
Abstract
Cutaneous bacteria may play an important role in the resistance of amphibians to the pathogenic fungus Batrachochytrium dendrobatidis (Bd). Microbial communities resident on hosts’ skin show topographical diversity mapping to skin features, as demonstrated by studies of the human microbiome. We examined skin microbiomes of wild and captive fire-bellied toads (Bombina orientalis) for differences across their body surface. We found that bacterial communities differed between ventral and dorsal skin. Wild toads showed slightly higher bacterial richness and diversity in the dorsal compared to the ventral region. On the other hand, captive toads hosted a higher richness and diversity of bacteria on their ventral than their dorsal skin. Microbial community composition and relative abundance of major bacterial taxonomic groups also differed between ventral and dorsal skin in all populations. Furthermore, microbiome diversity patterns varied as a function of their Bd infection status in wild toads. Bacterial richness and diversity was greater, and microbial community structure more complex, in wild than captive toads. The results suggest that bacterial community structure is influenced by microhabitats associated with skin regions. These local communities may be differentially modified when interacting with environmental bacteria and Bd. A better understanding of microbiome variation across skin regions will be needed to assess how the skin microbiota affects the abilities of amphibian hosts to resist Bd infection, especially in captive breeding programs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amphibian skin facilitates respiration, water and temperature regulation, excretion, reproduction, anti-predator defense, and immune responses [1]. A wide array of bacteria inhabits the skin of amphibians, some perhaps beneficial to their host. Multiple species of cutaneous bacteria inhibit the growth of the fungal pathogen Batrachochytrium dendrobatidis (Bd) in in vitro cultures [2–5]. These bacteria can contribute to the defenses of their amphibian hosts against the fungal pathogen [6]. Bd infects the skin of amphibians, creating an osmotic imbalance in susceptible individuals that inhibits rehydration [7] and finally leads to cardiac arrest [8]. Chytridiomycosis, the disease caused by Bd, is responsible for many amphibian population declines and species extinctions across the globe [9, 10].
Adding Bd-inhibiting bacteria to frog skin has been proposed as a “bioaugmentation” strategy to slow the spread of Bd in nature and to enhance the capacity of captive-bred amphibians to resist Bd infection when reintroduced into the wild [4]. However, current knowledge of amphibian microbial communities and their dynamics is insufficient to predict the success of such programs [11, 12]. Notably, an attempt to treat the critically endangered Panamanian golden frog (Atelopus zeteki) with Bd-inhibiting bacteria failed to mitigate the pathogen’s lethal effects [13]. At least for this species, skin bacterial community composition rather than particular probiotic treatments determines survival after experimental infection [14]. Bacterial community composition reflects complex interactions among species [15] which in turn are affected by environmental conditions [16, 17]. Bd infections can disrupt normal host skin bacterial communities [18] and, consequently, may compromise their protective capacity [14].
Technological advances, such as high-throughput 16S rRNA gene sequencing, make possible detailed studies of bacterial communities and their interactions with hosts [19]. Recent studies on amphibians have shown that their skin bacterial communities are characteristic of the host species, change dramatically during development, and vary in response to the environment [20, 21]. The composition and diversity of cutaneous bacterial communities change when amphibians are kept in captivity, perhaps as they are in contact with different environmental bacterial reservoirs [22, 23]. When salamanders are housed in sterile media, the diversity and richness of their skin microbiome decreases with time spent in captivity; yet, a core community of functionally important bacteria emerges independent of the host environment [23]. These community alterations may influence the outcome of Bd infection in captive-bred amphibians [13].
Microbiome diversity can differ widely among skin regions [24–26], but studies on amphibians have not considered this variation. Bd more readily infects the ventral pelvic region, which has a predominant role in water absorption [27]. For this reason, swab sampling to test for Bd infection concentrates on the ventral side and hind legs [28]. Thus, the bacterial community structure on the ventral skin should be more affected than other body regions by Bd infection. Also, adults with sublethal infection and those tolerant of—or resistant to—the disease may have localized Bd infections [29, 30]. Then, changes in bacterial community structure may be observable only in small areas of infection. Studies of the amphibian skin microbiome are generally based on whole-body swabbing or on ventral swabs taken to test Bd infection. These sampling methods may fail to detect area-specific changes, hindering our capacity to assess the dynamics of interactions between Bd and skin microbiota.
Here, we test for the first time whether skin microbial community structure differs between body regions in an amphibian species, the fire-bellied toad Bombina orientalis. We also study how patterns of variation are affected by captive rearing. Skin microbiomes of Asian amphibians have not been studied previously, and may provide novel evolutionary insights into protective roles played by symbiotic bacteria to their amphibian hosts. Unlike in areas of the world experiencing severe chytridiomycosis epizootics, Asian amphibians historically have been associated with endemic Bd strains [31–33]. Additionally, studies to date suggest that Asian amphibians are resilient to infection by emerging Bd strains [32, 34], and possibly also to other chytrid pathogens such as the newly discovered Batrachochytrium salamandrivorans [35].
Material and Methods
Sampling Strategy
Eleven oriental fire-bellied toads (Bombina orientalis) were captured by hand from a small pond (about 5 m2) in Chiak mountain, Gangwon Province, South Korea (37° 23.676′ N, 128° 03.221′ E). Individuals were sexed and measured (snout-to-vent length) using fresh latex gloves for every individual. Each individual was rinsed with 100 mL of sterile water before sampling to eliminate transient bacteria, i.e., those present in subjects’ environment but not otherwise associated with amphibian skin.
