
Abstract
Microbial function, composition, and distribution play a fundamental role in ecosystem ecology. The interaction between desert plants and their associated microbes is expected to greatly affect their response to changes in this harsh environment. Using comparative analyses, we studied the impact of three desert shrubs, Atriplex halimus (A), Artemisia herba-alba (AHA), and Hammada scoparia (HS), on soil- and leaf-associated microbial communities. DNA extracted from the leaf surface and soil samples collected beneath the shrubs were used to study associated microbial diversity using a sequencing survey of variable regions of bacterial 16S rRNA and fungal ribosomal internal transcribed spacer (ITS1). We found that the composition of bacterial and fungal orders is plant-type-specific, indicating that each plant type provides a suitable and unique microenvironment. The different adaptive ecophysiological properties of the three plant species and the differential effect on their associated microbial composition point to the role of adaptation in the shaping of microbial diversity. Overall, our findings suggest a link between plant ecophysiological adaptation as a “temporary host” and the biotic-community parameters in extreme xeric environments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In arid regions, aeolian particles are one of the major sources of nutrients and biotic diversity. These particles includes zooplankton, bacteria, algae, protists, and fungi [1, 2]. The growing interest in the impact of the aeolian dust biota and particulate-matter components was mainly considered in desert ecosystems either for their role in improving soil physical and chemical properties or their impact on human health [3, 4]. The relationship between the transferred aeolian biota and plant leaves is beneficial for both, embodying a widespread and ancient symbiosis [5] that greatly influence plant performance (e.g., hormone production, growth, pathogen infections, etc.) [6]. The phyllosphere (i.e., the above-ground parts of the plant, and the leaves, in particular) provides a “hostel” for the colonization of millions of bacteria, fungi, and other microscopic organisms [7]. The atmospheric deposition of particles originating from different sources, such as soil or other plants, facilitates microbial transfer to the phyllosphere [8, 9]. A positive correlation between chemical environmental factors and geographic distances with archeal and bacterial communities has been shown, suggesting that the different domains of life exhibit different correlations with environmental parameters [10]. The relative importance of each of the factors, e.g., environmental or geographic, varies between studies [10] and remains debatable. Many studies have focused on describing microbial communities in the laimosphere and phyllosphere; however, there are only a handful of studies that address the question of how the ecophysiological adaptation of the plant may affect the microbial community of the laimosphere and phyllosphere as a result of the alteration of the local microenvironment.
Scarce and unpredictable rainfall, high soil salinity, and high temperatures are the main limiting factors that shape the ecophysiological adaptation of desert organisms to the challenging desert environment [11–14]. Each perennial plant possesses a unique environmental niche impacted by the compilation of its adaptive characteristics, which are expected to influence the colonizing success of the microbial communities, e.g., it has been shown that bacteria inhabiting the Tamarix phyllosphere in arid regions are exposed to periods of extreme daytime desiccation interspersed with nighttime immersion in a highly saline solution (over 20 % dissolved salts) [15–17].
Most phyllosphere microorganisms, such as those inhabiting many other natural environments, are nonculturable [7, 18, 19]. Over the past decade, many studies have benefited from a variety of culture-independent methods, including PCR-based next-generation sequencing (NGS), shotgun metagenomics studies, and metaproteogenomics [20, 21]. Nowadays, the genomic analyses of individual microbial strains or metagenomics studies of whole microbial communities provide insight into the composition and physiological potential of plant- [22] and soil-associated microorganisms [23]. These NGS approaches directly link genetic diversity to species richness [22–24].
In our previous study, it was shown that functional diversity in the laimosphere and phyllosphere exhibited similar patterns for each individual shrub, where microbial functional diversity varied greatly across the shrubs in both the laimosphere and phyllosphere [25]. It was hypothesized that microbial structural diversity in the phyllosphere partly overlaps that of the laimosphere community, and is affected by perennial-shrub ecophysiological adaptations. In order to study the diversity of microbial communities colonizing the phyllosphere and the laimosphere habitats of the three desert shrubs in relation to their adaptations, we sought to determine microbial diversity using the NGS approach.
In the current study, we used NGS of 16S rRNA and internal transcribed spacer (ITS) amplicons to survey bacterial and fungal diversity in the phyllosphere and laimosphere of three morphologically and anatomically different species of perennial desert shrubs native to the Israeli desert: Atriplex halimus (A), Hammada scoparia (HS), and Artemisia herba-alba (AHA). Partial overlap between the microbial communities of each niche was identified. Niche-specific and plant-type-specific patterns that are likely dictated by plant ecophysiology were also identified. In addition, the microorganism communities were correlated with previously published data on abiotic parameters (e.g., moisture, pH, C/N ratio, organic matter content, and salinity) [13, 26] of the same environments.
Materials and Methods
Study Site and Plants
The study site was located in the northern Negev Desert at the M. Evenari Runoff Research Farm (30°47′N, 34°36′E), Avdat, Israel [11]. For a detailed description of the study site and shrub species, please refer to Martirosyan et al. [13, 26]. Three shrubs that share the same ecosystem and grow in the same vicinity (∼5–15 m), but differ in their ecophysiological properties, were sampled: Atriplex halimus (A) [(saltbush) shrub is halophytic with gray-green leaves (varying in size from 1 × 1 m to 2.5 m high) and extremely tolerant of salt content in the ground [11, 27]]; H. scoparia (HS) [shrub leaves are atrophied into scales and fused onto the segment that bears them. The presence of anabasine in the plant explains the plant’s toxicity [27, 28]]. Artemisia herba-alba (AHA) shrub (20–40 cm height) with strongly aromatic leaves exhibits an allelopathic effect on other plant species and has a high calcium content [29].
