
Abstract
In many vertebrates, social interactions and nutrition can affect the colonization of gut symbionts across generations. In the highly social honey bee, it is unknown to what extent the hive environment and older worker individuals contribute to the generational transmission of core gut bacteria. We used high-throughput sequencing to investigate the effect of nest materials and social contact on the colonization and succession of core hindgut microbiota in workers. With only brief exposure to hive materials following natural eclosion, gut bacterial communities at 3 and 7 days contained phylotypes typically found in the guts of mature adults regardless of treatment. Continuous exposure to nest materials or direct social interactions with mature adults did not affect the diversity or abundance of gut bacterial communities at the scale examined. Similarly, a common pollen supplement fed by beekeepers during pollen dearth had no effect. A consideration of unique OTUs revealed extensive microbial succession independent of treatment. The dominant Lactobacillus strain at 3 days was largely replaced by a different strain at day 7, revealing the colonization signature of a pioneer species. Similar but less pronounced patterns were evident in less abundant OTU’s, many of which may influence community succession via alteration of the gut environment. Our results indicate that the process of bacterial community colonization in the hindgut is resilient to changes in the nutritional, hive, and social environment. Greater taxonomic resolution is needed to accurately resolve questions of ecological succession and typical proportional variation within and between core members of the gut bacterial community.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gut bacteria may exhibit narrow environmental tolerances as evidenced by their strict niche fidelity within the host gut. For successful transmission between generations, gut microbes can take advantage of host behavior, and/or possess genes that permit transient survival between generations [1, 2]. Colonization by gut bacteria can be influenced by environmental (non-core) bacteria, artificial nutrition, and the degree and type of host exposure [3–6]. In mice, humans, and many herbivorous vertebrates, gut communities first organize as pioneer communities, likely altering the gut epithelial environment, then assemble into a climax community resembling that of other adults in taxonomy and structure [3]. During establishment, many different pioneer species may be introduced and subsequently selected by sites in the GI tract of newly eclosed adults. Colonization of the insect gut can be influenced by opportunist microbes introduced from the environment, food consumption, and different diets [7]. Unlike most organisms, however, social insects construct elaborate niches (nest structures) where they store and process food and raise their young. The social insect nest may thwart the growth of microbes vectored from the landscape but simultaneously support and facilitate the generational transfer of co-evolved hindgut microbiota [8].
Roughly six to eight bacterial phylotypes occur consistently in the hindgut lumen of Apis mellifera workers [9–11], but little is known about the mechanisms involved in the colonization and persistence of this microbial community [12]. Of these eight phylotypes, six are primarily associated with the adult hindgut [6, 13]. Snodgrassella alvi, Gilliamella apicola, and Frischella perrara tend to occupy the ileum, and Lactobacillus firm 5, Lactobacillus firm 4, and Bifidobacterium seem to prefer the rectum as a niche [6, 13, 14]. The remaining two “core” bacteria; Alpha 1 (related to Bartonella/Brucella), and Alpha 2.1 (Acetobacteraceae) are poorly understood, and more sporadic in occurrence [9–11, 13, 15]. Although individual bees are typically dominated by a single strain of each species, they can contain multiple strains of the same core species [9]. In most studies, the hindgut is dominated by Lactobacillus firm 5, a broadly defined phylogroup containing the greatest 16S sequence divergence of any core species cluster [8, 11, 16]. For a comprehensive review of core gut bacteria, see Moran [17].
When exploring honey bee microbiology, it is practical to distinguish the individual from the colony, and the colony (collective individuals) from the nest construct [18]. Individuals that comprise the colony interact continuously with one another, the hive environment, and the pollination environment, activities related to age, diet, and adult developmental state. The hive itself acts as a microbial buffer from the external landscape as evidenced by the existence of a characteristic hive microbiota distinct from both the hindgut microbiota and microbes of the pollination environment [8]. One niche dominated by the hive microbiota is the honeybee foregut (crop). This highly expandable gut section is a “social stomach” used in water and nectar collection, the conversion of nectar to honey via evaporation, and frequent food sharing via trophallaxis [8, 19]. The hive microbiota is found in the crop, honey, pollen stores, larval food, larval guts, and larval provisioning glands, highlighting the microbial connectedness of the colony with its hive environment. Bacteria typically found in the crop are transient, either en route to the hindgut niche or part of the hive bacterial community [8, 11, 17]. There are many components of the colony and established nest that may influence microbial exposure, colonization, and transmission. Substances collected or produced by honey bees are antimicrobial to varying degrees, including nectar, pollen, wax, honey, bee salivary excretions, royal jelly, tree resins, and propolis. Stored pollen is preserved via the same mechanisms as honey but acts to some degree as a reservoir for acid-tolerant bacteria including floral associates, environmental microbes and some groups characteristic of the hindgut. When blooming plants are limited or unavailable, beekeepers feed sugar syrup and pollen substitute diets thought to affect gut microbial establishment or balance [20, 21].
New colony members emerge as winged worker adults within an existing nest structure. Metamorphosis occurs in preconstructed wax cells that first contain highly nutritious worker glandular secretions and are then lined with the proteinaceous silk of larval cocoons [22, 23]. Larvae contain fewer bacteria than adults, but taxonomically similar communities [15, 24]. The honey bee larval gut and contents are shed as meconium during metamorphosis, and left in the bottom of the wax cell. Cell cleaning is the first task assumed by newly emerged workers that repeatedly probe the recently vacated cells of newly emerged bees (NEB) [25]. At 2–3 days of age, worker bees progress to nurse behavior that requires gorging on stored pollen to sustain royal jelly production by their hypopharyngeal glands. This lipid and protein-rich substance is fed to growing larvae and the queen. A portion of the core gut bacteria can thrive in royal jelly and is also found in larva of various stages of development-fed royal jelly secreted by adult nurse bees [15, 24, 26]. Bacteria characteristic of both the hindgut and the hive environment have also been detected in the hypopharyngeal gland, royal jelly, and the guts of larvae [24, 26]. Similarly, with the exception of alpha 1, sequences corresponding to all of the adult hindgut bacteria have been found in newly collected or stored pollen [27]. Middle-aged bees process nectar into honey within the hive, while the oldest bees leave the hive to forage for water and plant products (pollen, nectar, resins). Although taxon identity is roughly defined, relying on small 16S rDNA segments, adult foragers, nurses, and “in-hive” bees possess similar hindgut microbiota despite very different diets and tasks [9–11].
