
Abstract
Gastrointestinal microbiota is increasingly recognized as an important component of individual health, and therefore, our ability to quantify its diversity accurately is central for exploring different ways to improve health. Non-invasive sampling methods, such as cloaca swabs, are often used to measure gastrointestinal microbiota diversity within an individual. However, few studies have addressed to what degree differences exist in microbial community composition along the gastrointestinal tract, and measures obtained from the cloaca may not actually represent the diversity present elsewhere in the gastrointestinal tract. In this study, we systematically characterized the gastrointestinal microbial community of the critically endangered Attwater’s Prairie chicken (Tympanuchus cupido attwateri) by opportunistically sampling four different locations (ileum, cecum, large intestine, and cloaca) along the gastrointestinal tract of eight individuals. Spatial variation of microbial community was observed at different sampling locations within the gastrointestinal tract. The cecum harbored the most diverse and significantly different microbiota from the other locations, while the microbial α- and β-diversities were similar in the ileum, large intestine, and cloaca. The results of this study provide evidence that microbiota diversity can differ depending on sampling location and metric used to quantify diversity. As shown here, non-invasive cloacal sampling strategies may reflect microbiota diversity elsewhere in the gastrointestinal tract, yet caution is warranted when making generalizations in terms of the microbiota diversity correlations when samples are obtained from a single location within the gastrointestinal tract.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The gastrointestinal tract harbors a complex ecosystem of microbes that are essential to the health and wellness of the host [1–3]. These normal resident microbes comprise the “gut microbiome” and possess many vital metabolic capabilities that the host cannot otherwise perform such as synthesis of essential vitamins and short-chain fatty acids, as well as breakdown of host-indigestible material such as complex plant polysaccharides and other products [4, 5]. The host gut microbiome also affects a multitude of physiological properties, such as energy balance, pH, synthesis of vitamin K1 used in anabolic pathways, immune function, degradation of mucus produced by host goblet cells, and protection against pathogens [6–8]. The microbiome of the gastrointestinal tract reflects a co-evolutionary relationship with the host, but it can also be influenced by restricted migration of microorganisms, host diet, genotype, age, gender, antibiotic administration, and colonization history [3, 9].
Spatial variation in bacterial community structure has been demonstrated along the length of the gastrointestinal tract in humans [10], felines [5], chickens [8, 11, 12], quail [13], and pigs [14]. This is not surprising since there are distinctly different functions along the gastrointestinal tract of vertebrates and the microenvironment changes with different functions. For instance, the cecum is a unique morphological structure of the gastrointestinal tract, consisting of two blind pouches located at the junction of the small and large intestines [15] where fermentation of plant matter occurs, especially in herbivores and birds such as many galliformes that feed on plant material [16]. By contrast, the ileum is the last section of the small intestine, which typically maintains a more neutral pH and is responsible for absorption of the remaining products of digestion.
The gastrointestinal tract of the domestic chicken (Gallus gallus domesticus) has been extensively studied. Prior to molecular-based sequencing approaches, the dominant culturable bacteria in chicken ceca were found to contain primarily obligate anaerobes [17, 18], while facultatively anaerobic bacteria were greater in the ileum [18]. Later examination of partial 16S rRNA gene sequences determined a significant difference between bacterial communities in the ileum and cecum of broiler chickens consisting of primarily Lactobacillus (70 %) and Clostridiaceae (65 %) species, respectively [8].
Therefore, it is important to recognize that sampling biases may exist among studies investigating a correlation between microbiome diversity and fitness or behavior [1, 19–21]. For example, a study investigating the relationship of microbial composition to host fitness based upon a cloaca sampling strategy may yield vastly different results compared to a similar study derived from sampling of the ileum or cecum where the microbial diversity and composition may be substantially different. An increasing number of studies have reported microbial diversity levels for samples collected at the cloaca of birds [22], largely because such sampling is non-invasive; however, to what degree the cloaca diversity differs from other regions of the gastrointestinal tract deserves further investigation [23].
Here, we quantify and compare gastrointestinal tract microbial diversity based upon sampling location within the critically endangered Attwater’s Prairie chicken (Tympanuchus cupido attwateri). The Attwater’s Prairie chicken has experienced a significant decline in abundance over the past 100 years from a population of over one million individuals to fewer than 100 in the wild today [24]. Significant efforts have been made to prevent the extinction of the Attwater’s Prairie chicken, including a captive breeding and release program initiated in 1992. In an effort to identify factors limiting survivorship, we were interested to determine if cloacal microbial diversity correlated with survival among individuals. However, it was not known to what degree microbial diversity differs depending on gastrointestinal tract sampling location in the Attwater’s Prairie chicken. In this study, we investigate whether differences exist in the microbial community composition based on sequenced bacterial 16S rRNA gene libraries generated from samples obtained from the ileum, cecum, large intestine, and cloaca of eight individuals. Our findings provide valuable information about microbial diversity along the gastrointestinal tract of Attwater’s Prairie chicken and further insight on additional research required to investigate gut microbial diversity and fitness.