Sampling
At the end of the dry season, 200-g soil samples were randomly collected from the upper 0–10 cm soil layer beneath the individual shrubs (four replicates for each plant) and from open spaces acting as control (CO), placed in individual, sterile, plastic bags, and stored in a cool insulated box for transportation to the laboratory. Soil samples were sieved through a 2-mm mesh in order to remove stones, roots, and other organic debris, and stored at 4 °C for 2 days before processing. Additional samples were stored at −20 °C for further DNA extraction and microbial-community analysis. Twenty-gram leaf samples randomly collected from each shrub in four replicates were placed in sterile-paper envelopes, transported to the laboratory in a cool insulated box, and then stored at room temperature overnight for biotic analyses.
Molecular Microbiological Analyses
Genomic DNA was isolated from 0.25 g soil samples using the MoBio PowerSoil DNA Isolation Kit (MoBio Laboratories Inc., Carlsbad, CA). The microbe community colonizing the leaf surface was concentrated and collected by filtration on a 0.2-μm pore-size Millipore polycarbonate filter (GE Water & Process Technologies, Trevose, PA). The filters were then used for genomic DNA extraction using the MoBio PowerSoil® DNA Isolation Kit (MoBio Laboratories Inc.). PCR amplification of the 16S rRNA gene was performed using a universal forward primer 341(F) (5′-CCTACGGGAGGCAGCAG-3′) and reverse primer 700(R) (5′- CGMATTYCACYKCTACAC-3′) flanking the two variable regions, V3 and V4, of the 16S gene [30, 31]. For fungal-community analysis, the ITS1 fragment of the rRNA gene was amplified using ITS1(F) (5′- TCCGTAGGTGAACCTGCGG-3′) and ITS2(R) (5′-GCTGCGTTCTTCATCGATGC-3′) primer pair [32, 33]. PCRs were performed using Phusion® high-fidelity DNA polymerase (New England BioLabs, M0530S) with 35 amplification cycles, according to the manufacturer’s recommendations. Four soil and three leaf samples (each a mix of four replicates) were analyzed and each sample was PCR-amplified. PCR products were separated on gel and purified on Zymo-Spin™ IIN columns. Following purification, PCR amplicons were prepared for sequencing using the Ion Torrent Personal Genome Machine (PGM) (Life Technologies, Carlsbad, CA).
Ion Torrent™ Sequencing
Libraries from each sample were prepared using the Ion XpressTM Plus Fragment Library Kit (Life Technologies). In order to sequence all the samples, each sample had its own unique barcode from the Ion Xpress Barcode Adapters 1-16 Kit (Life Technologies). For each sample, the two PCR products were mixed together in equal molar amounts before the library preparation was started. Considering that the goal of the experiment was to comprehensively assess microbial diversity at each location, sequencing depth, not replication, was critical [34]. The libraries were quantified and qualified using a DNA 1000 Bioanalyzer chip (Agilent, Santa Clara, CA). The emulsion PCR was carried out on a OneTouch 2 system (Life Technologies) using the Ion PGM™ Template OT2 400 Kit (Life Technologies). The quality of the unenriched spheres was checked on a Qubit 2.0 using the Ion Sphere Quality Control Kit (Life Technologies). Sequencing of the amplicon libraries was carried out on the PGM system using the Ion Sequencing 400 Kit (Life Technologies), following the company’s protocol.
Bioinformatic Analysis
The sequencing data were analyzed using the quantitative insights into the microbial ecology pipeline v 1.7.0 (QIIME) [35]. QIIME is an open-source software pipeline built using the PyCogent toolkit [36] to address the problem of taking sequencing data from raw sequences for interpretation and database deposition. QIIME, available at http://qiime.sourceforge.net/, supports a wide range of microbial-community analyses and visualizations that have been central to several recent high-profile studies, including network analysis, histograms of within- or between-sample diversity, and analysis of whether “core” sets of organisms are consistently represented in certain habitats. Barcodes and primers were trimmed using the split library script available in QIIME. Reads shorter than 150 bases were discarded, as were reads whose barcode contained more than two errors. The sequences were then clustered and assigned to operational taxonomic units (OTUs) using the open-reference OTU picking with UCLUST algorithm in QIIME. The representative sequences, the most abundant sequences in the respective clusters, were extracted and classified taxonomically. Taxonomic classification was performed using the Ribosomal Database Project classifier in QIIME against the Greengenes (gg_12_10 version) database [37], preclustered at 97 % identity for the 16S rRNA samples, and the UNITE (its_12_11 version) database [38], preclustered at 97 % identity for the ITS1 samples. Chimeric OTUs were filtered out using ChimeraSlayer for the 16S rRNA samples, and BLAST for the ITS1 samples. Alpha diversity was calculated using the QIIME package. Rarefaction curves, Shannon indices, and Chao1 indices were calculated employing the QIIME alpha_rarefaction.py pipeline. When comparing multiple populations, beta diversity measures describe how many taxa are shared between them. Beta diversity was calculated using QIIME script beta_diversity_through_plots.py for computing beta diversity distance matrices and generating PCoA plots. Note that for the beta diversity analysis, we chose an even sampling depth of 6661 and 1666 for 16S rRNA and ITS1, respectively, based on the output of BIOM summary-table script. We used the UniFrac (weighted and unweighted) and Bray-Curtis metric for 16S rRNA and ITS1, respectively, to calculate the distance between samples.