NEB are colonized by bacteria following adult emergence, attaining their gut bacteria from older workers, the hive environment, or a combination of the two [6, 13, 28, 29]. Following adult emergence, worker bees perform a limited number of behaviors with the potential to affect bacterial colonization. They interact with hive materials and share food with other bees via oral trophallaxis. In particular, “cell cleaning” by NEB’s occurs immediately following emergence and has been described as a fixed developmental process, not as interchangeable as older adult tasks [30]. We hypothesize that cell cleaning and other early behaviors may serve to expose NEB’s to various microbial niches within the hive, promoting the inoculation and colonization of a healthy adult microbiota. In this study, we use deep sequencing and behavioral assays to explore the effect of both the social and hive environment on the early colonization of hindgut bacterial communities. Our experimental approach takes advantage of the hard-wired behavior of newly emerged bees, which do not interact with mature adults (perform trophallaxis) or consume food until 2 days of age [25, 30]. As an applied perspective, we also test the effects of a commonly used nutritional supplement fed by beekeepers.
Methods
Experimental Design and Sampling
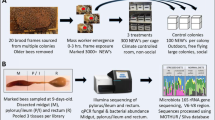
Experiments were performed at the USDA Carl Hayden Bee Research Center in Tucson, Arizona, USA. In the evening of September 23, 2013, frames of European honey bees containing sufficient brood were removed from six different hives and brushed clean of adult bees. These “brood” frames were placed in screened enclosures in a climate controlled room, and NEBs were allowed to emerge naturally overnight, exposed to hive components for 8–12 h. NEBs encountered the spectrum of inanimate components found on brood frames, including wax, stored pollen, royal jelly, capped and uncapped honey, and fringe environments like brood cell cappings, larval defecates, and silk cocoons. The following morning, NEB’s were homogenized in a large plastic tub, marked on the thorax with enamel paint, and divided into 24 groups of 50 bees each. Each group of 50 NEB’s was assigned to one of four experimental groups: (1) hive components, (2) artificial food, (3) hive components plus an older cohort of 300 bees, and (4) artificial food plus an older cohort of 300 bees. Thus, groups 1 and 2 remained as 50 individuals throughout the experiment, while groups 3 and 4 were increased to 350 individuals.
Each of the four treatment groups consisted of six replicates housed in flame-scorched boxes. One replicate from each treatment was placed in a 4-frame Plexiglas observation hive to survey behavior. Hive components consisted of two wax frames selected to provide natural food that had been collected and processed by honey bees and all substances associated with active brood rearing, including wax cells containing royal jelly, larvae of all ages, capped brood, and an abundance of stored pollen and honey (both open and capped).
Artificial food treatments lacked all typical hive components and contained plastic foundation frames void of wax cells. The artificial food treatment included both solids and liquids provided in sterile containers. The solid portion was a mixture (by weight) of granulated sugar (50 %), the commonly used pollen substitute “MegaBee” (32.5 %) and irradiated pollen (17.5 %). This solid food substitute was provided in a cylindrical falcon tube (15 ml) to limit spoilage potential. Using 20-ml plastic gravity drip-feeders, we supplied an autoclaved and ad libitum 50 % sucrose solution to each artificial food treatment.
The older cohort of worker bees was a random sample of 300 individual workers taken from around the brood nests of healthy active colonies. Thus, this group of bees likely contained the spectrum of functional cohorts including nurse workers with jelly production, and food processors and foragers that had defecated and then returned to the hive.
Experiments were performed in a climate controlled room at 33 °C and 50 % humidity. Autoclaved tap water was provided to all treatments in 20-ml sterile drip containers. Ten marked bees from each treatment were sampled at 3 and 7 days of age. Samples were immediately frozen on dry ice and stored at −80 °C until DNA extraction.
Behavioral Observations
We recorded the behavior of newly emerged bees using four separate observation hives. The frequency of behavioral acts was measured by visual scans with marked bees as focal subjects. We recorded acts from both sides of the frame. Scans were spaced at 10-min intervals amounting to 15 min per colony per day for the first 3 days postemergence. We used a chi-square analysis to compare the behavioral counts.
We defined six major behaviors: (1) cell cleaning: insertion of the head and thorax into empty cells where other workers had recently emerged; (2) trim caps: the chewing of brood cell capping material at the surface of the frame, around cells where workers had recently emerged; (3) lick surface: the extension of the proboscis to contact the surface of the frame; (4) eat pollen: insertion of the head into a cell or falcon tube that contained stored pollen or a mixture of pollen and protein supplement for a duration of >5 s; (5) eat nectar: insertion of the head into a cell that contained honey for a duration of >5 s; and (6) trophallaxis: the sharing of liquid food with another individual via proboscis extension and antennal contact.
When marked with paint for behavioral studies, newly emerged honeybees return invariably to the brood area and clean cells. Day 3 following adult emergence is typically associated with a behavioral shift from cell cleaning to brood care activities. It has been hypothesized that the newly emerged bee is acquiring adult physiological capability only available in this area of the nest [25, 30, 31].
Gut Bacteria Community Analysis
DNA Extraction and Pyrosequencing
DNA was extracted from the entire bee digestive tract excluding the foregut (crop). We used only guts that remained intact following dissection. For each DNA library (and extraction), we pooled the guts of five bees per replicate. Each pooled gut sample was placed into a sterile 2-mL centrifuge tube containing 350 μl of 0.5-mm silica beads and 1.5 ml TE/Triton X lysis buffer (20 mM Tris–HCl, 2 mM EDTA, 1.2 % Triton X-100, pH 8.0) and was subjected to bead beating for 30 s. The supernatant was transferred to a new 1.5-ml centrifuge tube and centrifuged for 5 min. The supernatant was removed, and 180-μl TE/Triton X lysis buffer (20 mM Tris–HCl, 2 mM EDTA, 1.2 % Triton X-100, pH 8.0; 20 mg/ml lysozyme added immediately before use) was added to the remaining pellet. Samples in lysis buffer were then subjected to genomic DNA extraction using the GeneJet Genomic DNA Purification Kit (Fermentas) following the protocol for gram-positive bacteria.
The V6–V8 region of the 16S rRNA was PCR amplified using universal 16S rRNA primers fitted with 454 FLX Titanium adapter sequences (926F 5′_CCATCTCATCCCTGCGTGTCTCCGACTCAG-NNNNNNNNNN-aaactyaaakgaattgacgg-3′; 1392R: 5′-CCTATCCCCTGTGTGCCTTGGCAGTCTCAG-acgggcggtgtgtrc-3′; uppercase letters denote the adapter sequences, Ns indicate library-specific barcodes, lowercase letters indicate universal 16S rRNA primers). Amplicons were sequenced using Roche 454 GS FLX + sequencing. All sequence data was deposited in the Sequence Read Archive under SRP057018.