Adults were swabbed on two different areas using two separate sterile cotton swabs (MW100, Medical Wire and Equipment, Corsham, Wiltshire, UK). A first swab was used to sample areas most infected by Bd in 30 strokes: ventral area (10 strokes), thighs (5 strokes/leg), and toe webbing (5 strokes/foot). The back of each individual was swabbed using a second cotton swab (30 strokes). Each swab was placed into a sterile vial and stored on dry ice. Within a few hours, each sample was transferred to a –80 °C freezer for storage until DNA extraction. Additionally, 250 mL of water were taken from the collection site. The water was later passed through 0.2-μm filters and stored at −80 °C until DNA extraction.
The same sampling strategy was applied to 18 B. orientalis individuals that had been collected previously from two localities and kept captive in our laboratory for different periods of time: Chuncheon, Gangwon Province (N = 9; date of capture, August 2013; time in captivity, 4 months; 37° 58.664′ N, 127° 36.146′ E; hereafter denoted “lab-4”) and Pocheon, Gyeonggi Province (N = 9; date of capture, August 2012; time in captivity, 16 months; 38° 03.003′ N, 127° 18.360′ E; denoted “lab-16”). During their time in captivity, the toads were housed in groups collected from the same locality, in polypropylene tanks (w: 80 cm, l: 40 cm, h: 45 cm), and maintained at 20 ± 2 °C under a LD 12:12 photoperiod. They were fed every second day with crickets (Gryllus bimaculatus) and mealworms (Tenebrio molitor). Water was changed weekly with fresh 0.5-μm filtered tap water, which was first UV sterilized and run through carbon filters. All individuals were sampled on the same day in December 2013, and 250 mL of water were collected from each tank, passed through 0.2-μm filters and stored at −80 °C until DNA extraction.
Animal husbandry and experimental protocols were approved by the Institutional Animal Care and Use Committee (SNU-121210-2) and the Institutional Biosafety Committee (SNUIBCP120725-2) of Seoul National University. Permits for fieldwork were issued by the mayors with jurisdiction over each locality in South Korea. The study species is not legally protected and not threatened in South Korea.
Molecular Laboratory Work
DNA was extracted from the swabs and water filters using PowerSoil DNA isolation kits (MoBio Laboratories, Carlsbad, CA, USA). Blank DNA extractions with a sterile swab and a sterile filter also were run in parallel to swabs and filters. These blank extractions were used in subsequent PCR amplification to test for potential contamination during the extraction process. Bacterial skin communities were determined using barcoded Illumina sequencing of the 16S rRNA gene with primers targeting a 200 bp portion of the V3 region. Sample-identifying barcode sequences were included at the 5′ ends on the forward primers (total of ten different barcoded forward primers, Table S1). PCR amplification was prepared in 50 μL reactions using 30–50 ng of template DNA under a laminar flow hood to minimize contamination (see Table S1 for detailed PCR protocol). Each run was accompanied by a negative control (1 μL of sterile water instead of DNA).
No PCR product was ever obtained from negative controls and blank extractions, confirming that no contamination occurred during the laboratory steps. Each sample was amplified in triplicate, and the three resulting PCR amplifications were pooled together. PCR amplicons from ten different swab samples, each amplified using a different barcoded primer, were pooled together at similar concentration (50 ng/μL per sample). These pooled samples were sequenced using a GAIIx Illumina sequencer at the Avison Biomedical Research Center (Seoul, South Korea).
Bd Screening
A portion of the DNA extracted from swabs was used to screen samples for Bd infection using a highly sensitive nested PCR method targeting specifically the 5.8S rDNA and the ribosomal internal transcribed spacer regions (ITS) of Bd [31]. The first PCR was run in a volume of 20 μL containing 1 μL of DNA sample, 0.2 μM of forward primer Bd18SF1 (5′-TTTGTACACACCGCCCGTCGC-3′) and reverse primer Bd28SR1 (5′-ATATGCTTAAGTTCAGCGGG-3′), 0.2 mM of each dNTP, 2 mM of MgCl2, and 1.0 unit of Takara Ex Taq DNA polymerase (Takara Bio, Otsu, Shiga, Japan). The PCR conditions consisted of an initial denaturation at 94 °C for 5 min, followed by 30 cycles of 30 s at 94 °C, 30 s at 50 °C, 2 min at 72 °C, and a final extension at 72 °C for 7 min. A second PCR was run in a volume of 20 μL containing 1 μL of products from the first PCR, 0.2 μM of forward primer, Bd1a (5′-CAGTGTGCCATATGTCACG-3′) and reverse primer, Bd2a (5′-CATGGTTCATATCTGTCCAG-3′), 0.2 mM of each dNTP, 2 mM of MgCl2, and 1.0 unit of Takara Ex Taq DNA polymerase. The conditions for the second PCR consisted of an initial denaturation at 94 °C for 5 min, followed by 30 cycles of 45 s at 94 °C, 45 s at 60 °C, 60 s at 72 °C, and a final extension at 72 °C for 7 min. Each sample was run in triplicate, with positive (DNA from Bd culture, strain AbercrombieNP-L.booroolongensis-09-LB-P7) and negative (1 μL sterile water) controls. Status of Bd infection was determined by presence/absence of a band after electrophoresis on an agarose gel. Samples were considered positive when finding at least one positive result out of the three replicates.