Statistical Analysis
Statistical analysis for the biotic parameters was conducted using the SAS statistical software package. The effects of both abiotic and biotic variables were determined using general linear-model (GLM) analysis. Duncan’s multiple range tests (MRTs) were used to determine differences between variables. Significance was defined at a level of p < 0.05.
Results
Abiotic and Biotic Analyses of the Laimosphere and Phyllosphere
The results of abiotic and microfloral community analysis [colony-forming units (CFUs)] of soil and leaf samples showed clear distinctions between the phyllosphere and laimosphere beneath the canopies of the three different shrubs [13, 26], suggesting distinct differences between population density (CFUs) of different shrubs and between the laimosphere and phyllosphere of the same shrub. Thus, both abiotic microenvironmental characteristics and bacterial- and fungal-value cultures were found to differ for different shrubs growing in the same desert environment, and were found to be affected by the ecophysiological adaptations of the shrub to the harsh environment, as hypothesized [13, 26, 39].
Microbial Composition Analysis
Microbial-community composition in the laimosphere and phyllosphere was examined via bacterial (16S rRNA) and fungal (ITS) genomic markers sequenced on an Ion Torrent™ sequencer. Average lengths of 300 bps (16S rRNA) and 235 bps for the fungal community (ITS1) were obtained with primers flanking the variable regions. Following removal of PGM linker primers, barcodes, and forward amplification primers during QIIME analysis (see “Experimental Procedures”), the read quality across the phylogenetically informative sequence length was used to determine the suitability of fragments for downstream analyses. Overall, we obtained 166,529 16S rRNA and 53,097 ITS1 sequences. After checking for sequencing errors and chimeras, we obtained 130,132 16S rRNA and 38,401 ITS1 reads (Tables 1 and 2).
The reads were clustered based on similarity to sequences in a reference database; reads that did not match any accession in the database were then clustered de novo, and the resulting OTU sets were merged. Clustering the extracted fragments resulted in 14,251 OTUs for 16S rRNA and 2079 OTUs for ITS1 at 97 % sequence similarity (Tables 1 and 2). While most of the 16S reads were identified, a large number of ITSs could not be classified; the 16S rRNA reads were assigned to 75 (61.5 %) and ITS1 reads were assigned to 33 (75 %) different classified orders. The taxonomic diversity of the different 16S rRNA and ITS1 gene OTUs showing greater-than-1 % relative abundance that were detected in the soil and in the leaf samples is represented in Figs. 1 and 2, respectively. Results revealed a high degree of diversity between all samples, with distinctive patterns of relative abundance of OTUs; laimosphere samples showed higher diversity compared with the phyllosphere samples.

The relative abundance of the 16S rRNA OTUs detected in soil [control (CO), Atriplex halimus (A), H. scoparia (HS), Artemisia herba-alba (AHA)] and leaf [leaf of Atriplex halimus (AL), leaf of H. scoparia (HSL), leaf of Artemisia herba-alba (AHAL)] samples. The highest-resolution taxonomic level that has been identified is order. Orders that comprised less than 1 % of each dataset are clustered together

The relative abundance of the ITS1 OTUs detected in soil [control (CO), Atriplex halimus (A), H. scoparia (HS), Artemisia herba-alba (AHA)] and leaf [leaf of Atriplex halimus (AL), leaf of H. scoparia (HSL), leaf of Artemisia herba-alba (AHAL)] samples. The highest-resolution taxonomic level that has been identified is order. Orders that comprised less than 1 % of each dataset are clustered together
While cluster analysis of the soil control samples showed greater diversity compared with the phyllosphere plant samples of both the bacterial and fungal communities, the degree of diversity between soil and leaf samples was not easily distinguishable (Figs. 1 and 2). The bacterial order of Sphingobacteriales (phylum Bacteroidetes) was dominant in all three laimospheres (Fig. 1), yet it was almost completely absent in leaf samples (≤0.1 %; Fig. 1). Moreover, Sphingobacteriales seem to thrive under the canopy of all three plant species, as this order was 3–4-fold more abundant than in the control open-space samples. In contrast, no dominant bacterial order was found to be common to all phyllospheres but absent in the laimosphere; Burkholderiales and Streptophyta were the two predominant orders on Artemisia herba-alba leaves (AHAL), covering 53 and 41 % of the total OTUs, respectively. On H. scoparia leaf (HSL) samples, an undetermined order belonging to the Bacteroidetes phylum made up 80 % of the population, and on Atriplex halimus leaves (AL), Streptophyta and Bacillales make up 44 and 35 % of the population, respectively. Interestingly, Burkholderiales (of the Proteobacteria phylum) was the only order common to all soil and leaf samples, with >1 % abundance (Fig. 1).
