuenzel S, Baines JF, Wegner KM (2016) The role of tissue-specific microbiota in initial establishment success of Pacific oysters. Environ Microbiol 18(3):970–987
Samain JF, McCombie H (eds.) (2008) Summer mortality of Pacific oyster Crassostrea gigas, the Morest project. Ifremer/Quæ Éditions, Versailles,
Pernet F, Lagarde F, Le Gall P, D’Orbcastel ER (2014) Associations between farming practices and disease mortality of Pacific oyster Crassostrea gigas in a Mediterranean lagoon. Aquac Environ Interact 5:99–106
Labreuche Y, Soudant P, Goncalves M, Lambert C, Nicolas JL (2006) Effects of extracellular products from the pathogenic vibrio aestuarianus strain 01/32 on lethality and cellular immune responses of the oyster Crassostrea gigas. Dev Comp Immunol 30:367e79
Craft JA, Gilbert JA, Temperton B, Dempsey KE, Ashelford K, Tiwari B, et al. (2010) Pyrosequencing of Mytilus Galloprovincialis cDNAs: tissue-specific expression patterns. PLoS One 5(1):e8875
Pezzati E, Canesi L, Damonte G, Salis A, Marsano F, Grande C, et al. (2015) Susceptibility of Vibrio aestuarianus 01/032 to the antibacterial activity of Mytilus hemolymph: identification of a serum opsonin involved in mannose-sensitive interactions. Environ Microbiol 17(11):4271–4279
Canesi L, Grande C, Pezzati E, Balbi T, Vezzulli L, Pruzzo C (2016) Killing of Vibrio cholerae and Escherichia coli strains carrying D-mannose-sensitive ligands by Mytilus hemocytes is promoted by a multifunctional hemolymph serum protein. Microb Ecol 72(4):759–762
Penders J, Vink C, Driessen C, London N, ThiisC SEE (2005) Quantification of Bifidobacterium spp., Escherichia coli and Clostridium difficile in fecal samples of breast-fed and formula-fed infants by real time PCR. FEMS Microbiol Ecol 24:141–147
Luna GM, Dell'Anno A, Pietrangeli B, Danovaro R (2012) A new molecular approach based on qPCR for the quantification of fecal bacteria in contaminated marine sediments. 157(4):446–453
IFREMER report (2013) Vibrio splendidus et V. aestuarianus detection by real time polymerase chain reaction. European union reference laboratory for molluscs diseases. Edition n 1, Laboratoire de Génétique et Pathologie des Mollusques Marins, Av. de Mus de Loup, 17390 La Tremblade France. URL http://www.eurlmollusc.eu/content/download/72924/948279/file/Vsplendidus&aestuarianus%20_RealTimePCR.pdf
Wilson B, Muirhead A, Bazanella M, Huete-Stauffer C, Vezzulli L, Bourne DG (2013) An improved detection and quantification method for the coral PathogenVibrio coralliilyticus. PLoS One 812:e81800
Vezzulli L, Stauder M, Grande C, Pezzati E, Verheye HM, Owens NJP, et al. (2015) gbpA as a novel qPCR target for the species-specific detection of vibrio cholerae O1, O139, non-O1/non-O139 in environmental, stool, and historical continuous plankton recorder samples. PLoS One 10(4):e0123983
Campbell MS, Wright AC (2003) Real-time PCR analysis of Vibrio vulnificus from oysters. Appl Environ Microbiol 69(12):7137–7144
Nordstrom JL, Vickery MC, Blackstone GM, Murray SL, DePaola A (2007) Development of a multiplex real-time PCR assay with an internal amplification control for the detection of total and pathogenic Vibrio parahaemolyticus bacteria in oysters. Appl Environ Microbiol 73(18):5840–5847
Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR (2006) Microbial diversity in the deep sea and the underexplored ‘rare biosphere’. Proc Natl Acad Sci U S A 103:12115–12120
Prieur D, Nicolas JL, Plusquellec A, Vigneulle M (1990) Interactions between bivalve mollusks and bacteria in the marine-environment. Oceanogr Mar Biol 28:277–352