Materials and Methods
Sample Collection
A total of 32 samples were obtained from four sampling locations within the gastrointestinal tract: ileum, cecum, large intestine, and cloaca among the eight individual captive-born Attwater’s Prairie chickens at the Abilene Zoo. Five female and three male birds were available for opportunistic sampling because of required culling due to space limitations. Age varied from 13 to 74 months of age (mean ± std dev = 27.3 ± 22.8 months; Table S1), but all were fed the same diet and maintained in similar environments. A cloaca sample from each bird was obtained using a sterile polyester-tipped applicator swab prior to euthanasia, and a staff veterinarian obtained the samples from the ileum, cecum, and large intestine at approximately the same location at each region using a sterile swab immediately post mortem. Each sample was immersed in TE buffer and placed on ice before storage at −80 °C prior to DNA extraction.
DNA Isolation
The FastDNA™ SPIN Kit for Soil (MP Biomedicals) was used to extract DNA from each sample following the manufacturer’s protocol. Briefly, the samples were thawed on ice, vortexed, and centrifuged at 6000×g for 3 min, and 300 μl of the supernatant was then removed to allow for space in the lysing matrix tube. Sodium phosphate buffer was added to maximize recovery of DNA. Each sample was homogenized using a FastPrep®-24 Instrument (MP Biomedicals). Two ethanol washes were used, and DNA was eluted in 100 μl of nuclease-free molecular biology-grade water after a 5-min incubation at 55 °C. DNA was quantified using a NanoDrop-1000 spectrophotometer (ThermoFisher Scientific Inc., Waltham, MA, USA).
16S rRNA Gene Amplicon Library Preparation
16S rRNA gene amplicon library for each sample was constructed by amplifying the V4 hypervariable region with universal bacterial primers as previously described [25]. The forward primer was barcoded with a 10-base code unique to each sample following the IonXpress barcode design (Life Technologies, Carlsbad, CA, USA). Each sample was prepared in triplicate, 25-μl reactions, using 20 ng of original DNA template, 0.5 μM of each barcoded forward primers IonA-515F and IonP1-806R reverse primer, 0.5U Phusion® High Fidelity DNA Polymerase (New England BioLabs®), 5 μl of 5X Phusion® HF reaction buffer, 200 μM of dNTPs, and 3 % (vol:vol) DMSO. The cycling conditions used were 98 °C for 3 min, followed by 30 cycles of 98 °C for 10 s, and 52 °C for 15 s, 72 °C for 15 s, with a final extension time of 5 min at 72 °C. PCR products were examined following electrophoresis on a 1 % agarose gel, and then, the remaining volumes for each replicate were pooled into one tube for each sample. Each pooled amplicon library was purified using Agencourt® AMPure® XP (Beckman Coulter) magnetic beads. Purified amplicon libraries were quantified using the Agilent DNA 7500 kit with a Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). The libraries were then diluted to the appropriate concentration according to the manufacturer’s recommendation for emulsion PCR using the Ion OneTouch™ 2 instrument (Life Technologies, Carlsbad, CA, USA).
Semiconductor Sequencing
Emulsion PCR was performed on the Ion OneTouch™ 2 instrument (Life Technologies, Carlsbad, CA, USA) using the Ion OneTouch 200 Template Kit v2 DL following the manufacturer’s instructions. The template-positive Ion Sphere particles (ISPs) were enriched using the OneTouch ES instrument. The quality of both unenriched and enriched ISPs was determined on the Qubit 2.0 fluorometer using the Ion Sphere quality control kit (Life Technologies). The enriched ISPs were loaded onto a 316 chip and sequenced on the Ion Torrent Personal Genome Machine (PGM) using the Ion Sequencing 200 kit v2 (Life Technologies) as per the manufacturer’s instructions.
Sequence Analysis
Sequences generated from the Ion Torrent PGM were processed through the mothur v.1.32.1 pipeline as previously described [26]. DNA sequences originating from the different samples were identified based on the sample-specific barcodes. Primers and barcodes were removed, and short sequences (<100 bp) and low quality sequences (average quality score <20 and homopolymers >8) were excluded from the dataset. Sequence alignments were performed using reference sequences from the SILVA database. The sequences that could not be aligned were removed, and the aligned sequences were further filtered to remove gaps. Redundant sequences were reduced using the unique.seqs command and a precluster (diffs = 2) algorithm, and chimeras were removed after identification using UCHIME [27].
Operational taxonomic units (OTUs) were assigned with the average neighbor clustering algorithm based on 97 % sequence similarity. Taxonomic classification was conducted using the Ribosomal Database Project (RDP) classifier with a minimum of 80 % confidence [28]. Sequences classified as mitochondria, chloroplast, archaea, and eukaryote, as well as unknown sequences, were removed from the data set.
To investigate differences in microbial community diversity among the sampling locations, both α-diversity and β-diversity were calculated. α-Diversity, or the diversity within communities, was compared between the gastrointestinal tract sampling locations. Diversity indices (Shannon diversity and evenness) and richness (Chao1 and abundance coverage-based estimator (ACE)) estimators were generated based on OTU grouped at 97 % sequences similarity for species-level classification [29–31]. β-Diversity, or the degree by which membership or structure is shared between microbial communities, in the different gastrointestinal tract locations was investigated using UniFrac distances and principle coordinate analysis (PCoA) [32]. Analysis of similarities (ANOSIM) was performed to assess the overall similarity among the different sampling locations. ANOSIM generates a value of R measuring how separate groups are reviewed by Clark [33]. R = 1 means significantly different; R = 0 means no difference; R > 0.75 suggests good separation; R > 0.5 indicates differences with some overlapping; and R < 0.25 means almost no differences. Diversity estimators, UniFrac, principal coordinate analysis (PCoA), analysis of molecular variance (AMOVA) analyses, and ANOSIM were performed using mothur.