As mentioned above, many of the ITS1 reads were not classified. These unclassified reads constituted the majority or a large proportion of all samples except HSL; however, significant diversity in fungal communities could still be observed between samples (Fig. 2). Although the distinction between laimosphere and phyllosphere clusters was less obvious than for 16S rRNA clusters (Fig. 1), only one order, Pleosporales, was abundant in all soil and leaf samples. Notable distinctions between soil and leaf samples were observed for orders of Ascomycota, Agaricales, Pezizales, and two undetermined orders found almost exclusively in the soil (Fig. 2). Moreover, all leaf samples hosted the epiphytic order of Capnodiales, which constitutes the majority of the fungal communities on HSL (81 %), while on AL and AHAL, it made up 23 and 9.8 % of the fungal communities, respectively. In contrast, Capnodiales was marginally represented in the soil, making up less than 1 % of the fungal communities in the open-space control and in Atriplex halimus (Fig. 2).
Comparison Between the Bacterial and Fungal Communities in the Phyllosphere and Laimosphere of Each Plant Species
Differences in microbial diversity were most distinguishable between soil and leaf samples, as can be expected (Figs. 1 and 2). However, the clear distinction between the bacterial diversity of the open-space control and the clusters of the three laimosphere samples demonstrates the effect of the plants. The phylum Acidobacteria was most dominant in the soil samples, about 600 times higher than in the leaves, which is in agreement with a previous report on its abundance within soil [40]. The order Bacillales of the Firmicutes phylum showed relatively higher abundance in the shrubs than in the corresponding soil beneath them, elucidating the plant impact, similar to the findings of Bulgarelli et al. [41] for the Firmicutes phylum.
In contrast to the corresponding similar frequencies of Pleosporales (between 17 and 20 %) in the vicinity of and on Atriplex halimus and H. scorparia, it made up almost 50 % of the fungal community beneath the Artemisia herba-alba shrub, while above the shrub—only 11.9 %. The Pleosporales is the largest order in the fungal class Dothideomycetes. Most of the species are saprophytes commonly found on decaying plant material in fresh water, marine, or terrestrial environments, but several species are also associated with living plants, such as parasites, epiphytes, or endophytes.
Alpha Diversity Within Samples and Rarefaction Curves
To better show the mean species diversity in sites or habitats, OTU-based alpha diversity or Shannon index was calculated using QIIME software (Fig. 3). These results also confirmed that the sequencing depth [34] was sufficient to accurately describe bacterial and fungal diversities in our tests (Fig. 3), and eventually allow for detailed diversity analyses that account for rare taxa such as Chao1. According to the Shannon index, the information regarding the entropy of the observed OTU abundances accounting for both richness and evenness, the bacterial community is more diverse in the soil samples than in the leaf samples. The observed order of diversity is as follows: HS > AHA > A > CO > AL > AHAL > HSL and CO > AHA > HS > A > HSL > AHAL > AL for the bacterial and fungal communities, respectively. Our data suggest that all three plant laimospheres similarly support rich and diverse bacterial communities compared with the control soil sample (R = 0.8 and p = 0.015) (Fig. 3a). In contrast, a similar effect does not exist for fungal communities (R = 0.467 and p = 0.0675) (Fig. 3b). This means that diversity rises in that order. It should be mentioned that for the calculation of α-diversity in QIIME, two metrics were also used: (a) Chao1 metric estimated species richness (Fig. 3a, b); and (b) the observed species metric, which is simply the count of unique OTUs found in the samples (Fig. 4a, b).

Diversity as a function of sampling depth (16S rRNA, a) and fungi (ITS1, b), as indicated by the Shannon index (H′) in different soil treatments [control (CO), Atriplex halimus (A), H. scoparia (HS), Artemisia herba-alba (AHA)] and leaf [leaf of Atriplex halimus (AL), leaf of H. scoparia (HSL), leaf of Artemisia herba-alba (AHAL)] samples

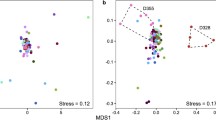
Principal coordinate analysis of the microbial communities via 16S rRNA (a) and ITS1 (b) in the soil [control (CO), Atriplex halimus (A), H. scoparia (HS), Artemisia herba-alba (AHA)] and leaf [leaf of Atriplex halimus (AL), leaf of H. scoparia (HSL), leaf of Artemisia herba-alba (AHAL)] samples. The first two principal component axes are shown
Beta Diversity (Pairwise Sample Dissimilarity) Between Samples and Plots
β-Diversity analysis was carried out to compare OTU distribution between the different soil and leaf samples and to each other. QIIME was used for the calculation of β-diversity for each of the seven microbial communities (four soil and three leaves). Principal coordinate analysis (PCoA) plots exhibited similarity between soil samples beneath the different shrubs (A, HS, AHA), which were relatively distant from the CO sample (Fig. 4a); this is consistent with previous observations [25]. Regarding the fungal laimosphere community, CO, AHA, and HS exhibited similar structure, and A was distant from the soil samples (Fig. 4b), indicating that the fungal community structure in soil beneath Atriplex halimus was different compared to other soil samples. On the other hand, phyllosphere beta diversity analysis showed similarity between AHAL and HSL samples for fungal and bacterial communities, but it showed some distance for AL (Fig. 4a, b). Two metrics were used to show quantitative (e.g., weighted UniFrac) and qualitative (e.g., unweighted unifrac UniFrac) measures of beta diversity for 16S rRNA, and non-phylogenetic Bray-Curtis metric for ITS1.