For each sample found to be positive for Bd infection, the intensity of infection was estimated in terms of Bd zoospore genomic equivalents (ZGE) in the swab samples using a qPCR assay [28]. The qPCR assay was performed on an Illumina Eco Real-Time PCR system (Illumina, San Diego, CA, USA) in a volume of 10 μL containing 1× SYBR green quantitative PCR reagent kit (PhileKorea Technology, Seoul, South Korea), 0.25 mM of both ITS1-3 Chytr (5′-CCTTGATATAATACAGTGTGCCATATGTC-3′) and 5.8S Chytr (5′-AGCCAAGAGATCCGTTGTCAA-3′) primers, and 2 μL of DNA. The PCR conditions consisted of an initial denaturation at 95 °C for 10 min, followed by 50 cycles of 10 s at 95 °C and 1 min at 58 °C. Each sample was assayed in duplicate, along with standards of known Bd quantity (100, 10, 1, and 0.1 zoospores, strain AbercrombieNP-L.booroolongensis-09-LB-P7) and negative controls (5 μL sterile water). We estimated Bd zoospore genomic equivalents (ZGEs) per swab, averaged over the two replicates, from threshold cycle (Ct) values after corrections for dilutions following DNA extraction and PCR procedures.
Sequence Data Processing
Illumina sequencing data (total number of reads, 1,026,343) were pair-assembled using PANDAseq [36] with an assembly quality score of 0.9, the most stringent option to reduce errors. Pair-assembled sequences were trimmed, aligned, and filtered using the mothur pipeline [37]. Alignment was done using the EzTaxon database version 2014 [38]. Additionally, chimeras detected using the chimera.uchime command within mothur were removed (total number of clean reads, 984,104). The sequence dataset was subsampled (rarefied) to 10,085 reads, which corresponds to the minimum number of reads obtained across samples, including soil and water samples (Table S2). Any sequence present only once across samples (singletons) was removed from the dataset to improve the comparability of the sequence data and the resolution of the analyses [39, 40]. This filtered dataset was subsampled again to 7972 reads per sample to have comparable estimates of richness and diversity among samples. The EzTaxon training set was used with Bayesian classifier algorithm implemented in mothur for taxonomic classification of operational taxonomic units (OTUs), clustered at ≥97 % sequence similarity. No sequences matching chloroplast, mitochondria, eukaryote, nor archaea were observed among the classified sequences. Richness (i.e., number of OTUs and Chao Index) and diversity indices (i.e., nonparametric Shannon and inverse Simpson) were estimated using calculators within mothur [37]. Faith’s phylogenetic diversity (PD) [41] was estimated using the phylo.diversity command within mothur from a phylogenetic tree built using FastTreeMP [42].
Statistical Analyses
All statistical analyses were performed on the subsampled dataset of 7972 reads per sample. We assessed whether species richness (number of OTUs and Chao index) and diversity (nonparametric Shannon, inverse Simpson, and Faith’s PD indices) differed in relation to body region. We used linear mixed models for all indices except number of OTUs, for which we used a generalized linear mixed model. Fixed terms in the models included the variables body region, evironment (wild versus laboratory), Bd infection intensity, and the interaction of environment with body region. Subject identification number (ID) was included as a random term.
Further analyses were run on data from captive and wild toads separately, following similar procedures. For analyses on wild toads, body region, Bd infection status (negative vs positive), and their interaction were included as factors. Bd infection status was used as a factor instead of infection intensity in this model because half of the wild individuals were not infected by Bd (see “Results”). For analyses on captive toads, body region, population (lab-4 vs lab-16), the interaction between body region and population, and Bd infection intensity were used as factors. Subject ID was included as a random factor in all these models. To account for overdispersion, we included an observational random effect in the models. Subject ID was removed from the model if its standard deviation was less than that of the observational term, except in the cases where including it showed a better fit of the data to the model. Likelihood ratio tests were used to calculate the predictive power of each variable. Nonsignificant interactions and terms were removed sequentially.
Permutational multivariate analyses (PERMANOVA) [43] with 999 iterations were performed to assess whether community composition differed in relation to: body region and environment in all toads, body region and Bd infection status in wild toads, and body region and population in captive toads. The Bray-Curtis (based on abundance of OTUs) and the Unifrac (based on phylogenetic relationships of OTUs) distance matrices were used as measures of community structure for these analyses. Differences in community structure were visualized with nonmetric multidimensional scaling (NMDS) plots based on the distance matrices. All statistical analyses were run with R v3.0.2 [44].
Phyla, Order, and Core Community Analyses
The core community was defined as those OTUs that were present on at least 90 % of all the samples [23]. The relative abundance of these OTUs, of phyla, and of orders within Proteobacteria (by far the most abundant phylum, see Results) were calculated by dividing the number of sequences of each OTU/phylum/order by the total number of reads per sample (i.e., 7972). Phyla and orders within Proteobacteria were retained for further analyses if their mean relative abundance in wild or captive toads was over 0.5 %. This cutoff value was chosen so that over 60 % of the OTUs within each phylum and order were represented. In most cases, 80–100 % of the OTUs were included (see “Results”).