A linear discriminant analysis (LDA) was performed to test whether the microbial taxa among the four sampling locations can be differentiated from each other by using the bacterial taxa as biomarkers. Bacterial families with relative abundance larger than 2 % in at least one sample were selected for this analysis. Kruskal-Wallis test and post hoc Tukey’s test (P < 0.05 with 1000 Monte-Carlo permutations) were used to screen the significantly different bacterial families among the four sampling locations. False positive discovery rate (FDR) for multiple hypothesis testing was controlled using the Benjamini-Hochberg method [34]. Analysis of variance (ANOVA) with post-hoc Tukey honestly significant difference (HSD) test, LDA, Kruskal-Wallis test, and Tukey’s test were performed using MASS and coin packages in the R program v3.1.3. The sequences were deposited in the NCBI Sequence Read Archive (SRA) under Accession PRJNA311230.
Results
Taxonomic Composition of Gastrointestinal Microbiome
After removing short low quality sequences as well as chimera sequences, a total of 1,397,097 high quality sequences with an average length of 239 bp were obtained (mean ± standard deviation 43,659 ± 13,867 per individual sample; n = 32). At 80 % confidence level for taxonomic classification, 98.4 % of those sequences were assigned to a total of 17 bacterial phyla, including Firmicutes (34.3 %), Actinobacteria (34.2 %), Proteobacteria (22.3 %), Bacteroidetes (7.5 %), and multiple rare phyla (0.1 %). A total of 92.0 % of the sequences were assigned to 113 known bacterial families, and 47.1 % to 226 known bacterial genera. The top five most abundant bacterial families were Micrococcaceae (29.8 %), Planococcaceae (7.2 %), Pseudomonadaceae (7.2 %), Enterobacteriaceae (5.4 %), and Erysipelotrichaceae (4.2 %). Sequences of Micrococcaceae and most of Enterobacteriaceae were unclassified at the genus level. A few Enterobacteriaceae, however, were identified as Escherichia/Shigella genera. Sequences of Planococcaceae, Pseudomonadaceae, and Erysipelotrichaceae were further assigned to the genera of Sporosarcina, Planococcaceae Incertae Sedis, Pseudomonas, and Turicibacter.
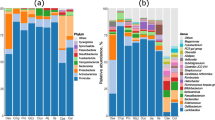
Differences in microbial community composition among the gastrointestinal sampling locations were observed at phylum-level classification (Fig. 1, Table 1). The ileum, large intestine, and cloaca were dominated by Firmicutes, Actinobacteria, and Proteobacteria, while the cecum also possessed Bacteroidetes. Heterogeneity for each individual and differences in community structure were observed at the family and genus levels (Table 1). The ileum, large intestine, and cloaca were predominated by one or two highly abundant families that accounted for 39.4 to 98.8 % of each community. Micrococcaceae was the most dominant bacterial family in half of the ileum (4/8), large intestine (4/8), and cloaca (5/8) samples. The microbial communities in the cecum were dominated by five to seven relatively high abundant (5.5–37.8 %) families including Enterobacteriaceae (13.9 % ± 13.4 %), Ruminococcaceae (12.9 % ± 7.6 %), Lachnospiraceae (10.6 % ± 4.5 %), Coriobacteriaceae (7.2 % ± 4.7 %), and Erysipelotrichaceae (3.9 % ± 6.2 %). Sequences from Ruminococcaceae were further assigned to the genus Faecalibacterium. Most sequences from Lachnospiraceae could not be classified to the genus level, whereas a few were classified to the genus Blautia. Sequences from Coriobacteriaceae were mainly classified to the genus of Collinsella and Olsenella.

Attwater’s Prairie chicken microbial community at phylum level at the four sampling locations in the gastrointestinal tract (ileum, cecum, large intestine, and cloaca). Only phyla with >1 % relative abundance are shown
Core Microbiota Shared by Sampling Location and Individual
A large number of microbial taxa were present in all the four sampling locations, referred herein as the “core microbiota.” A total of 50 microbial families and 70 genera occurred in all the four sampling locations (Fig. 2a, b). The core microbiota accounted for 99.3, 80.2, 80.2, and 84.7 % of total sequences in the ileum, cecum, large intestine, and cloaca samples, respectively (Fig. 2c). There were six microbial families with an average relative abundance >1 % in at least three locations including Micrococcaceae, Burkholderiaceae, Halomonadaceae, Planococcaceae, Streptococcaceae, and Erysipelotrichaceae. Few Micrococcaceae could be classified at the genus level with 80 % or greater confidence. With a lower confidence level (50 %), most unclassified sequences were assigned to the genus of Sinomonas (90.5 %), Kocuria (5.9 %), and Arthrobacter (3.5 %). The other shared bacterial genera were further identified as Ralstonia, Halomonas, Planococcaceae Incertae Sedis, Streptococcus, and Turicibacter.