Interactions of Biotic and Abiotic Parameters
Previous studies have shown a correlation between microbial-community structure and abiotic variables, such as pH or moisture content, particularly in soil, in culture-free high-throughput surveys [25]. To correlate between abiotic parameters and microbial structure, we assigned the prevalent phyla for both bacteria and fungi: Acidobacteria (9.27 %), Bacteroidetes (32.37 %), Firmicutes (9.84 %), Cyanobacteria (28.08 %), and Proteobacteria (16.51 %) for 16S rRNA, and Basidiomycota (11.05 %) and Ascomycota (87.45 %) for ITS1 (Figs. 1 and 2). Linear regression analysis was conducted to show the relationship between microbial communities (dominant phyla) and abiotic parameters [pH, C/N ratio, total organic carbon (TOC), soil moisture, and electrical conductivity (EC)], as described previously by Guo et al. [25].
The results showed no significant correlation between pH and all dominant bacterial and fungal phyla for soil or leaves (p > 0.5). Only two dominant bacterial phyla, Bacteroidetes (ANOVA, p < 0.02) and Cyanobacteria (ANOVA, p < 0.04), showed a relationship with the soil TOC (Fig. 5a, b). No significant correlation was found between microbial communities and the C/N ratio, water content, or EC.

The relationships between the relative abundance of two dominant bacterial [Bacteroidetes (a) and Cyanobacteria (b)] phyla and soil TOC. Linear regressions were used to test the correlation between the relative microbial abundance and TOC
Discussion
The current work aims to investigate the role of perennial-plant ecophysiological adaptations in determining the leaf-coat phyllosphere microbial diversity and the below-canopy laimosphere microbial diversity in the shrubs, present in the same macroenvironment. The uniqueness of the present study is that it focused on three co-located perennial plant species, all well adapted to the Negev Desert and, therefore, subjected to the same environmental conditions. This allowed us to pinpoint the effect of the perennial-plant ecophysiological adaptations on the microbial-community structure of the laimosphere and phyllosphere. Comparison between the three phyllospheres revealed microbial composition that is plant-type-specific, particularly for bacteria, and distinct diversities for leaves, soil under the plant, and open-space control samples. However, our data obtained by culture-independent methods differ somewhat from the previously published data on the microbial-community structure of the same plants and/or soil samples from the same environment. A previous study on the abundance of ground bacteria in arid Negev soil [42] reported that most of the community consisted of (1) Actinobacteria, most commonly detected in the bulk soil; (2) Proteobacteria, typically found under the plant canopy; and (3) Acidobacteria, generally found underneath the canopy in the arid sites (Supplementary Fig. 1). In contrast, our culture-free sequencing data revealed that none of the above-mentioned phyla were very common in the laimosphere, and the orders belonging to these phyla were below 1 %.
In addition, a previous culture-based investigation of the microfungal community in the root zone of H. scoparia in the Negev Desert identified a number of fungal species belonging mainly to Ascomycota, although some Zygomycota were also identified [43]. Our results identified orders of the Zygomycota only in the soil samples and only in the HS samples showing >1 % frequency (Supplementary Fig. 2).
Previous data on microbial-community structure in the laimosphere and phyllosphere suggest that a microorganism community is highly correlated with edaphic factors, such as moisture, pH, climate, parent-rock material, temperature, and nutrient and organic matter content [44, 45]. Microorganisms residing on plant leaves are exposed to highly variable environmental factors. Relative to fungi and archaea, bacteria are the most prevalent phyllosphere-colonizing microbes, with bacterial titers averaging approximately 106–107 microbial cells per square centimeter of leaf area [46]. The shape of the leaves and their position, as well as the leaf-surface chemistry, constitute a unique microenvironment. Thus, the surface of leaves may offer distinct habitats, even on closely co-located plants, differentially affecting microbial growth and development. In general, the phyllosphere-associated bacterial communities found on the three plants accommodated relatively few phyla. Other recent comparisons of plant-associated bacterial communities, e.g., sampled for various trees, also identified relatively few abundant bacterial phyla, among them Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria [41]. Except for Bacteroidetes, which dominated the leaves of H. scoparia, these bacterial phyla were not abundant in the phyllosphere of the three desert plants reported here.
The defined phylogenetic structure of the low-complexity phyllosphere communities raises questions regarding the source of its inoculum. This is most relevant for the phyllosphere-specific or plant-type-specific orders abundant on leaves but scarce or absent in the soil; this was the case for most bacterial communities identified on the three plants and for Capnodiales fungi. The typical bacterial titer in air, as determined by different methods, ranges from 101 to 105 cells per cubic meter [47], which is several orders of magnitude lower than the typical titer in soil, commonly 106 to 109 cells per gram [48]. Neighboring plants and plant debris constitute another important colonization source, as these bacteria are already adapted to phyllosphere conditions [49].