GLMMs were used to assess whether the relative abundance of phyla, orders, and core OTUs differed in relation to body region and environment in the full dataset, in relation to body region or Bd infection status in wild toads, or in relation to body region and population in captive toads, following the same procedure as described above. P values were corrected for multiple comparisons using the false discovery rate method [45].
Core OTUs that were not classified further than the class or family during sequence processing were aligned to GenBank sequences of representative genera within their class/family using Clustal W [46]. The alignment was manually refined using BioEdit [47]. Phylogenetic relationships among sequences were inferred using a neighbor-joining method conducted in MEGA v5.0 [48] and a maximum likelihood approach conducted in Treefinder [49], with support for each node estimated by bootstrap analysis of 5000 replicates.
Results
Five out of 11 wild and all captive B. orientalis individuals were infected by Bd with low infection loads (range 1.5–48.7 ZGEs per swab, Table S2). Fifty-nine B. orientalis swab samples and all environmental samples were sequenced successfully (Table S2). No sequence could successfully be pair-assembled using the most stringent quality score in PANDAseq for two ventral swabs from wild individuals (BO251.v and BO253.v), and thus, these data were excluded.
The number of OTUs at 97 % similarity from skin swabs was on average 255 ± 26 (mean ± SE). Wild toads had on average 399 ± 52 OTUs, whereas captive toads had 176 ± 18 OTUs (combining data from the two captive populations; Fig. 1). The soil and water samples from the field site contained 1819 and 823 OTUs, respectively. The water samples from the 4-months and 16-months captive populations had 232 and 203 OTUs, respectively (Table S2).

Rarefaction curves of the number of bacterial OTUs (means with standard error bars) as a function of the number of sequences on the skin of wild and captive adult B. orientalis. Criteria for OTU membership is ≥97 % sequence similarity
Richness and Diversity
Wild vs Captive Toads
Numbers of OTUs and Chao richness indices were higher in samples from wild than captive toads (OTUs: LRT1 = 19.92, P < 0.0001, Chao: LRT1 = 12.26, P < 0.0001; Fig. 1). Also bacterial richness was higher in ventral samples than in dorsal ones for captive toads, but not for wild toads (body region-environment interaction, OTUs: LRT1 = 5.33, P = 0.02; Chao: LRT1 = 8.52, P = 0.003; Fig. 1). Diversity did not differ between wild and captive toads (nonparametric Shannon: LRT1 = 2.15, P = 0.14; inverse Simpson: LRT1 = 1.61, P = 0.20). Faith’s PD index was higher in samples from wild than captive toads (LRT1 = 15.14, P < 0.0001). Bd infection intensity was correlated neither with richness nor diversity indices, probably owing to the low Bd loads observed (OTUs: LRT1 = 2.59, P = 0.11; Chao: LRT1 = 3.05, P = 0.08; npShannon: LRT1 = 0.75, P = 0.37; invSimpson: LRT1 = 1.70, P = 0.15; PD: LRT1 = 0.08, P = 0.78).
Wild Toads Only
For wild toads, the number of OTUs did not differ as a function of Bd infection status (LRT1 = 0.86, P = 0.35) nor body region (LRT1 = 0.81, P = 0.81), but the Chao index was higher for dorsal samples (LRT1 = 5.32, P = 0.02) and for Bd-positive samples (LRT1 = 4.45, P = 0.03). The nonparametric Shannon diversity index was higher in ventral but not dorsal samples from Bd-positive individuals (body region-Bd interaction, LRT1 = 6.05, P = 0.01; Fig. 2). For the inverse Simpson index, ventral samples were marginally more diverse than dorsal ones (LRT1 = 3.68, P = 0.05), but Bd had no effect (LRT1 = 2.56, P = 0.11). Faith’s PD was higher on dorsal than ventral samples (LRT1 = 5.63, P = 0.02). Bd infection status did not affect Faith’s PD for wild toads (LRT1 = 2.74, P = 0.10).

Nonparametric Shannon diversity index in dorsal and ventral samples of wild B. orientalis in relation to Bd infection (mean ± SE)
Captive Toads Only
For captive toads, the number of OTUs and Chao index were higher in ventral than in dorsal samples (OTUs: LRT1 = 10.21, P = 0.001; Chao: LRT1 = 9.36, P = 0.001). Ventral swabs were also more diverse than dorsal ones (nonparametric Shannon: LRT1 = 8.2, P = 0.004; inverse Simpson: LRT1 = 5.68, P = 0.02), and lab-16 samples were more diverse than lab-4 ones (nonparametric Shannon: LRT1 = 11.39, P = 0.0007; inverse Simpson: LRT1 = 21.18, P < 0.0001). Faith’s PD was lower on dorsal than ventral samples (LRT1 = 7.85, P = 0.005). The two captive populations did not differ in their Faith’s PD (LRT1 = 3.32, P = 0.07). Bd infection intensity was correlated neither with richness nor diversity indices (OTUs: LRT1 = 0.32, P = 0.57; Chao: LRT1 = 0.32, P = 0.57; npShannon: LRT1 = 1.59, P = 0.21; invSimpson: LRT1 = 1.10, P = 0.29; PD: LRT1 = 0.63, P = 0.43).