Sharing of core microbiota families (a) and genera (b) at the four sampling locations. The number in each field depicts the number of shared taxa present in the corresponding sampling location. c Relative abundance of core microbiota at each sampling location. Bacterial families with relative abundance >1 % in at least one location are shown
Variation in microbial composition existed among individual birds depending on taxonomic level and sampling location. However, 94.6 % of the sequences from the cecum samples were assigned to 50 microbial families and 92 genera shared among the eight sampled birds. In contrast, the eight birds shared nine genera from nine families for the cloaca samples, representing 80.1 % of the total cloaca sequences. The ileum and large intestine showed more variation among the sampled birds. Only seven genera in the ileum samples from six families were observed in common among the sampled birds, representing 9.5 % of the total ileum sequences. Similarly, the eight sampled birds shared only five genera from six families representing 17.0 % of the total large intestine sequences. Sample sizes were insufficient to determine if the patterns observed were correlated with age or sex.
Microbial Diversity Comparisons
Significant differences in microbial α-diversity were observed among the sampling locations depending on the metric used to quantify diversity within each location. Differences in Shannon diversity (ANONA, P = 0.001), evenness (P = 0.023), and ACE (P < 0.001) estimates were observed among the sampling locations, while the Chao1 richness estimator was not significant (P = 0.051). Pairwise tests (Tukey’s HSD) showed that Shannon diversity and evenness estimates for the cecum samples were significantly higher than those observed in the ileum (P < 0.001), large intestine (P < 0.006), and cloaca (P < 0.003) (Fig. 3). ACE and Chao1 richness estimates in the cecum were also significantly higher than those observed in the large intestine (P = 0.019 and 0.047, respectively). However, differences in cecum microbial richness were not significant (ACE, P > 0.083; Chao1, P > 0.131) from the ileum and cloaca sampling locations. None of the diversity and richness measures were significantly different (P > 0. 415) among the pairwise comparisons between the ileum, large intestine, and cloaca sampling locations.

Microbial community diversity (a Shannon diversity; b Shannon evenness) and richness (c ACE; d Chao1) indices for the four gastrointestinal tract sampling locations (ileum, cecum, large intestine, and cloaca). *Tukey HSD P < 0.05, **P < 0.01
Microbial community β-diversity of the cecum samples clustered separately from the other sample locations based on the unweighted UniFrac distances (Fig. 4a). When the relative abundance of each microbial population was considered (i.e., weighted PCoA), the cecum microbial communities still clustered separately from the other sampling locations along PCo1 and PCo3 axis (Fig. 4b). However, considerable overlap in distribution was observed among microbial communities in the ileum, large intestine, and cloaca. AMOVA tests based on both unweighted and weighted UniFrac analyses showed that microbial community variances within the same location were greater than the variances among the different locations (P < 0.001). Further, pairwise comparisons based on both unweighted and weighted UniFrac analyses also showed that bacterial communities in the cecum were significantly different from the other three locations (P < 0.001). No significant differences were observed for microbial communities among the ileum, large intestine, and cloaca. ANOSIM tests based on both weighted and unweighted UniFrac distances also revealed clear separation between cecum microbial community and the other locations (P < 0.001), but no significant differences between the ileum, large intestine, and cloaca sampling locations (Table S2).

Principal coordinate analysis (PCoA) based on unweighted (a) and weighted (b) UniFrac distances showed that gut microbial community changed at the different gastrointestinal tract sampling locations
Since 92.0 % of the total sequences were classified at the family level, abundant bacterial families were selected for LDA analysis. In the LDA chart, the cecum and cloaca microbiota clearly separated from that of the ileum and large intestine through the first and second discriminant functions, which explained 99.67 and 0.2917 % of total variances (Fig. 5). The third discriminant function only represented 0.0383 % of total variances separated the ileum and large intestine microbiota. The results indicate that the cecum microbiota can be easily differentiated from the other sampling locations. The microbiota in the other three locations can be differentiated according to the best linear combinations of the abundant bacterial families; however, the variances between each other were comparatively small.

Linear discriminant analysis (LDA) based on abundant bacterial families. Only bacterial families with relative abundant larger than 2 % in at least one sample were used
Results of the Kruskal-Wallis test showed that twenty-three out of thirty-eight tested families had significantly different relative abundance in at least one sampling location (P < 0.05) (Fig. 6). Relative abundances of ten out of twenty-three bacterial families were significantly different in the cecum compared to those in the other three locations (Fig. 6a). There were eight bacterial families that showed significantly different relative abundances between the cecum and both ileum and cloaca (Fig. 6b). Four bacterial families had significantly different relative abundance between the cecum and cloaca, and one bacterial family, Burkholderiaceae, showed significantly difference between the cecum and the other two sample sites: large intestine and cloaca (Fig. 6c). Only two taxonomic families (Bifidobacteriaceae and Erysipelotrichaceae) had significant differences between the large intestine (P = 0.031) and cloaca (P = 0.027) (Fig. 6b, c). None of the tested bacterial families had significantly different relative abundances between the ileum and large intestine or between the ileum and cloaca.