Based on our data, we suggest that plant species and, thus, their ecophysiological adaptation play a key role in determining the microbial diversity of phyllosphere communities. These results agree with the findings of Redford et al. [50], who studied the phyllosphere of 56 tree species representing 14 different plant orders, and who showed that the interspecific geographic variability of a microbial community exceeded the intraspecific variability. The results indicate that the plant species is important for the makeup of microbial communities. Comparison between bacterial communities found in the phyllosphere of tropical trees showed a significant tendency to follow host-plant phylogeny, where similarities between communities increased as hosts were more closely related [51]. We show distinctions in microbial communities, particularly for bacteria, colonizing three phylogenetically divergent plant species sharing the same geographic location. Corresponding with the findings of Redford et al. [50] and Kim et al. [51], our findings also support, if not confirm, the idea that the impact of the host plant on a microbial community is much greater than the effects of geographic location.
In contrast, other research demonstrated that site-specific factors and seasons had a stronger impact on the Methylobacterium-community composition of plant leaves than did plant species-specific factors [52, 53]. This indicates a seasonal, abiotic-induced variability in the alpha diversity. Differences observed between the abiotic parameters measured beneath the shrubs [26] indicate that microenvironmental conditions are impacted by the plant. Furthermore, because the A, HS, and AHA sampled in this study share the same macroenvironment, the distinct microbial composition strengthens the hypothesis that the host-guest relationship is plant-type specific. Consequently, it can be further hypothesized that plant ecophysiological adaptation has an impact on the development and composition of microbial communities. Taken together, our current results demonstrate specific interactions between each plant species and its soil environment that likely arise from the distinct deposition of organic material and the subsequent effects on the microbial soil communities. Even though a high proportion of fungi and bacteria function as decomposers in the soil, they degrade plant residues differently and play different roles in the recycling of nutrients [25]. This is partly due to their different choices of habitats within the soil and the different types of organic matter they consume. Material with a lower C/N ratio, which is more labile, would be much more easily decomposed by the microorganisms [25]. Although abiotic and biotic parameters have shown some interactions and the shrubs were observed to have a major impact on the microbial community, a more detailed study will be necessary in order to be able to predict the microbial phyllosphere-community structure based on plant species ecophysiology and, also, to elucidate the role of the temporal and spatial (vertical) components. Moreover, Bowers et al. [19] showed that despite the importance of diversity and biogeography of the microbial community in the atmosphere, which is represented by known and unknown sources, this subject has been understudied. The obvious follow-up question is what the specific roles are played by microbes and/or microbial communities in the adaptation of desert plants to their harsh environment.
References
Offer ZY, Steinberger Y (1994) Some data on elemental composition of airborne particulate matter in the northern Negev Desert, Israel. Environ Conserv 21:342–346
Acosta-Martinez V, Van Pelt S, Moore-Kucera J, Baddock MC, Zobeck TM (2015) Microbiology of wind-eroded sediments: current knowledge and future research directions. Aeolian Res 18:99–113
Zaady E, Ben-David EA, Sher Y, Tzirkin R, Nejidat A (2010) Inferring biological soil crust successional stage using combined PLFA, DGGE, physical and biophysiological analyses. Soil Biol Biochem 42:842–849. doi:10.1016/j.soilbio.2010.02.002
Goudie AS (2014) Desert dust and human health disorders. Environ Int 63:101–113
Fedorov DN, Doronina NV, Trotsenko YA (2011) Phytosymbiosis of aerobic methylobacteria: new facts and views. Microbiology 80:443–454
Reed SC, Townsend AR, Cleveland CC, Nemergut DR (2010) Microbial community shifts influence patterns in tropical forest nitrogen fixation. Oecologia 164:521–531
Vorholt JA (2012) Microbial life in the phyllosphere. Nat Rev Microbiol 10:828–840
Beattie GA, Lindow SE (1994) Comparison of the behavior of epiphytic fitness mutants of Pseudomonas syringae under controlled and field conditions. Appl Environ Microbiol 60:3799–3808
Wilson M, Hirano SS, Lindow SE (1999) Location and survival of leaf-associated bacteria in relation to pathogenicity and potential for growth within the leaf. Appl Environ Microbiol 65:1435–1443