Pyrotagged Sequence Analysis
Sequence data was processed using Mothur v.1.31.1 [32]. Sequences in the .sff files were quality filtered using the trim.seqs command (with no ambiguities or barcode differences, ≤8 homopolymers, ≤2 primer differences, window size = 50, average quality within window = 35). Pyrotags were removed as well as sequences that did not align to the V6–V8 16S region (using the align.seqs command; Silva SSURef database (v102)) or were < 184 bp in length so as to encompass at least the entire 16S V6 region. Chimeras were removed using UCHIME [33], in addition to any sequences that were mitochondrial, chloroplast, Archaeal, Eukaryote, or of unknown origin. A distance matrix was constructed for the aligned sequences using the dist.seqs command and the default parameters. Sequences were then grouped into operational taxonomic units (OTUs) based on 97 % similarity. Representative sequences from each OTU were then characterized in two ways. First, the sequences were used to query the NCBI nucleotide database. Second, these sequences were classified using the RDP Naïve Bayesian Classifier [34] using a manually constructed training set that contained sequences from the Greengenes 16S rRNA database, the RDP version 8 training set, and all full-length honey bee associated gut microbiota listed in NCBI trimmed to the V6–V8 region of the 16S rRNA gene. Any remaining sequences that were of chloroplast or mitochondrial origin, that were classified with less than 80 % confidence at the Phylum level according to the RDP Naïve Bayesian Classifier, or that were present in <2 libraries at a frequency of <2 sequences per library [9] were removed.
Multivariate Analysis of Gut Community Composition
The processed sequences were subjected to an analysis of the experimental factors. As above, a distance matrix was constructed for the aligned sequences using the dist.seqs command and the default parameters. Seven low-abundance libraries containing an average of nine sequences per library were removed leaving a total of 39 libraries for analysis. Representative sequences from each 100 % OTU were obtained and aligned using the align.seqs command. This alignment was filtered using the filter.seqs command (vertical = T, trump = .), and the resulting alignment was analyzed using the phangorn, seqinr, ape, and pegas packages in R [35, 36]. A pairwise distance matrix was constructed from the sequence alignment and a midpoint-rooted neighbor-joining phylogeny was constructed. Rarefied estimates of the number of sequences per library per OTU were generated using the GUniFrac package [37] in R. Unweighted, weighted, variance adjusted weighted (VAW), and generalized (α = 0.5) pairwise UniFrac distances were then calculated, and a permutational multivariate analysis of variance was used to determine whether time (3 vs. 7 days), older bees (present vs. not present) nested within time, and hive materials (natural vs. artificial) nested within older bees and time affected gut community diversity (i.e., the combined UniFrac distances between libraries).
Post hoc Analysis
To discover patterns within and among our experimental groups, we further compared the treatment groups deemed significant by our multivariate analysis. We used univariate testing to compare the relative abundances of the top 25 unique OTUs, calculated by dividing the absolute abundances by the total sequence count per sample. To account for compositional constraints (the compared frequencies violate the assumption of independence because read numbers are drawn from the same pool of finite sequence reads), we used the Kruskal-Wallis test followed by the false discovery rate (FDR) correction for multiple testing [38].
Co-occurrence of Lactobacillus firm 5
We further explored our data set by comparing OTU occurrence patterns of Lactobacillus firm 5 using simulations generated in ECOSIM [39]. More specifically, we tested the hypothesis that the occurrences of unique Lactobacillus firm 5 OTUs were non-independent, reflecting biological interaction in the early gut environment. For this analysis, we compiled the 20 most common Lactobacillus firm 5 OTUs accounting for 80.7 % of all reads and 98 % of all firm 5 reads. We constructed a matrix with rows as unique OTUs (presence or absence) and columns as independent gut samples (39 amplicon libraries). We used ECOSIM to evaluate two different aspect of firm 5 OTU co-occurrence, calculating the C-score [40] as the average of all pairwise values for a matrix, and the V-index as the ratio of the variance of the column sums to the sum of the row variances [41]. In constructing the null model for both metrics, the rows were iterated with fixed sums (default), to avoid Type I errors [39]. The sites (columns) were set at equiprobable to reflect an equal chance for each gut to be colonized by different firm 5 strains. Simulations were performed with and without singletons in the matrix.
Results
Gut Bacteria Community Analysis
Reads Analyzed and Taxa Generated
A total of 884,149 reads were generated across the 48 sample libraries. Following initial quality trimming, we retained 39 libraries and 461,229 reads. Removing suspect or low-abundance OTUs (349 sequences) yielded 460,880 sequences in the final analysis. A total of 32 operational taxonomic units (OTUs) were resolved across the 39 libraries at the 97 % level of similarity, and 418 OTUs were resolved at the 100 % level of similarity (Table S1). Although 82 % of all sequences were classified as Lactobacillus sp. firm 5, the remaining members of the core honey bee microbiota (Lactobacillus sp. firm 4, Bifidobacterium sp., Gilliamella apicola, Snodgrassella alvi, and Frischella perrara) were all represented within the top 25 unique OTUs (Table 1). G. apicola was represented by three unique OTUs, S. alvi by three, and F. perrara by one. Considered by some studies as core to the hindgut, the relatively unknown phylotypes alpha 1 and alpha 2.1 were detected in 33 and 15 % of the 39 libraries, respectively.
Now considered a hive bacteria common in the crop, larvae, and food stores, the recently named Parasaccharibacter apium (alpha 2.2) was detected at very low levels, confirming its exclusion from the core gut group. In contrast, Lactobacillus kunkeei, another non-gut bacterium found throughout the hive and pollination environment, was somewhat more abundant and present in a majority of libraries irrespective of treatment or sampled age.
Multivariate Analysis of Gut Community Composition
We used PermANOVA to determine whether time (3 vs. 7 days), hive components (natural vs. artificial), and older worker cohorts (presence vs. absence) affected gut community diversity. In the full analysis, only the effect of time (3 vs. 7 days) was significant (F 1,38 = 8.24, p = 0.001). When the data from each time period (3 or 7 days) was analyzed separately for the effect of hive components and older worker cohorts, neither effect was significant.