Statistically significant changes in bacterial family abundance at the different sampling locations (P < 0.05 with 1000 Monte-Carlo permutation). Bacterial families that showed significant difference between the cecum and all the other three locations (a), ileum and cloaca (b), and cloaca or large intestine (c). Error bars indicate standard error of the mean (n = 8)
Discussion
The aim of this study was to investigate if differences in microbiota exist among the sampling locations within the gastrointestinal tract in order to ascertain whether non-invasive sampling strategies, specifically at the cloaca, are sufficient to represent microbial diversity elsewhere in the intestine (e.g., ileum, large intestine, and cecum). The cloaca is located at the end of the gastrointestinal tract after the large intestine and easily accessible for non-invasive sampling methods, making it a convenient and preferred approach when working with endangered species or when more invasive sampling strategies are undesirable. Concern exists, however, that sampling at the cloaca may not be the best approach for quantifying microbial diversity and abundance within an individual because spatial heterogeneity in microbiota among gastrointestinal regions has been observed among bird species [12, 13, 35], particularly when compared with the cecum ([36]. Our study showed that spatial variation within the microbial community does exist along the gastrointestinal tract of captive Attwater’s Prairie chickens. These results therefore suggest that employing a sampling strategy from a single location within the gastrointestinal tract may not fully represent microbial diversity or abundance in other locations. The extent of the difference is dependent upon the taxonomic level used for characterization (e.g., phylum or family) and the metric used for comparison, especially when comparing between the cecum and the other sample locations (ileum, large intestine, and cloaca). These results have important practical implications for studies interested in identifying correlations between microbiota diversity and fitness [37, 38], particularly those working to identify better management strategies to increase survival among endangered species, such as the Attwater’s Prairie chicken.
The microbial community composition is influenced by the local physiological conditions and function of the particular sampled location within the gastrointestinal tract. In this study, microbial community diversity and abundance had fewer differences among three of the four sampling locations (cloaca, ileum, and large intestine) than observed with the cecum. The ANOSIM analysis results showed that R values between the ileum, large intestine, and cloaca were all less than 0.25, indicating that microbial communities in the cloaca strongly overlapped and barely separated from the ileum and large intestine (Table S2). Further, after excluding the microbial taxa shared by all the four sampling locations, the cloaca shared more microbiota at the family and genus taxonomic levels with the large intestine (31 and 47 taxa, respectively) and ileum (27 and 37 taxa, respectively), than it did with the cecum (8 and 9 taxa, respectively; Fig. 2). The cloaca samples also possessed the majority of highly abundant bacterial families (those comprising >2 % of the total population in at least one sample) identified within the ileum and large intestine.
Although the cloaca microbial communities overlap extensively with those of the ileum and large intestine, it is important to recognize that sampling of the cloaca did not fully represent all of the taxa observed in the other two locations. Because our results were largely based on 38 highly abundant families (>2 %) that represented 94.4–99.7 % of the populations in the samples, rare taxa were found to comprise only a small proportion of the observed microbiota. Among abundant taxa, two bacterial families showed significant differences between the cloaca and the large intestine. Therefore, when quantifying rare taxa (defined here as those comprising <2 % of the total population in at least one sample by sequence count), cloaca sampling may not represent the gastrointestinal tract overall. The resident microflora may also be affected by secretions of other digestive organs located in the proximal end of the gut. In contrast to our results, Choi et al. [35] found that broiler chicken microbiota of the large intestine were clustered together with the cecum, but separate from the ileum. Interestingly, broiler chickens empty their cecal contents into the large intestine several times per day, which may result in the higher similarity between the microbial communities of the large intestine and cecum in those birds [39].
The different gastrointestinal sections have different physiological functions, which may also affect their microflora composition. The ileum is part of the lower small intestine where food nutrients are mainly absorbed, while the primary function of the avian large intestine is absorption of water and electrolytes. The avian cloaca is both an excretory and a copulatory organ; therefore, its microbial community can be affected by both the digestive system and mating behavior [40, 41]. Micrococcaceae were the most dominant and prevalent family in the ileum, cloaca, and large intestine of the sampled individual Attwater’s Prairie chicken. Micrococcaceae were similarly observed in broiler chicken ileum, but present in lower levels and suppressed by a medium chain fatty acid diet [42]. Micrococcaceae were also observed in other avian cloaca as well as eggshells [22, 43, 44]. Members of the Micrococcaceae family are capable of producing acid and spermidine, which might inhibit the growth of other bacteria including pathogens. The function of Micrococcaceae in Attwater’s Prairie chicken is not clear, and further research is needed to determine whether Micrococcaceae are commensal and beneficial bacteria.
In contrast to the digestive functions of the ileum, large intestine, and cloaca, the cecum is unique in that it is the location of carbohydrate fermentation and recycling uric acid for amino acid synthesis [45–47], which has likely influenced the dominant bacterial families observed in the cecum. Taxa within the Enterobacteriaceae are associated with hemicellulose degradation [48], and Ruminococcaceae (Faecalibacterium) and Lachnospiraceae are butyrate producers in the gut [49, 50]. Coriobacteriaceae have been associated with steroid and bile salt metabolism in the mammalian gut [2], and Collinsella are prevalent in the human gut and contribute to bile salt biotransformation [51, 52]. Olsenella are observed in the broiler chicken cecum and human subgingival plaque [53, 54].