Pagaling E, Wang H, Venables M, Wallace A, Grant WD, Cowan DA, Jones BE, Ma Y, Ventosa A, Heaphy S (2009) Microbial biogeography of six salt lakes in Inner Mongolia, China, and a salt lake in Argentina. Appl Environ Microbiol 75:5750–5760. doi:10.1128/AEM.00040-09
Evenari ME, Shanan L, Tadmor W (1982) The Negev: the challenge of a desert, 2nd edn. Harvard University Press, Cambridge, p 345
Guenaoui C, Gorai M, Smiti S, Neffati M (2008) Biochemical and physiological changes in Artemisia herba-alba plants under water stress conditions. Middle-East J Sci Res 3:156–163
Martirosyan V, Steinberger Y (2014) Microbial functional diversity in the phyllosphere and laimosphere of different desert plants. J Arid Environ 107:26–33. doi:10.1016/j.jaridenv.2014.04.002
Whitford WG (2002) Ecology of desert systems. Academic Press, New York, p 343
Qvit-Raz N, Jurkevitch E, Belkin S (2008) Drop-size soda lakes: transient microbial habitats on a salt-secreting desert tree. Genetics 178:1615–1622. doi:10.1534/genetics.107.082164
Finkel OM, Burch AY, Lindow SE, Post AF, Belkin S (2011) Geographical location determines the population structure in phyllosphere microbial communities of a salt-excreting desert tree. Appl Environ Microbiol 77:7647–7655. doi:10.1128/aem.05565-11
Finkel OM, Burch AY, Elad T, Huse SM, Lindow SE, Post AF, Belkin S (2012) Distance-decay relationships partially determine diversity patterns of phyllosphere bacteria on Tamarix trees across the Sonoran Desert. Appl Environ Microbiol 78:6187–6193
Muller T, Ruppel S (2014) Progress in cultivation-independent phyllosphere microbiology. FEMS Microbiol Ecol 87:2–17
Bowers RM, Sullivan AP, Costello EK, Collett JL, Knight R, Fierer N (2011) Sources of bacteria in outdoor air across cities in the Midwestern United States. Appl Environ Microbiol 77:6350–6356. doi:10.1128/Aem.05498-11
Lozupone CA, Knight R (2007) Global patterns in bacterial diversity. Proc Natl Acad Sci U S A 104:11436–11440
Rothberg JM, Hinz W, Rearick TM, Schultz J, Mileski W, Davey M, Leamon JH, Johnson K, Milgrew MJ, Edwards M, Hoon J, Simons JF, Marran D, Myers JW, Davidson JF, Branting A, Nobile JR, Puc BP, Light D, Clark TA, Huber M, Branciforte JT, Stoner IB, Cawley SE, Lyons M, Fu YT, Homer N, Sedova M, Miao X, Reed B, Sabina J, Feierstein E, Schorn M, Alanjary M, Dimalanta E, Dressman D, Kasinskas R, Sokolsky T, Fidanza JA, Namsaraev E, McKernan KJ, Williams A, Roth GT, Bustillo J (2011) An integrated semiconductor device enabling non-optical genome sequencing. Nature 475:348–352
Knief C (2014) Analysis of plant microbe interactions in the era of next generation sequencing technologies. Front Plant Sci 5:216. doi:10.3389/fpls.2014.00216
Barriuso J, Valverde JR, Mellado RP (2011) Estimation of bacterial diversity using next generation sequencing of 16S rDNA: a comparison of different workflows. BMC Bioinformatics 12:473. doi:10.1186/1471-2105-12-473
Unterseher M, Jumpponen A, Opik M, Tedersoo L, Moora M, Dormann CF, Schnittler M (2011) Species abundance distributions and richness estimations in fungal metagenomics—lessons learned from community ecology. Mol Ecol 20:275–285
Guo SZ, Lu ZM, Kang GY, Chen Z, Luo H (2012) A tree-structured deterministic small-world network. IEICE Trans Inform Syst E95d:1536–1538. doi:10.1587/transinf.E95.D.1536
Martirosyan V, Steinberger Y, Doniger T, Wachtel C, Miller G (2014) Structural and functional microbial diversity determined by shrubs ecophysiological adaptation in the Negev Desert. The 99th Annual Meeting of the Ecological Society of America, August 10–15, 2014, Sacramento, CA
Barness G, Zaragoza S, Shmueli I, Steinberger Y (2009) Vertical distribution of a soil microbial community as affected by plant ecophysiological adaptation in a desert system. Microb Ecol 57:36–49. doi:10.1007/s00248-008-9396-5
Berg N, Steinberger Y (2008) Role of perennial plants in determining the activity of the microbial community in the Negev Desert ecosystem. Soil Biol Biochem 40:2686–2695. doi:10.1016/j.soilbio.2008.07.019
Mohamed AEHH, El-Sayed MA, Hegazy ME, Helaly SE, Esmail AM, Mohamed NS (2010) Chemical constituents and biological activities of Artemisia herba-alba. Rec Nat Prod 4:1–25
Muyzer G, Ramsing NB (1995) Molecular methods to study the organization of microbial communities. Water Sci Technol 32:1–9
Vasileiadis S, Puglisi E, Arena M, Cappa F, Cocconcelli PS, Trevisan M (2012) Soil bacterial diversity screening using single 16S rRNA gene V regions coupled with multi-million read generating sequencing technologies. PLoS ONE 7:e42671
Gardes M, Bruns TD (1993) ITS primers with enhanced specificity for basidiomycetes—application to the identification of mycorrhizae and rusts. Mol Ecol 2:113–118. doi:10.1111/j.1365-294X.1993.tb00005.x
White TJ, Bruns T, Lee S, Taylor JW (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR protocols: a guide to methods and applications. Academic Press, San Diego, pp 315–322
Smith DP, Peay KG (2014) Sequence depth, not PCR replication, improves ecological inference from next generation DNA sequencing. PLoS ONE 9:e90234