Community Structure
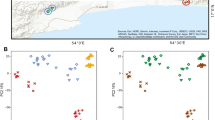
The microbial community structure was different among the three populations (i.e., wild, lab-4, and lab-16; PERMANOVA with Bray-Curtis distances: pseudo-F = 37.74, P = 0.001; with Unifrac distances: pseudo-F = 10.13, P = 0.001; Figs. 3 and S1A). For the wild toads, microbial community was not structured in relation to Bd infection status (Bray-Curtis: pseudo-F = 1.47, P = 0.17; Unifrac: pseudo-F = 1.01, P = 0.38) nor to body region (Bray-Curtis: pseudo-F = 0.86, P = 0.53; Unifrac: pseudo-F = 0.77, P = 0.96). For captive toad populations, the microbial community composition significantly differed between body regions according to Unifrac but not Bray-Curtis distances (Bray-Curtis: pseudo-F = 0.53, P = 0.56; Unifrac: pseudo-F = 1.65, P = 0.02; Fig. S1B-C). Microbial community composition also differed significantly between the two captive populations (Bray-Curtis: pseudo-F = 79.39, P = 0.001; Unifrac: pseudo-F = 4.95, P = 0.001), with the composition of individuals from the population captive for 4 months showing the highest homogeneity (Figs. 3 and S1B-C).

Nonmetric multidimensional scaling (NMDS) plot of Bray-Curtis bacterial community composition in ventral and dorsal samples of wild and captive B. orientalis
Wild toads shared 28.2 and 45.1 % of OTUs with soil and water, respectively, whereas captive toads shared 18.7 % (lab-4) and 11.2 % (lab-16) with their aqueous environment. In wild toads, 39.8 % of OTUs were unique to dorsal regions and 12.1 % were unique to ventral regions. In contrast, in captive individuals, only 5 and 7 % of OTUs were unique to the dorsal region, whereas 46 and 57 % were unique to ventral regions in lab-4 and lab-16 populations, respectively.
Relative Abundance of Major Phyla
The relative abundance of four out of six major bacterial phyla (mean relative abundance over 0.5 %) significantly differed between wild and captive toads, even after correction for multiple comparisons (Table 1; Fig. 4). Proteobacteria, already the most abundant in wild individuals, was significantly more abundant in the captive populations. On the other hand, the relative abundance of Actinobacteria, Firmicutes, and Fusobacteria were much reduced in the captive populations compared to the wild population (Table 1; Fig. 4).

Average relative abundance of sequences assigned to major bacteria phyla, pooled by population of origin and sampled body region. Only phyla with abundance over 0.5 % in at least one of the pool of samples are represented
In wild toads, relative abundance of the major phyla did not differ between body regions nor between Bd-positive and Bd-negative samples. In captive amphibians, mean relative abundance of Firmicutes, Acidobacteria, and Actinobacteria was higher on the ventral region than on the dorsal region, but the opposite was observed for Proteobacteria (Table 1; Fig 4.). These differences were more pronounced in the lab-16 population (Fig. 4).
Relative Abundance of Orders Within Proteobacteria
The mean relative abundance of all 8 major orders within Proteobacteria significantly differed between wild and captive B. orientalis populations (Table 1; Fig. 5). Notably, the relative abundance of Pseudomonodales was much reduced in captive compared to wild individuals, whereas Burkholderiales and especially Methylophilales were more abundant in captive populations (Table 1; Fig. 5). In wild toads, Burkholderiales was more abundant on the dorsal than on the ventral region with the difference more marked in Bd-negative samples (Table 1; Fig. 6a). Also, the relative abundance of Xanthomonadales was higher on the dorsal region for Bd-negative samples, but higher on ventral region for Bd-positive samples (Table 1; Fig. 6b).

Average relative abundance of sequences assigned to major orders within the phylum Proteobacteria, pooled by population of origin and sampled body region. Only orders with abundance over 0.5 % in at least one of the three populations are represented

Difference in mean relative abundance of Burkholderiales (a) and Xanthomonadales (b) in wild B. orientalis in relation to body region and Bd infection status (mean ± SE)
Composition and Abundance of Core Bacterial Community
Wild vs Captive Toads
The core community of bacteria shared by all wild and captive toads was represented by 13 OTUs (Fig. 7). Three of these OTUs could only be classified to their family group, and two others up to their class. Further phylogenetic analyses could only determine that the OTUs classified as Actinobacteria_c and Microbacteriaceae_f probably were related to the genus Microbacterium (Fig. S2A-E). The core community corresponded on average to 58 ± 6 % of the whole community in wild toads, 23 ± 1 % in lab-4, and 20 ± 1 % in lab-16 (Fig. 7). The total relative abundance of core OTUs differed among populations (LRT2 = 21.12, P < 0.0001). In captive toads, the total relative abundance of core OTUs was higher for the lab-4 population (LRT1 = 5.76, P = 0.02). Several noncore OTUs were very abundant in captive populations, representing 5 to 53 % of the whole community, but were rare or absent in wild individuals (Fig. S3). The relative abundance for all but three core OTUs significantly differed between wild and captive toads (Table S3, Fig. 7).

Heatmap showing the mean relative abundance of 13 OTUs forming the core bacterial community in wild and captive populations of B. orientalis. Samples are pooled by population, by body region within populations (V = ventral, D = dorsal), and by Bd infection status in wild individuals (N = Bd-negative, P = Bd-positive)
Wild Toads Only
In wild toads, ventral and dorsal samples did not differ in relative abundance of core OTUs, except for the Pseudomonas parafulva and Microbacteriaceae_f OTUs (Table 2, S3; Fig.7). Both OTUs had a higher relative abundance on dorsal samples for Bd-negative toads, but the opposite was observed for Bd-positive toads. Both the Chryseobacterium shigense and Rhizobiales_c OTUs showed differences in their relative abundance as a function of their Bd infection status (Table 2, S3; Fig.7).