Large variations in microbial diversity and composition among each individual was also observed in this study, especially within the ileum, large intestine, and cloaca (Fig. 3). Micrococcaceae dominated a total of four out of eight ileum and five out of eight cloaca samples; however, the other four ileum and three cloaca samples, respectively, were each dominated by different bacterial families (Table 1). Microbial composition variations were also observed in the large intestine samples, where different bacterial families dominated six of the eight individuals. Intestinal microbiota among individuals are affected by many factors including diet, age, gender, mating activities, and health conditions [13, 22, 41, 55]. In this study, the Attwater’s Prairie chickens were held in the same captive facility and fed similar diets, which likely minimized differences among individuals with respect to environment. However, it should be noted that the ages of the birds ranged from 13 to 74 months at time of sampling and both male and female birds were sampled, which may result in increased microbial variation among individuals. Inter-individual variation may also have limited our ability to identify differences between the samples obtained at the ileum, large intestine, and cloaca. Increased sampling effort would likely help control for differences due to age and sex and possibly reveal other patterns, but our ability to obtain a larger sample size was limited due to the species’ classification as an endangered species. Additional research is warranted to investigate how the gastrointestinal diversity and composition differ based on age and sex using a larger sample size than used in this study.
Several studies have suggested that the fitness of the host is linked to the microbiome in the gastrointestinal tract [1, 46, 56]. Understanding microbial community composition variation and diversity will provide valuable background knowledge for captive breeding of Attwater’s Prairie chicken and other endangered species to improve management strategies and increase survival. In this study, we defined the core microbiome at the family level among captive Attwater’s Prairie chickens, and both α- and β-diversity estimates were similar between the cloaca and other regions of the gastrointestinal tract such as in the ileum and large intestine. However, significant differences were observed with the cecum. α-Diversity measures the diversity within a sample, while β-diversity measures the distance or dissimilarity between the samples. The results from this study, therefore, suggest that non-invasive sampling techniques, such as at the cloaca, can be used to also assess family-level diversity levels elsewhere within the gastrointestinal tract, but not with the cecum. To what degree similar results exist with other species, particular in wild populations that possess a more varied diet, deserves further study. Likewise, our ability to resolve microbial taxonomic affinities depends on the length of the generated diagnostic DNA sequence, e.g., 16S rRNA, and a trade-off exists between coverage and the length of sequence produced using current short-read sequencing technologies [57]. As next-generation sequencing technologies continue to improve producing longer read lengths, more work is needed to compare microbial diversity measures at multiple locations within the gastrointestinal tract using both genus- and species-level analyses.
References
Clemente Jose C, Ursell Luke K, Parfrey Laura W, Knight R (2012) The impact of the gut microbiota on human health: an integrative view. Cell 148:1258–1270. doi:10.1016/j.cell.2012.01.035
Clavel T, Desmarchelier C, Haller D, Gérard P, Rohn S, Lepage P, Daniel H (2014) Intestinal microbiota in metabolic diseases. Gut Microbes 5:544–551. doi:10.4161/gmic.29331
Dethlefsen L, McFall-Ngai M, Relman D (2007) An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 449:811–818
Gill SR, Pop M, DeBoy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE (2006) Metagenomic analysis of the human distal gut microbiome. Science 312:1355–1359. doi:10.1126/science.1124234
Ritchie LE, Steiner JM, Suchodolski JS (2008) Assessment of microbial diversity along the feline intestinal tract using 16S rRNA gene analysis. FEMS Microbiol Ecol 66:590–598. doi:10.1111/j.1574-6941.2008.00609.x
Macpherson A, Harris N (2004) Interactions between commensal intestinal bacteria and the immune system. Nat Rev Immunol 4:478–485
Kurokawa K, Itoh T, Kuwahara T, Oshima K, Toh H, Toyoda A, Takami H, Morita H, Sharma VK, Srivastava TP, Taylor TD, Noguchi H, Mori H, Ogura Y, Ehrlich DS, Itoh K, Takagi T, Sakaki Y, Hayashi T, Hattori M (2007) Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res 14:169–181. doi:10.1093/dnares/dsm018
Lu J, Idris U, Harmon B, Hofacre C, Maurer JJ, Lee MD (2003) Diversity and succession of the intestinal bacterial community of the maturing broiler chicken. Appl Environ Microbiol 69:6816–6824. doi:10.1128/aem.69.11.6816-6824.2003