Post hoc Analysis
We examined the mean relative abundances of the 25 major OTUs (Table 1), detecting five OTUs that differed significantly across the sampled time period. All five corresponded to core hindgut Lactobacillus species. The primary OTU shift from 3 to 7 days involved an increase in the first and third most abundant OTUs, Lactobacillus firm 5 and Lactobacillus firm 4, with a concomitant decrease in the second most abundant OTU, Lactobacillus firm 5 (Fig. 2). Although a magnitude less abundant, another OTU classified as Lactobacillus firm 4 increased significantly over the sampled time period. We detected similar but non-significant trends for core bacterial species with less read abundance including Bifidobacterium and Frischella perrara.
Different patterns were evident for other core OTUs. The most numerous Bifidobacterium OTU was present in 100 % of the libraries, at the same relative abundance (1.5 %) regardless of sampled day. Similarly, the fourth and fifth most abundant OTUs, Lactobacillus firm 5 and Snodgrassella alvi showed little change over time (Table 1).
Accounting for 1.2 % of the sequences in Table 1, only three of the 25 most abundant OTUs are considered environmental or hive bacteria, as opposed to core hindgut bacteria. All three decreased in relative abundance over the sampled time period. Many of the moderately abundant Lactobacillus firm 5 showed a similar pattern or were sporadic in abundance among libraries (Fig. 3).
Co-occurrence of Lactobacillus firm 5
Across all gut environments, Lactobacillus firm 5 was represented by total of 162 unique OTUs, while the other eight core species combined were represented by a total of 21 OTUs. A diversity metric is not needed to describe this as a dominance environment. Across all libraries, Lactobacillus firm 5 accounted for 81 % of total read number. As a proportion of total reads, sequences corresponding to Lactobacillus firm 5 decreased significantly from 3 to 7 days (paired t test, t 16 = 3.75, P = 0.002), a change due primarily to the decrease in OTU2 over the measured time period (Figs. 2 and 3). Using ECOSIM, we determined the C-score and V-index as indications of biological processes occurring among Lactobacillus firm 5 OTUs. At 3 days, both the C-score and V-index were significant, a pattern disappearing by day 7 (Table 2). The C-score was significantly smaller than expected by chance indicating greater than average species co-occurrence. The V-ratio was significantly larger than expected by chance indicating positive covariance between species pairs. Robust to noise, both metrics were largely unaffected following removal of 43 singletons. The relative abundance of singletons did not differ significantly between the two age groups (Chi-square = 0.21, P = 0.65), averaging 1.1 singletons per OTU, and accounting for only 5 % of matrix values.
Behavioral Observations
Consistent with Seeley [25], our behavioral results demonstrate that despite being removed from their typical colony environment, newly emerged bees perform tasks typical of 1–3-day-old bees when given access to hive components (Fig. 1). The artificial food treatments had no hive components; hence, the only behaviors that could be compared were eating artificial food or trophallaxis. We recorded more food consumption in the treatment lacking both hive components and older bees, but this effect was likely due to decreased potential for alternate behaviors, and the consistent aggregation of individuals in and around the tube containing artificial food. Comparing the presence vs. absence of older workers in the treatments containing hive components, instances of trophallaxis were low relative to cell cleaning behaviors, and no difference in the frequency of trophallaxis was evident between these groups. The proportional occurrence of NEB that were trimming caps was increased significantly in the presence of 300 older bees. The lack of older bees was associated with a significantly greater frequency of NEB cleaning cells (χ 2 = 47.5, df = 5, P < 0.0001).

Behaviors recorded when observing 1–3-day-old worker bees given typical hive materials. Groups are presence or absence of 300 older worker bees introduced from other colonies (older cohorts). Observed counts are based on focal scans performed 15 min/day per colony, for a grand total of 45 min per treatment
Discussion
We explored effects of the social and hive environment on bacterial colonization of the gut in newly emerged honey bees. Exposure to natural hive materials for an average of 8–12 h posteclosion was sufficient to inoculate workers with all core members of the gut bacterial community. Contact with older worker cohorts was unnecessary. Following natural eclosion, the establishment of gut microbial communities was unaffected by continuous exposure to the hive environment, interaction with older worker cohorts, or the consumption of artificial food. These results are robust because the potential sources of bacterial inoculate were multiple and varied. The introduced social environment of 300 older bees likely contained all adult age cohorts and key physiological caste differences including “post-defecation” workers, nurses producing royal jelly, food processors, and foragers with mature salivary enzyme profiles. Similarly, the introduced hive environment included wax frames, honey, royal jelly, stored pollen, propolis, developing individuals, and newly emerging adults. Conversely, the artificial food environment contained only plastic (empty) frames, sterile tubes containing artificial pollen substitute used by beekeepers, 50 % sucrose solution, and water. Despite these prevalent differences, bacterial communities were statistically indistinguishable across treatments. These results indicate that all eight major groups of gut bacteria were acquired directly from natural hive components in 8–12 h post-emergence. Measured at 3 and 7 days, subsequent colonization was largely unaffected by continuous contact with nest materials, artificial diet, or the presence of older colony members. These findings demonstrate that brief initial exposure, followed by selection in the host gut, are strong determinants of gut bacterial colonization in honey bees [42]. Our results also demonstrate that Lactobacillus firm 5 dominates early gut colonization perhaps due to its survival in the hive environment [6, 8, 27].
Colonization Differs by Species and Strain
We found that colonization occurs in a species and strain-specific manner. Stark differences from 3 to 7 days indicate that early colonizers may dramatically alter the gut niche (Fig. 2). As a proportion of the total gut community, the relative abundance of Lactobacillus firm 5 decreased by 7.3 % over the assessed time period due almost exclusively to a concurrent increase in the relative abundance of Lactobacillus firm 4 (Table 1). As bacterial numbers in the hindgut are well established by day 3 [6], firm 4 may have displaced firm 5 as opposed to colonizing an unoccupied niche. It is also possible that the ingestion of pollen is more suited to firm 4 growth or establishment. A striking population shift from 3 to 7 days occurred within Lactobacillus firm 5, a group representing the majority of reads and more than half of the unique OTUs recovered among the top 25 most abundant (Fig. 3, Table 1). Within firm 5, the replacement of OTU2 by OTU1 occurred in a uniform fashion across all replicates and treatments (Fig. 3). This early and massive shift in bacterial communities suggests that OTU2 may function as a pioneer species for both Lactobacillus firm 4 and other strains of Lactobacillus firm 5. Like environmental bacteria in the guts of vertebrates, particular strains of firm 5 may prime the gut by lowering the pH or generating metabolic byproducts [3]. Sequences with 100 % similarity to firm 5 OTU2 have been cultured and/or cloned from a variety of environments including the honey bee foregut, stored pollen, floral nectar, and the guts of other plant associated insects [8], suggesting that OTU2 is a generalist firm 5, capable of multiplying in a variety of hive associated environments. We hypothesize that bacteria performing the role of pioneer species in honey bee guts reside within the hive on a consistent basis and include many strains of Lactobacillus firm 5 most adapted to the hive environment and a couple atypical taxa more typically labeled as pioneer species during gut colonization: Enterococcaceae and Enterobacteriacreae [3]. Although rare in our study, such “atypical” bacteria were more prevalent in the gut at day 3 and virtually undetected by day 7.