Captive Toads Only
In captive toads, ventral and dorsal samples differed in relative abundance of the Sphaerotilus sp. and Aeromonas sp. OTUs (Table 2, S3; Fig. 7). The Pseudomonadaceae_f and Microbacteriaceae_f OTUs also showed differences in relative abundance between body regions, but opposite trends were observed in the two captive populations. Lab-4 and lab-16 populations had different relative abundance for the Pseudomonas sp., Chryseobacterium shigense, Chryseobacterium sp., and Acinetobacter sp. OTUs (Table 2, S3; Fig. 7).
Discussion
Our results suggest that the bacterial community of the fire-bellied toad Bombina orientalis varies between ventral and dorsal skin in different ways in wild and captive populations. Wild toads showed slightly higher bacterial richness (Chao index) and diversity (PD) in the dorsal compared to the ventral region. On the other hand, captive toads hosted a higher richness and diversity of bacteria on their ventral than their dorsal skin, and these body regions also differed in microbial community composition analyzed by phylogenetic similarity (i.e., Unifrac distance). Additionally, the relative abundance of major bacterial phyla and orders, and of specific OTUs that comprise the core bacterial community living on the skin, differed between ventral and dorsal skin in both captive and wild individuals. Further work with a larger number of captive and wild populations will be necessary to determine whether the different trends observed in this study between wild and captive populations represent a common effect of captivity on amphibian skin microbiota.
The differences observed among body regions may reflect the wider range of sampling on the ventral skin, as it included thighs and toe webbing. However, if sampling technique were the main reason for the differences observed, we would expect to find similar trends for both wild and captive individuals. Furthermore, differences observed between body regions were not limited to richness and diversity indices but included differences in community composition and relative abundance of major bacterial taxa dependent on Bd infection status, suggesting that the observed variation in skin microbiota has complex origins.
The rugged dorsal skin surface of the toads and the smooth ventral skin provide microhabitats with different characteristics that may be more or less favorable for particular bacteria. For example, toads often rest on wet substrate so their ventral skin tends to be moister than dorsal skin. Furthermore, dorsal skin often is covered with glands that secrete antimicrobial peptides [50, 51]. These peptides likely alter microbial community structure on the skin. Comparative studies including species with different glandular characteristics may be useful in assessing the generality of our findings.
An earlier study had determined that the diversity and richness of amphibian skin microbiomes decrease in captivity [23]. Our results suggest that the bacterial community is disrupted by captivity more on dorsal than ventral skin. Notably, bacterial phylogenetic diversity was higher on dorsal than on ventral skin in wild toads, with almost 40 % of OTUs unique to the dorsal region. In captive toads, bacterial phylogenetic diversity was lower on dorsal than ventral skin, with only 5–7 % of OTUs unique to the dorsal region. Our study is based on only two captive populations kept in similar conditions, so the sample size is too small to make general conclusions about effects of captivity on skin microbiota heterogeneity. However, these results clearly warrant additional research on the issue.
As in humans [25, 26], variation in microbiome diversity likely exists at a much finer scale on the skin of amphibians. Nevertheless, the demonstration of variation of microbiomes even over large sections of amphibian skin may be important for amphibian conservation as many pathogens, including Bd, more readily infect skin in the ventral pelvic region [27]. Our results suggest that more targeted sampling of amphibian skin may be needed to better assess the possible role of skin microbiota in conferring resistance against Bd infection.
When amphibians are heavily infected by Bd, their skin bacterial community can be dramatically disrupted [18]. However, these changes may be better resolved by studying local skin microbiota, especially with individuals that bear only low infection loads or those at critical early stages of infection. In this study, B. orientalis infected by Bd presented only low levels of infection, as commonly observed in South Korean amphibians [32].
We found differences in the skin microbiome of subjects between Bd-infected and uninfected subjects, with higher bacterial diversity on the ventral than dorsal skin only of infected individuals. Abundance of OTUs within the Burkholderiales and Xanthomonadales orders, and of core OTUs within Pseudomonas parafulva and Microbacteriaceae_f, also differed between body regions in infected and uninfected samples. These results suggest that the microbiome of the two body regions reacts differently to Bd infection. Possibly, even more localized sampling is needed to better understand the dynamics of interactions between Bd and skin microbiota.
We also observed differences in the richness, diversity, and composition of skin bacteria between the two captive populations, despite their being kept in the same conditions and sampled at the same time. The skin microbiome of toads kept in captivity for 4 months was less rich and more homogenous than that of toads kept captive for 16 months. Abundance of some major phyla, orders, and core bacteria differed between the two populations. These differences may be attributable to the different origins of the two populations [52], the length of their captivity, or other factors of the captive environment [23]. However, compared to the wild population sampled, levels of bacterial richness and diversity, and bacterial community composition, were more similar among the captive populations. Thus, we believe that microbiome convergence occurred in captivity, although sampling of additional captive populations may be necessary to confirm this hypothesis. Other studies also have demonstrated that the bacterial communities of captive amphibians become more similar to their environment with time spent in captivity [22, 23]. Nonetheless, our results suggest that, despite interactions with environmental bacteria, differences in microbial community between body regions remain after 16 months in captivity, with both regions far from the level of bacterial richness and diversity observed in wild populations.