Markle JG, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, von Bergen M, McCoy KD, Macpherson AJ, Danska JS (2013) Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 339:1084–1088
Wang M, Ahrné S, Jeppsson B, Molin G (2005) Comparison of bacterial diversity along the human intestinal tract by direct cloning and sequencing of 16S rRNA genes. FEMS Microbiol Ecol 54:219–231. doi:10.1016/j.femsec.2005.03.012
Yeoman CJ, Chia N, Jeraldo P, Sipos M, Goldenfeld ND, White BA (2012) The microbiome of the chicken gastrointestinal tract. Anim Health Res Rev 13:89–99
van der Wielen PWJJ, Keuzenkamp DA, Lipman LJA, van Knapen F, Biesterveld S (2002) Spatial and temporal variation of the intestinal bacterial community in commercially raised broiler chickens during growth. Microb Ecol 44:286–293. doi:10.1007/s00248-002-2015-y
Su H, McKelvey J, Rollins D, Zhang M, Brightsmith DJ, Derr J, Zhang S (2014) Cultivable bacterial microbiota of northern bobwhite (Colinus virginianus): a new reservoir of antimicrobial resistance? PLoS One 9:e99826. doi:10.1371/journal.pone.0099826
Pryde SE, Richardson AJ, Stewart CS, Flint HJ (1999) Molecular analysis of the microbial diversity present in the colonic wall, colonic lumen, and cecal lumen of a pig. Appl Environ Microbiol 65:5372–5377
McLelland J (1989) Anatomy of the avian cecum. J Exp Zool 252:2–9. doi:10.1002/jez.1402520503
Gelis S (2006) Evaluating and treating the gastrointestinal system. Spix Publishing, Inc., Palm Beach
Barnes EM, Mead GC, Barnum DA, Harry EG (1972) The intestinal flora of the chicken in the period 2 to 6 weeks of age, with particular reference to the anaerobic bacteria. Br Poult Sci 13:311–326
Salanitro JP, Fairchilds IG, Zgornicki YD (1974) Isolation, culture characteristics, and identification of anaerobic bacteria from the chicken cecum. Appl Microbiol 27:678–687
Ruiz-RodrÍGuez M, Lucas FS, Heeb P, Soler JJ (2009) Differences in intestinal microbiota between avian brood parasites and their hosts. Biol J Linn Soc 96:406–414. doi:10.1111/j.1095-8312.2008.01127.x
Ezenwa VO, Gerardo NM, Inouye DW, Medina M, Xavier JB (2012) Animal behavior and the microbiome. Science 338:198–199
Ruiz-Rodríguez M, Soler JJ, Lucas FS, Heeb P, José Palacios M, Martín-Gálvez D, De Neve L, Pérez-Contreras T, Martínez JG, Soler M (2009) Bacterial diversity at the cloaca relates to an immune response in magpie Pica pica and to body condition of great spotted cuckoo Clamator glandarius nestlings. J Avian Biol 40:42–48. doi:10.1111/j.1600-048X.2008.04471.x
van Dongen WF, White J, Brandl H, Moodley Y, Merkling T, Leclaire S, Blanchard P, Danchin E, Hatch S, Wagner R (2013) Age-related differences in the cloacal microbiota of a wild bird species. BMC Ecol 13:11
Leticia Mirón AM, Rocha-Ramírez V, Belda-Ferre P, Cabrera-Rubio R, Folch-Mallol J, Cardénas-Vázquez R, DeLuna A, Lilia Hernández A, Maya-Elizarrarás E, and Schondube JE (2014) Gut bacterial diversity of the house sparrow (Passer domesticus) inferred by 16S rRNA sequence analysis. Metagenomics 3
Morrow ME, Rossignol TA, Silvy NJ (2004) Federal listing of prairie grouse: lessons from the Attwater’s prairie-chicken. Wildl Soc Bull 32:112–118. doi:10.2193/0091-7648(2004)32[112:FLOPGL]2.0.CO;2
Caporaso J, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R (2011) Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci 108:4516–4522
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi:10.1128/aem.01541-09
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. doi:10.1093/bioinformatics/btr381
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi:10.1128/aem.00062-07
Shannon CE, Weaver W (1949) The mathematical theory of communication. University of Illinois Press, Urbana
Chao A (1984) Nonparametric estimation of the number of classes in a population. Scand J Stat 11:265–270
Chazdon RL, Colwell RK, Denslow JS, Guariguata MR (1998) Statistical methods for estimating species richness of woody regeneration in primary and secondary rain forests of Northeastern Costa Rica. In: Dallmeier F, Comiskey JA (eds) Forest biodiversity research, monitoring and modeling: conceptual background and old world case studies. Parthenon Publishing, Paris, France., pp 285–309
Lozupone C, Knight R (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228–8235. doi:10.1128/aem.71.12.8228-8235.2005
Clarke KR (1993) Non-parametric multivariate analyses of changes in community structure. Aust J Ecol 18:117–143. doi:10.1111/j.1442-9993.1993.tb00438.x
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc Ser B (Stat Method) 57:289–300. doi:10.2307/2346101
Choi JH, Kim GB, Cha CJ (2014) Spatial heterogeneity and stability of bacterial community in the gastrointestinal tracts of broiler chickens. Poult Sci 93:1942–1950. doi:10.3382/ps.2014-03974
Rehman HU, Vahjen W, Awad WA, Zentek J (2007) Indigenous bacteria and bacterial metabolic products in the gastrointestinal tract of broiler chickens. Arch Anim Nutr 61:319–335. doi:10.1080/17450390701556817