Olafsen JA, Mikkelsen HV, Glaever HM, Hansen GH (1993) Indigenous bacteria in hemolymph and tissues of marine bivalves at low-temperatures. Appl Environ Microbiol 59:1848–1854
Pruzzo C, Gallo G, Canesi L (2005) Persistence of vibrios in marine bivalves: the role of interactions with haemolymph components. Environ Microbiol 7(6):761–772
Holmström C, Kjelleberg S (1999) Marine Pseudoalteromonas species are associated with higher organisms and produce biologically active extracellular agents. FEMS Microbiol Ecol 30(4):285–293
Engel S, Jensen PR, Fenical W (2002) Chemical ecology of marine microbial defense. J Chem Ecol 28(10):1971–1985
Balbi T, Fabbri R, Cortese K, Smerilli A, Ciacci C, Grande C, et al. (2013) Interactions between Mytilus galloprovincialis hemocytes and the bivalve pathogens Vibrio aestuarianus 01/032 and Vibrio splendidus LGP32. Fish Shellfish Immunol 35(6):1906–1915
Bayne BL, Bayne CJ, Carefoot TC, Thompson RJ (1976) The physiological ecology of Mytilus californianus Conrad. 1. Metabolism and energy balance. Oecologia 22:211–228
Matias PM, Pereira IAC, Soares CM, Carrondo MA (2005) Sulphate respiration from hydrogen in Desulfovibrio bacteria: a structural biology overview. Prog Biophys Mol Biol 89:292–329
Wang WX, Widdows J (1993) Metabolic responses of the common mussel Mytilus edulis to hypoxia and anoxia. Mar Ecol Prog Ser 95:205–214
Lee R, Lovatelli A, Ababouch L (2008) Bivalve depuration: fundamental and practical aspects. FAO Fisheries Technical Paper. No. 511. FAO, Rome, p 139
Di Cesare A, Eckert EM, Teruggi A, Fontaneto D, Bertoni R, Callieri C, et al. (2015) Constitutive presence of antibiotic resistance genes within the bacterial community of a large subalpine lake. Mol Ecol 24(15):3888–3900
Di Cesare A, Pasquaroli S, Vignaroli C, Paroncini P, Luna GM, Manso E, et al. (2014) The marine environment as a reservoir of enterococci carrying resistance and virulence genes strongly associated with clinical strains. Environ Microbiol Rep 6(2):184–190
Neave MJ, Apprill A, Ferrier-Pagès C, Voolstra CR (2016) Diversity and function of prevalent symbiotic marine bacteria in the genus Endozoicomonas. Appl Microbiol Biotechnol 100(19):8315–8324
Chauhan A, Green S, Pathak A, Thomas J, Venkatramananc R (2013) Whole-genome sequences of five oyster-associated bacteria show potential for crude oil hydrocarbon degradation. Genome Announc 1(5):e00802–e00813
Richards GP (2014) Bacteriophage remediation of bacterial pathogens in aquaculture: a review of the technology. Bacteriophage 4(4):e975540
Acknowledgements
We wish to thank the director and staff of Coop. Mitilicoltori Spezzini A.R.L. (La Spezia, Italy) for their invaluable collaboration during the sampling activity and for kindly allowing the present study. We are particularly indebted to Prof. Luigi Pane and Dr. Guido Bonello (University of Genoa) for helpful assistance during bivalve sampling. We are also kindly grateful to Dr. Adriana Amaro (University of Genoa) for precious help with pyrosequencing analysis. This work was supported by the HORIZON2020 project “Preventing and mitigating farmed bivalve disease—VIVALDI (grant number 678589)”.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Table S1
(DOCX 16 kb)
Rights and permissions
About this article

Cite this article
Vezzulli, L., Stagnaro, L., Grande, C. et al. Comparative 16SrDNA Gene-Based Microbiota Profiles of the Pacific Oyster (Crassostrea gigas) and the Mediterranean Mussel (Mytilus galloprovincialis) from a Shellfish Farm (Ligurian Sea, Italy). Microb Ecol 75, 495–504 (2018). https://doi.org/10.1007/s00248-017-1051-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-017-1051-6