Proportional change in the three most abundant OTU’s in the gut from 3 to 7 days postemergence. Shown in parentheses, OTU’s are composed of unique sequences (100 %) and represent Lactobacillus species commonly associated with the rectum. Groups labeled firm 4 and firm 5 are phylogenetic designations based on 16S rRNA sequence monophyly [16, 57]

Diversity and succession of the 20 most common Lactobacillus firm 5 OTUs accounting for 80.7 % of total amplicon reads, and 98 % of all reads classified as firm 5. Each color represents a unique OTU (unique sequence). The upper section represents gut communities at 3 days of age, the lower section at 7 days of age. Values at the right of each horizontal bar are read numbers representing the 20 most abundant Lactobacillus firm 5, and the proportion (in parentheses) of the total community represented by the top 20 Lactobacillus firm 5
Gut colonization patterns likely represent many different community-level processes including interstrain competition, cross-feeding, population growth, and niche selection [43]. For example, honey bee associated Bifidobacterium is found at highly predictable proportions across studies regardless of adult developmental state [9–11, 13, 44]. Unlike any Bifidobacterium described thus far, it is equipped to endure the hive environment, possessing genes for oxidative metabolism and defense against reactive oxygen species [45, 46]. Although relatively rare in stored pollen, we detected Bifidobacterium in every replicate gut library (Table 1). While one of the Bifidobacterium detected by our study (Table 1; OTU23) had been largely selected out of the gut niche by day 7, the more prevalent Bifidobacterium (OTU7) had apparently established in a well-defined gut niche by day 3, and remained unaltered in relative abundance at day 7, a pattern suggestive of extreme niche fidelity (Table 1). At the other end of the spectrum, the core bacterium alpha 2.1 was barely detected at 3 or 7 days of age but is abundant in queens and much older pollen foragers (Table S1), two groups of bees that receive the protein in their diets solely from royal jelly [6, 11, 47]. Unlike Bifidobacterium, alpha 2.1 may respond to environmental factors experienced by foragers, but not commonly found in the early gut environment of workers.
Gram-negative core species that predominate in the adult ileum occurred in the majority of newly emerged bees. Unlike gram positive species, they were absent or undetected in some replicate libraries. At 7 days of age, S. alvi was present in 95 % of libraries, while F. perrara and G. apicola occurred in 68 and 50 % of libraries respectively. The abundance of these same groups in corbicular and stored pollen shows the inverse of this pattern with S. alvi detected at the lowest levels [27]. Thus, early colonization dynamics likely reflect species-specific growth or transmission differences (see [29]), and/or our sampling approach. Consistent with differences in epithelial surface area, the rectum tends to contain approximately 10× more bacteria than the ileum [6, 13]. We performed DNA extractions on whole guts, a method favoring the subsequent PCR amplification of rectum-specific species. Thus, core species that tend to grow slowly or are highly specific to the ileum may be underrepresented by our approach. If we account for this bias when interpreting Table 1, colonization patterns would be amplified for the ileum inhabitants S. alvi (OTU17), G. apicola (OTU19), and F. perrara (OTU21). Although the ileum is likely the primary niche of S. alvi, this species can be found in the rectum with some regularity and abundance [6, 13]. Further, it has been suggested that the evolution of S. alvi strains was fashioned in part by competition within the gut niche [14]. In support of this claim, the two most abundant S. alvi strains show a negative co-occurrence pattern consistent with competitive exclusion from the same niche (Table S1).
Lactobacillus Firm 5 Co-occurrence
The Lactobacillus firm 5 phylotype dominates the early honey bee gut comprising a large portion of the pioneer (cell cleaning) and primary succession (nurse development) communities, a pattern confirmed in other studies of honey bees [6, 29]. Results demonstrate that firm 5 is typically the most abundant group of bacteria in the honey bee hindgut, and also the core gut member most commonly found throughout the hive environment in honey, beebread, and the foregut [8–11, 15, 27]. Combined with these findings, our results suggest that the degree to which a core gut bacterium can endure the hive environment affects inoculation and colonization in the guts of newly emerged bees. To further identify colonization differences within Lactobacillus firm 5, we analyzed strain co-occurrence. To simplify this analysis, we included only the 20 most abundant firm 5 OTUs accounting for 98 % of all reads classified as firm 5. Removing the remaining 2 % of unique firm 5 reads (142 OTUs) did not affect subsequent analysis.
Community metrics revealed significant patterns among the top 20 firm 5 strains (unique OTU’s) at day 3 (Table 2). Relative to a randomly assembled community, the V-ratio was large and C-score small, the pattern expect of a community not yet structured by competition. Specifically, the V-ratio at 3 days indicates significant heterogeneity in strain richness by gut, suggesting relaxed constraints on the number of co-existing firm 5 strains per gut, perhaps a result of scramble competition or stochastic strain introduction. Consistent with the V-ratio, the C-score at 3 days reveals a significant tendency for firm 5 strains to co-occur, again indicating a lack of competition. Over the course of colonization, the co-variance in species number narrowed significantly indicating a refinement in the number of firm 5 strains inhabiting the gut niche. More informative than the statistical significance of these metrics at day 3, the direction of change over the sampled time course suggests that interstrain competition begins to shape community structure by day 7. Collectively, the direction of change suggests a hypothesis for firm 5 colonization in the adult gut. Day three is reflective of stochastic or “pioneer” species introduction, while day 7 reflects greater density-dependent effects as the gut becomes fully colonized.