The relative abundance of the core bacterial community was much reduced in captive compared to wild toads. This is in stark contrast with results of the experiment by Loudon et al. [22], where the core community in salamanders comprised up to 93.5 % of the total community after 21 days in captivity. Instead, other OTUs of the orders Burkholderiales and Methylophilales, not present or rare in wild toads, became dominant in captive B. orientalis individuals (Figs. 4 and S3). These differences may have resulted from environmental bacteria in our study, whereas the salamanders were housed in a sterile environment.
Whether the 13 OTU members of the core community on B. orientalis are functionally important to their host, or represent an artifact of the method used to identify them, is unclear. More than half of the OTUs were rare (<1 %, Fig. 7) in both captive and wild populations, but rarity does not preclude an important role in the community [53]. Of the 13 core OTUs, four belonged to the Pseudomonas and Chryseobacterium genera (or five, if we count Pseudomonadaceae_f; Fig. S2C). These genera comprise many species with Bd-inhibiting properties [2, 54]. Differences in abundance of these OTUs between infected and uninfected samples in wild toads would suggest that these OTUs are affected by Bd infection, although cause and effect cannot be easily disentangled. Isolation of these bacteria and inhibition tests would be necessary to demonstrate anti-Bd properties of these bacteria.
The reduced abundance of Bd-inhibiting bacteria in captive amphibians may affect their health, including their susceptibility to Bd [3]. All captive B. orientalis were infected by Bd, which might indicate that they were more susceptible to Bd than wild individuals. The captive individuals were in closer contact, however, which may have resulted in the accumulation of more Bd zoospores in limited space and fostered disease transmission. Captive toads all showed low Bd loads and did not present any clinical signs of chytridiomycosis. Possibly, the modified bacterial community still provided an effective defense against Bd infection. Additionally, this species likely possesses strong immune defenses against Bd, owing to its historical association with endemic Bd strains in Korea [32].
Skin bacteria composition can affect the likelihood of survival of amphibians bred in ex situ facilities for reintroduction into natural environments [14]. Our study shows that the skin of amphibians kept in captivity hosts a microbial community that may become very heterogeneous across regions of the body. Furthermore, these local communities may be differentially modified when interacting with environmental bacteria and may respond in different ways to infection by the pathogenic amphibian chytrid fungus. Life-history characteristics of amphibian species need to be considered when examining how microbial communities differ among skin regions. For example, we would expect more marked differences in terrestrial than in aquatic amphibian species, corresponding to more variable microenvironments to which terrestrial frogs are exposed. To ensure their effectiveness, bioaugmentation plans should focus on ensuring homogeneity of bacteria composition across the body regions most susceptible to Bd infection.
The skin comprises a complex ecosystem, with diverse microhabitats, hosting a wide variety of microorganisms [24]. The extent of this topographical microbiome diversity has been well studied in humans but not in other animals. Our study represents a first exploration of these local differences on the skin of a wild vertebrate. The skin microbiome most likely plays an important role for the health of many wild animals, e.g., whales [55] and birds [56]. Defensive symbioses in invertebrates are also a major area of research, with important implications for human health [57, 58]. Studies of wildlife host-pathogen interactions need to consider the potential impacts of microhabitat variability on microbiome distribution and diversity. Understanding the dynamics of skin microbiome heterogeneity may be especially important for amphibians, as skin microbiota may act as an important first line of defense against the amphibian chytrid fungus pathogen.
References
Clarke BT (1997) The natural history of amphibian skin secretions, their normal functioning and potential medical applications. Biol Rev 72:365–379
Woodhams DC, Vredenburg VT, Simon M-A, Billheimer D, Shakhtour B, Shyr Y, Briggs CJ, Rollins-Smith LA, Harris RN (2007) Symbiotic bacteria contribute to innate immune defenses of the threatened mountain yellow-legged frog, Rana muscosa. Biol Cons 138:390–398
Becker MH, Harris RN (2010) Cutaneous bacteria of the redback salamander prevent morbidity associated with a lethal disease. PLoS One 5, e10957
Harris RN, Brucker RM, Walke JB, Becker MH, Schwantes CR, Flaherty DC, Lam BA, Woodhams DC, Briggs CJ, Vredenburg VT, Minbiole KPC (2009) Skin microbes on frogs prevent morbidity and mortality caused by a lethal skin fungus. ISME J 3:818–824