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R (2011) Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 108:4516–4522. doi:10.1073/pnas.1000080107
Knight R, Maxwell P, Birmingham A, Carnes J, Caporaso JG, Easton BC, Eaton M, Hamady M, Lindsay H, Liu ZZ, Lozupone C, McDonald D, Robeson M, Sammut R, Smit S, Wakefield MJ, Widmann J, Wikman S, Wilson S, Ying H, Huttley GA (2007) PyCogent: a toolkit for making sense from sequence. Genome Biol 8:R171
McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P (2012) An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6:610–618
Koljalg U, Nilsson RH, Abarenkov K, Tedersoo L, Taylor AFS, Bahram M, Bates ST, Bruns TD, Bengtsson-Palme J, Callaghan TM, Douglas B, Drenkhan T, Eberhardt U, Duenas M, Grebenc T, Griffith GW, Hartmann M, Kirk PM, Kohout P, Larsson E, Lindahl BD, Luecking R, Martin MP, Matheny PB, Nguyen NH, Niskanen T, Oja J, Peay KG, Peintner U, Peterson M, Poldmaa K, Saag L, Saar I, Schuessler A, Scott JA, Senes C, Smith ME, Suija A, Taylor DL, Telleria MT, Weiss M, Larsson KH (2013) Towards a unified paradigm for sequence-based identification of fungi. Mol Ecol 22:5271–5277
Berg N, Steinberger Y (2010) Are biological effects of desert shrubs more important than physical effects on soil microorganisms? Microb Ecol 59:121–129. doi:10.1007/s00248-009-9599-4
Schrempf H (2013) Actinobacteria within soils: capacities for mutualism, symbiosis and pathogenesis. FEMS Microbiol Lett 342:77–78
Bulgarelli D, Schlaeppi K, Spaepen S, van Themaat EVL, Schulze-Lefert P (2013) Structure and functions of the bacterial microbiota of plants. Annu Rev Plant Biol 64:807–838
Bachar A, Soares MIM, Gillor O (2012) The effect of resource islands on abundance and diversity of bacteria in arid soils. Microb Ecol 63:694–700
Yu J, Grishkan I, Steinberger Y (2013) Microfungal-community diversity in Zygophyllum dumosum and Hammada scoparia root zones in the northern Negev Desert. J Basic Microbiol 53:390–400
Lau JA, Lennon JT (2011) Evolutionary ecology of plant-microbe interactions: soil microbial structure alters selection on plant traits. New Phytol 192:215–224
Brockett BFT, Prescott CE, Grayston SJ (2012) Soil moisture is the major factor influencing microbial community structure and enzyme activities across seven biogeoclimatic zones in western Canada. Soil Biol Biochem 44:9–20
Lindow SE, Brandl MT (2003) Microbiology of the phyllosphere. Appl Environ Microbiol 69:1875–1883
Fahlgren C, Hagstrom A, Nilsson D, Zweifel UL (2010) Annual variations in the diversity, viability, and origin of airborne bacteria. Appl Environ Microbiol 76:3015–3025
Whitman WB, Coleman DC, Wiebe WJ (1998) Prokaryotes: the unseen majority. Proc Natl Acad Sci U S A 95:6578–6583
Vokou D, Vareli K, Zarali E, Karamanoli K, Constantinidou HIA, Monokrousos N, Halley JM, Sainis I (2012) Exploring biodiversity in the bacterial community of the Mediterranean phyllosphere and its relationship with airborne bacteria. Microb Ecol 64:714–724
Redford AJ, Bowers RM, Knight R, Linhart Y, Fierer N (2010) The ecology of the phyllosphere: geographic and phylogenetic variability in the distribution of bacteria on tree leaves. Environ Microbiol 12:2885–2893. doi:10.1111/j.1462-2920.2010.02258.x
Kim SK, Gignoux CR, Wall JD, Lum-Jones A, Wang HS, Haiman CA, Chen GK, Henderson BE, Kolonel LN, Le Marchand L, Stram DO, Saxena R, Cheng I (2012) Population genetic structure and origins of native Hawaiians in the multiethnic cohort study. PLoS ONE 7:e47881
Knief C, Ramette A, Frances L, Alonso-Blanco C, Vorholt JA (2010) Site and plant species are important determinants of the Methylobacterium community composition in the plant phyllosphere. ISME J 4:719–728. doi:10.1038/Ismej.2010.9
van Overbeek L, van Elsas JD (2008) Effects of plant genotype and growth stage on the structure of bacterial communities associated with potato (Solanum tuberosum L.). FEMS Microbiol Ecol 64:283–296. doi:10.1111/j.1574-6941.2008.00469.x
Acknowledgments
We wish to thank Ms. Sharon Victor for her helpful comments. This study was part of a research program funded by the UNESCO/Japan Young Researchers’ Fellowship Program, 2012 (UNESCO/Keizo Obuchi Research Fellowships Program), awarded to Dr. Varsik Martirosyan, and which covered her stay in Israel. The sponsor played no role in the study design, collection, analysis, data interpretation, writing of the paper, or the decision to submit it for publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Funding
This study was part of a research program funded by the UNESCO/Japan Young Researchers’ Fellowship Program, 2012 (UNESCO/Keizo Obuchi Research Fellowships Program), awarded to Dr. Varsik Martirosyan, and which covered her stay in Israel.
Rights and permissions
About this article

Cite this article
Martirosyan, V., Unc, A., Miller, G. et al. Desert Perennial Shrubs Shape the Microbial-Community Miscellany in Laimosphere and Phyllosphere Space. Microb Ecol 72, 659–668 (2016). https://doi.org/10.1007/s00248-016-0822-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-016-0822-9