Sources of Inoculation
Concerning inoculation in general, and particularly of the gram-negative ileum-associated strains, our study should be carefully distinguished from a recent and related study designed to control for microbial exposure post-eclosion (see [6]). This study removed pre-emergence pupae from their wax cells, allowed them to eclose in a sterile petri dish, and then applied various treatments using sterile cups. The control group eclosed like the others but was exposed to no treatment. The control produced 106 16S gene copies with qPCR, but unfortunately failed to amplify using the barcoded 16S amplicon primers. Thus, it remained unclear what portion of the bacterial communities resulting from this sterile cup assay could be ascribed to experimental error or bacteria clinging to the bodies of eclosed pupae. This is a critical point because larvae contain core gut bacteria and defecate within their wax cells prior to pupation [24]. In the absence of a negative control, effects were compared to a “standard” community stressing evenness [9]. As a positive inoculation control, the hindguts of mature adults were macerated and fed to newly emerged workers. Communities resulting from the macerated hindgut treatment differed significantly from all others, revealing greater average relative proportions of the three gram-negative bacteria that colonize the ileum. Based on this result, a general lack of gram-negative species from other treatments, and similarity in evenness with the chosen standard community, it was concluded that the gram-negative bacteria appear to be acquired through non-trophallaxis contact with older (nurse) bees. Applied to our study, this conclusion suggests that the gram-negative core bacteria we identified at relatively low levels may not be the most successful colonizers of the ileum, and/or that the ileum associated core bacteria were underrepresented by our PCR assay. It is also possible that a standard gut community has yet to emerge from studies of honey bee gut bacteria due to methodological or other differences [10].
Newly emerged workers were frequently seen cleaning the interior of recently vacated brood cells the first 3 days following emergence [25, 30]. Further, we recorded many instances of NEB trimming irregular cap remnants of this same cell type (Fig. 1). We hypothesize that cell cleaning behavior may be important for gut bacterial inoculation. It is possible that all of the characteristic bacteria found at low levels in larval guts may survive within brood cells following larval defecation. Although a comprehensive study is lacking, a combination of culture-dependent and culture-independent studies indicates that larvae contain all core bacterial groups found in adults, with last instar larvae harboring the greatest core bacterial diversity [15, 24]. Just prior to pupation, larvae deposit meconium at the bottom of the cell, a substance composed of shed epithelial cells, >100,000 pollen grains, uric acid crystals, and other sources of nutrition that may temporarily support core bacteria [48]. Silk proteins are abundant in larval cells and may also provide sustenance for transitional bacteria [49]. However, it is unlikely that all of the major groups of gut bacteria employ the same transmission strategy. As suggested by Powell [6], transmission may be multimodal as there are many fringe environments and interaction networks within the hive and colony that may support bacteria.
We detected core gram-positive species in all gut libraries at both 3 and 7 days (Table 1), but the gram-negative core species, primarily G. apicola and F. perrara, were conspicuously absent from many libraries. It has been hypothesized that inoculation with the core gram-negative species may require contact with adult hindgut defecates [6, 29]. The addition of macerated adult hindguts directly to food resulted in 8-day-old gut communities with relatively greater prevalence of gram-negative core bacteria, although a few libraries still lacked G. apicola and F. perrara [6]. While this artificial inoculation (a form of conventionalization) does not occur naturally, it indicates that the gram-negative species are more quickly assimilated when abundant in the environment. Given the high concentration of bacteria in the adult hindgut, incidental contact of the adult abdomen with hive materials may be one natural transmission route. But, bacteria that occupy the more anterior ileum may have different transmission frequencies. As a result of incidental contact, all members of the core gut bacterial community can be found in newly collected (corbicular) pollen but decrease in abundance with pollen storage time [27]. Newly emerged bees do not consume stored pollen on the day they emerge [25, 30], but the pollen results suggest that hindgut bacteria occur commonly on the outer surface of the forager abdomen. In actively foraging colonies, the abdominal tip of adult workers that have recently defecated and then returned to the hive frequently contacts the wax surface and cell cappings of developing individuals. Given the high frequency of self and other grooming, it is likely that such inoculates are constantly spread throughout the hive with bacterial survival time dependent on the particular species/strain/microenvironment. Because many of the gut bacteria may not survive for extended periods in the hive environment, fresh and somewhat continuous inoculate may be needed for an effective transmission route. Such colony-level inoculation dynamics may be diminished by cage studies [50].
Typical colony interactions are altered by cup experiments containing very few bees [6], or much larger frames housed in small hive (nuclear) boxes containing 50–350 bees (this experiment). Even in small-scale sterile cup experiments, the high degree of community variation attributable to replicate “cups” indicates stochastic inoculation followed by social transmission [6]. Honey bees groom one another and transfer liquid food to other individuals at high frequency, social behaviors that continue when worker bees are placed in artificial cages or small hives that lack a queen and developing brood [51]. In contrast with the conclusions of Powell et al. [6], we found no difference attributable to the social environment that included 300 adult bees and the spectrum of adult development including “nurse age” bees. These adult bees were applied 8–12 h posteclosion and given ample time to affect the microbiota. Although we observed trophallaxis at low frequency relative to other recorded behaviors (Fig. 1), our findings indicate that grooming others and oral trophallaxis are unnecessary for inoculation with core gut bacteria, because NEB acquired the same bacteria regardless of exposure to older cohorts. However, different strains may colonize when resources are abundant, and all interactive components of the colony are functioning in unison. Cage studies exclude or limit many potential inoculation routes including worker orientation and defecation flights, incoming and outgoing foragers, fresh pollen and nectar, and associated signals and social interactions like nursing behavior. Cage studies can also introduce stress, a factor known to affect gut bacterial diversity in bees and other organisms [52, 53].
Perspective
A greater understanding of the developing gut ecosystem holds promise for disease prevention, immune status, and environmental detoxification in managed honey bees. Our results suggest that the colonization and persistence of core hindgut bacterial species are highly resilient to alterations in typical hive function including diet and social contact. With only a brief exposure to hive materials, honey bees were able to acquire a microbiota indistinguishable from those with continuous exposure to hive materials and older individuals. The consumption of stored pollen known to contain gut bacteria did not alter gut colonization dynamics, but we cannot exclude the development of the hypopharyngeal gland in the colonization process [26, 27]. Diet changes can rapidly alter gut bacterial communities in both insects and vertebrates [54, 55]. In contrast, our results indicate that the use of pollen substitutes by beekeepers does not affect bacterial establishment in newly emerged bees. Similarly, bumblebees show great resilience in gut microbial community composition when the food supply is altered [56]. The hindgut bacterial community of the honey bee has clearly developed in accord with host-associated tissues of the ileum and rectum. While most studies reveal the continuous presence of six broad taxonomic groups, the characteristic or “core” community proportions by gut segment are still unknown, as is the occurrence/combination of species/strains that are beneficial, detrimental, or commensal. As each major honey bee behavioral transition is accompanied by changes in diet, activity, and physiology, bacterial abundance and function may vary in accordance with adult developmental state [29]. Future experiments including taxon-specific qPCR and RT-qPCR can link community composition with biological function, providing further insight to the patterns reported here.