Dillon RJ, Vennard CT, Buckling A, Charnley AK (2005) Diversity of locust gut bacteria protects against pathogen invasion. Ecol Lett 8:1291–1298. doi:10.1111/j.1461-0248.2005.00828.x
Girvan MS, Campbell CD, Killham K, Prosser JI, Glover LA (2005) Bacterial diversity promotes community stability and functional resilience after perturbation. Environ Microbiol 7:301–313. doi:10.1111/j.1462-2920.2005.00695.x
Ussery H (2011) The small-scale poultry flock: an all-natural approach to raising chickens and other fowl for home and market growers. Chelsea Green Publishing, Chelsea
Stewart R, Rambo TB (2000) Cloacal microbes in house sparrows. Condor 102:679–684. doi:10.1650/0010-5422(2000)102[0679:CMIHS]2.0.CO;2
White J, Mirleau P, Danchin E, Mulard H, Hatch SA, Heeb P, Wagner RH (2010) Sexually transmitted bacteria affect female cloacal assemblages in a wild bird. Ecol Lett 13:1515–1524. doi:10.1111/j.1461-0248.2010.01542.x
van der Hoeven-Hangoor E, van der Vossen JMBM, Schuren FHJ, Verstegen MWA, de Oliveira JE, Montijn RC, Hendriks WH (2013) Ileal microbiota composition of broilers fed various commercial diet compositions. Poult Sci 92:2713–2723. doi:10.3382/ps.2013-03017
Ruiz-de-Castañda R, Vela AI, Lobato E, Briones V, Moreno J (2011) Prevalence of potentially pathogenic culturable bacteria on eggshells and in cloacae of female Pied Flycatchers in a temperate habitat in central Spain. J Field Ornithol 82:215–224
Shawkey MD, Firestone MK, Brodie EL, Beissinger SR (2009) Avian incubation inhibits growth and diversification of bacterial assemblages on eggs. PLoS One 4:e4522. doi:10.1371/journal.pone.0004522
Mead GC (1989) Microbes of the avian cecum: types present and substrates utilized. J Exp Zool Suppl 3:48–54
Kohl K (2012) Diversity and function of the avian gut microbiota. J Comp Physiol B 182:591–602. doi:10.1007/s00360-012-0645-z
Józefiak D, Rutkowski A, Martin SA (2004) Carbohydrate fermentation in the avian ceca: a review. Anim Feed Sci Technol 113:1–15. doi:10.1016/j.anifeedsci.2003.09.007
Waite DW, Taylor MW (2014) Characterizing the avian gut microbiota: membership, driving influences, and potential function. Front Microbiol 5:223. doi:10.3389/fmicb.2014.00223
Yang J (2012) Influence of dietary fibers and whole grains on fecal microbiota during in vitro fermentation. Thesis, the University of Nebraska
Dethlefsen L, Relman DA (2011) Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci 108:4554–4561. doi:10.1073/pnas.1000087107
Ridlon JM, Kang D-J, Hylemon PB (2006) Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47:241–259. doi:10.1194/jlr.R500013-JLR200
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto J-M, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Dore J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, Bork P, Ehrlich SD, Wang J (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464:59–65. doi:10.1038/nature08821
Kita K, Ken IR, Akamine C, Kawada W, Shimura Y, Inamoto T (2014) Influence of propolis residue on the bacterial flora in the cecum of Nanbu Kashiwa. J Poult Sci 51:275–280. doi:10.2141/jpsa.0130137
Dewhirst FE, Paster BJ, Tzellas N, Coleman B, Downes J, Spratt DA, Wade WG (2001) Characterization of novel human oral isolates and cloned 16S rDNA sequences that fall in the family Coriobacteriaceae: description of olsenella gen. nov., reclassification of Lactobacillus uli as Olsenella uli comb. nov. and description of Olsenella profusa sp. nov. Int J Syst Evol Microbiol 51:1797–1804. doi:10.1099/00207713-51-5-1797
Apajalahti JHA, Kettunen A, Bedford MR, Holben WE (2001) Percent G+C profiling accurately reveals diet-related differences in the gastrointestinal microbial community of broiler chickens. Appl Environ Microbiol 67:5656–5667. doi:10.1128/aem.67.12.5656-5667.2001
Oakley BB, Lillehoj HS, Kogut MH, Kim WK, Maurer JJ, Pedroso A, Lee MD, Collett SR, Johnson TJ, Cox NA (2014) The chicken gastrointestinal microbiome. FEMS Microbiol Lett 360:100–112. doi:10.1111/1574-6968.12608
Mizrahi-Man O, Davenport ER, Gilad Y (2013) Taxonomic classification of bacterial 16S rRNA genes using short sequencing reads: evaluation of effective study designs. PLoS One 8:e53608. doi:10.1371/journal.pone.0053608
Acknowledgments
We thank Susan Hammerly for early discussions concerning the feasibility of this study and Shannon Nodolf, DVM, for conducting the sampling. This work was supported by the National Science Foundation (DEB 0948787) to JAJ and U.S. Fish and Wildlife Service to JAJ.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 20 kb)
Rights and permissions
About this article

Cite this article
Zhang, Y., Simon, S.E., Johnson, J.A. et al. Spatial Microbial Composition Along the Gastrointestinal Tract of Captive Attwater’s Prairie Chicken. Microb Ecol 73, 966–977 (2017). https://doi.org/10.1007/s00248-016-0870-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-016-0870-1