References
Troyer K (1984) Microbes, herbivory and the evolution of social behavior. J Theor Biol 106:157–169. doi:10.1016/0022-5193(84)90016-X
Lombardo MP (2008) Access to mutualistic endosymbiotic microbes : an underappreciated benefit of group living. Behav Ecol Sociobiol 62:479–497. doi:10.1007/s00265-007-0428-9
Gillilland MG, Erb-Downward JR, Bassis CM et al (2012) Ecological succession of bacterial communities during conventionalization of germ-free mice. Appl Environ Microbiol 78:2359–2366. doi:10.1128/AEM.05239-11
Wall R, Hussey SG, Ryan CA et al (2008) Presence of two Lactobacillus and Bifidobacterium probiotic strains in the neonatal ileum. ISME J 2:83–91. doi:10.1038/ismej.2007.69
Blum JE, Fischer CN, Miles J, Handelsman J (2013) Frequent replenishment sustains the beneficial microbiome of Drosophila melanogaster. MBio. doi:10.1128/mBio.00860-13
Powell JE, Martinson VG, Urban-Mead K, Moran NA (2014) Routes of acquisition of the Gut microbiota of the honey Bee Apis mellifera. Appl Environ Microbiol 80:7378–7387. doi:10.1128/AEM.01861-14
Pernice M, Simpson SJ, Ponton F (2014) Towards an integrated understanding of gut microbiota using insects as model systems. J Insect Physiol. doi:10.1016/j.jinsphys.2014.05.016
Anderson KE, Sheehan TH, Mott BM et al (2013) Microbial ecology of the hive and pollination landscape: bacterial associates from floral nectar, the alimentary tract and stored food of honey bees (Apis mellifera). PLoS One 8:e83125. doi:10.1371/journal.pone.0083125
Moran NA, Hansen AK, Powell JE, Sabree ZL (2012) Distinctive Gut microbiota of honey bees assessed using deep sampling from individual worker bees. PLoS One 7:e36393. doi:10.1371/journal.pone.0036393
Sabree ZL, Hansen AK, Moran NA (2012) Independent studies using deep sequencing resolve the same set of core bacterial species dominating Gut communities of honey bees. PLoS One 7:e41250. doi:10.1371/journal.pone.0041250
Corby-Harris V, Maes P, Anderson KE (2014) The bacterial communities associated with honey Bee (Apis mellifera) foragers. PLoS One 9:e95056. doi:10.1371/journal.pone.0095056
Kwong WK, Moran NA (2015) Evolution of host specialization in gut microbes: the bee gut as a model. Gut Microbes 6:214–220. doi:10.1080/19490976.2015.1047129
Martinson VG, Moy J, Moran NA (2012) Establishment of characteristic gut bacteria during development of the honey bee worker. Appl Environ Microbiol 78:2830–2840. doi:10.1128/AEM.07810-11
Engel P, Stepanauskas R, Moran NA (2014) Hidden diversity in honey bee Gut symbionts detected by single-cell genomics. PLoS Genet 10:e1004596. doi:10.1371/journal.pgen.1004596
Ahn J-H, Hong I-P, Bok J-I et al (2012) Pyrosequencing analysis of the bacterial communities in the guts of honey bees Apis cerana and Apis mellifera in Korea. J Microbiol 50:735–45. doi:10.1007/s12275-012-2188-0
Martinson VG, Danforth BN, Minckley RL et al (2011) A simple and distinctive microbiota associated with honey bees and bumble bees. Mol Ecol 20:619–628. doi:10.1111/j.1365-294X.2010.04959.x
Moran NA (2015) Genomics of the honey bee microbiome. Curr Opin Insect Sci 10:22–28. doi:10.1016/j.cois.2015.04.003
Evans JD, Spivak M (2010) Socialized medicine: individual and communal disease barriers in honey bees. J Invertebr Pathol 103(Suppl):S62–S72
Crailsheim K (1988) Regulation of food passage in the intestine of the honeybee (Apis mellifera L.). J Insect Physiol 34:85–90. doi:10.1016/0022-1910(88)90158-8
Anderson KE, Sheehan TH, Eckholm BJ et al (2011) An emerging paradigm of colony health: microbial balance of the honey bee and hive (Apis mellifera). Insect Soc 58:431–444. doi:10.1007/s00040-011-0194-6
Disayathanoowat T, Young JPW, Helgason T, Chantawannakul P (2011) T-RFLP analysis of bacterial communities in the midguts of Apis mellifera and apis cerana honey bees in Thailand. FEMS Microbiol Ecol 79:273–281. doi:10.1111/j.1574-6941.2011.01216.x
Zhang K, Duan H, Karihaloo BL, Wang J (2010) Hierarchical, multilayered cell walls reinforced by recycled silk cocoons enhance the structural integrity of honeybee combs. Proc Natl Acad Sci U S A 107:9502–9506. doi:10.1073/pnas.0912066107
Kamakura M (2011) Royalactin induces queen differentiation in honeybees. Nature 473:478–83. doi:10.1038/nature10093
Vojvodic S, Rehan SM, Anderson KE (2013) Microbial Gut diversity of Africanized and European honey Bee larval instars. PLoS One 8:e72106. doi:10.1371/journal.pone.0072106
Seeley TD (1982) Adaptive significance of the age polyeithism schedule in honeybee colonies. Behav Ecol Sociobiol 11:287–293
Corby-Harris V, Snyder LA, Schwan MR et al (2014) Origin and effect of alpha 2.2 Acetobacteraceae in honey bee larvae and description of Parasaccharibacter apium gen. nov., sp. nov. Appl Environ Microbiol 80:7460–7472. doi:10.1128/AEM.02043-14
Anderson KE, Carroll MJ, Sheehan TIM, Mott BM (2014) Hive-stored pollen of honey bees: many lines of evidence are consistent with pollen preservation, not nutrient conversion. Mol Ecol. doi:10.1111/mec.12966
Koch H, Schmid-Hempel P (2011) Bacterial communities in central European bumblebees: low diversity and high specificity. Microb Ecol 62:121–133. doi:10.1007/s00248-011-9854-3
Guo J, Wu J, Chen Y et al (2015) Characterization of gut bacteria at different developmental stages of Asian honey bees, Apis cerana. J Invertebr Pathol 127:110–114. doi:10.1016/j.jip.2015.03.010
Johnson BR (2010) Division of labor in honeybees: form, function, and proximate mechanisms. Behav Ecol Sociobiol 64:305–316. doi:10.1007/s00265-009-0874-7
Seeley D, Kolmes A (1991) Age polyethism for hive duties in honey bees - illusion or reality? Ethology 87:284–297
Schloss PD, Westcott SL, Ryabin T et al (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–41. doi:10.1128/AEM.01541-09
Edgar RC, Haas BJ, Clemente JC et al (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–200. doi:10.1093/bioinformatics/btr381
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi:10.1128/AEM.00062-07
Schliep KP (2011) Phangorn: phylogenetic analysis in R. Bioinformatics 27:592–3. doi:10.1093/bioinformatics/btq706
Paradis E (2010) Pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics 26:419–20. doi:10.1093/bioinformatics/btp696
Chen J, Bittinger K, Charlson ES et al (2012) Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics 28:2106–13. doi:10.1093/bioinformatics/bts342
Benjamini YHY (1995) Benjamini and Y FDR.pdf. J R Stat Soc Ser B 57:289–300
Gotelli NJ and GLE (2002) EcoSim: null models software for ecology. Version 7.0. Jericho VT, USA
Stone L, Roberts A (1990) The checkerboard score and species distributions. Oecologia 85:74–79. doi:10.1007/BF00317345
Schluter D (1984) A variance test for detecting species associations with some example applications. Ecology 65:998–1005
Schluter J, Foster KR (2012) The evolution of mutualism in Gut microbiota Via host epithelial selection. PLoS Biol. doi:10.1371/journal.pbio.1001424
El Aidy S, Van Den Abbeele P, Van De Wiele T et al (2013) Intestinal colonization: how key microbial players become established in this dynamic process: microbial metabolic activities and the interplay between the host and microbes prospects & overviews S E. Aidy et al. BioEssays 35:913–923. doi:10.1002/bies.201300073
Lee FJ, Rusch DB, Stewart FJ et al (2014) Saccharide breakdown and fermentation by the honey bee gut microbiome. Environ Microbiol. doi:10.1111/1462-2920.12526
Bottacini F, Milani C, Turroni F et al (2012) Bifidobacterium asteroides PRL2011 genome analysis reveals clues for colonization of the insect Gut. PLoS One 7:e44229. doi:10.1371/journal.pone.0044229
Anderson KE, Johansson A, Sheehan TH et al (2013) Draft genome sequences of two Bifidobacterium sp. From the honey bee (Apis mellifera). Gut Pathog 5:42. doi:10.1186/1757-4749-5-42
Tarpy DR, Mattila HR, Newton ILG (2015) Characterization of the honey bee microbiome throughout the queen-rearing process. Appl Environ Microbiol. doi:10.1128/AEM.00307-15, AEM.00307–15
Simpson J, Simpson BJ (1955) The significance of the presence of pollen in the food of worker larvae of the honey-bee. Q J Microsc Sci 96:117–120
Sutherland TD, Campbell PM, Weisman S et al (2006) A highly divergent gene cluster in honey bees encodes a novel silk family a highly divergent gene cluster in honey bees encodes a novel silk family. Genome Res 16:1414–1421. doi:10.1101/gr.5052606
Lass A, Crailsheim K (1996) Influence of age and caging upon protein metabolism, hypopharyngeal glands and trophallactic behavior in the honey bee (Apis mellifera L.). Insect Soc 43:347–358. doi:10.1007/BF01258408
Crailsheim K (1998) Trophallactic interactions in the adult honeybee (Apis mellifera L.). Apidologie 29:97–112. doi:10.1051/apido:19980106
Gilliam M (1997) Identification and roles of non-pathogenic microflora associated with honey bees. FEMS Microbiol Lett 155:1–10
Archie EA, Theis KR (2011) Animal behaviour meets microbial ecology. Anim Behav 82:425–436. doi:10.1016/j.anbehav.2011.05.029
Wang Y, Gilbreath TM, Kukutla P et al (2011) Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS One 6:e24767. doi:10.1371/journal.pone.0024767
David LA, Maurice CF, Carmody RN et al (2014) Diet rapidly and reproducibly alters the human gut microbiome. Nature 505:559–63. doi:10.1038/nature12820
Koch H, Cisarovsky G, Schmid-Hempel P (2012) Ecological effects on gut bacterial communities in wild bumblebee colonies. J Anim Ecol 81:1202–1210. doi:10.1111/j.1365-2656.2012.02004.x
Babendreier D, Joller D, Romeis J et al (2007) Bacterial community structures in honeybee intestines and their response to two insecticidal proteins. FEMS Microbiol Ecol 59:600–610. doi:10.1111/j.1574-6941.2006.00249.x
Engel P, Martinson VG, Moran NA (2012) Functional diversity within the simple gut microbiota of the honey bee. Proc Natl Acad Sci U S A 109:1–6. doi:10.1073/pnas.1202970109
Acknowledgments
The authors acknowledge Jay Evans and Randy Oliver for comments on a previous version of the manuscript and the members of the Anderson lab for technical assistance. The corresponding author thanks Belynda Starr, Ariel Calypso, and Isaak Edward for their valuable input. We thank the Bio5 Institute at the University of Arizona Genomics Core for the generation of 454 amplicons. The USDA is an equal opportunity employer and provider.
Author Contributions
PAPR, BMM, and KEA conceived and designed research, PAPR, PM, and KEA performed research, PAPR, VCH, and KEA analyzed data, and KEA wrote the paper.
Author information
Authors and Affiliations
Corresponding author
Additional information
The first authorship is shared between Kirk E. Anderson and Pedro A. P. Rodrigues
Kirk E. Anderson and Pedro A. P. Rodrigues contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(XLSX 96 kb)
Rights and permissions
About this article

Cite this article
Anderson, K.E., Rodrigues, P.A.P., Mott, B.M. et al. Ecological Succession in the Honey Bee Gut: Shift in Lactobacillus Strain Dominance During Early Adult Development. Microb Ecol 71, 1008–1019 (2016). https://doi.org/10.1007/s00248-015-0716-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-015-0716-2