Woodhams DC, Alford RA, Antwis RE, Archer H, Becker MH, Belden LK, Bell SC, Bletz M, Daskin JH, Davis LR, Flechas SV, Lauer A, Gonzalez A, Harris RN, Holden WM, Hughey MC, Ibanez R, Knight R, Kueneman J, Rabemananjara F, Reinert LK, Rollins-Smith LA, Roman-Rodriguez F, Shaw SD, Walke JB, McKenzie V (2015) Antifungal isolates database of amphibian skin-associated bacteria and function against emerging fungal pathogens. Ecology 96:595–595
Rollins-Smith LA, Ramsey JP, Pask JD, Reinert LK, Woodhams DC (2011) Amphibian immune defenses against chytridiomycosis: impacts of changing environments. Integr Comp Biol 51:552–562
Carver S, Bell BD, Waldman B (2010) Does chytridiomycosis disrupt amphibian skin function? Copeia 2010:487–495
Voyles J, Young S, Berger L, Campbell C, Voyles WF, Dinudom A, Cook D, Webb R, Alford RA, Skerratt LF, Speare R (2009) Pathogenesis of chytridiomycosis, a cause of catastrophic amphibian declines. Science 326:582–585
Fisher MC, Garner TWJ, Walker SF (2009) Global emergence of Batrachochytrium dendrobatidis and amphibian chytridiomycosis in space, time, and host. Annu Rev Microbiol 63:291–310
Skerratt L, Berger L, Speare R, Cashins S, McDonald K, Phillott A, Hines H, Kenyon N (2007) Spread of chytridiomycosis has caused the rapid global decline and extinction of frogs. EcoHealth 4:125–134
Bletz MC, Loudon AH, Becker MH, Bell SC, Woodhams DC, Minbiole KPC, Harris RN (2013) Mitigating amphibian chytridiomycosis with bioaugmentation: characteristics of effective probiotics and strategies for their selection and use. Ecol Lett 16:807–820
Woodhams D, Bosch J, Briggs C, Cashins S, Davis L, Lauer A, Muths E, Puschendorf R, Schmidt B, Sheafor B, Voyles J (2011) Mitigating amphibian disease: strategies to maintain wild populations and control chytridiomycosis. Front Zool 8:8
Becker M, Harris R, Minbiole K, Schwantes C, Rollins-Smith L, Reinert L, Brucker R, Domangue R, Gratwicke B (2011) Towards a better understanding of the use of probiotics for preventing chytridiomycosis in Panamanian golden frogs. EcoHealth 8:501–506
Becker MH, Walke JB, Cikanek S, Savage AE, Mattheus N, Santiago CN, Minbiole KPC, Harris RN, Belden LK, Gratwicke B (2015) Composition of symbiotic bacteria predicts survival in Panamanian golden frogs infected with a lethal fungus. Proc R Soc B 282:20142881
Kung D, Bigler L, Davis LR, Gratwicke B, Griffith E, Woodhams DC (2014) Stability of microbiota facilitated by host immune regulation: informing probiotic strategies to manage amphibian disease. PLoS One 9, e87101
Daskin JH, Bell SC, Schwarzkopf L, Alford RA (2014) Cool temperatures reduce antifungal activity of symbiotic bacteria of threatened amphibians - implications for disease management and patterns of decline. PLoS One 9, e100378
Daskin JH, Alford RA (2012) Context-dependent symbioses and their potential roles in wildlife diseases. Proc R Soc B 279:1457–1465
Jani AJ, Briggs CJ (2014) The pathogen Batrachochytrium dendrobatidis disturbs the frog skin microbiome during a natural epidemic and experimental infection. Proc Natl Acad Sci U S A 11:5049–5058
McFall-Ngai M, Hadfield MG, Bosch TCG, Carey HV, Domazet-Loso T, Douglas AE, Dubilier N, Eberl G, Fukami T, Gilbert SF, Hentschel U, King N, Kjelleberg S, Knoll AH, Kremer N, Mazmanian SK, Metcalf JL, Nealson K, Pierce NE, Rawls JF, Reid A, Ruby EG, Rumpho M, Sanders JG, Tautz D, Wernegreen JJ (2013) Animals in a bacterial world, a new imperative for the life sciences. Proc Natl Acad Sci U S A 110:3229–3236
Kueneman JG, Parfrey LW, Woodhams DC, Archer HM, Knight R, McKenzie VJ (2013) The amphibian skin-associated microbiome across species, space and life history stages. Mol Ecol 23:1238–1250
McKenzie VJ, Bowers RM, Fierer N, Knight R, Lauber CL (2011) Co-habiting amphibian species harbor unique skin bacterial communities in wild populations. ISME J 6:588–596
Becker MH, Richards-Zawacki CL, Gratwicke B, Belden LK (2014) The effect of captivity on the cutaneous bacterial community of the critically endangered Panamanian golden frog (Atelopus zeteki). Biol Cons 176:199–206
Loudon AH, Woodhams DC, Parfrey LW, Archer H, Knight R, McKenzie V, Harris RN (2013) Microbial community dynamics and effect of environmental microbial reservoirs on red-backed salamanders (Plethodon cinereus). ISME J 8:830–840
Grice EA, Segre JA (2011) The skin microbiome. Nat Rev Micro 9:244–253
Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, Program NCS, Bouffard GG, Blakesley RW, Murray PR, Green ED, Turner ML, Segre JA (2009) Topographical and temporal diversity of the human skin microbiome. Science 324:1190–1192
Bouslimani A, Porto C, Rath CM, Wang M, Guo Y, Gonzalez A, Berg-Lyon D, Ackermann G, Moeller Christensen GJ, Nakatsuji T, Zhang L, Borkowski AW, Meehan MJ, Dorrestein K, Gallo RL, Bandeira N, Knight R, Alexandrov T, Dorrestein PC (2015) Molecular cartography of the human skin surface in 3D. Proc Natl Acad Sci U S A 112:E2120–E2129
Pessier AP, Nichols DK, Longcore JE, Fuller MS (1999) Cutaneous chytridiomycosis in poison dart frogs (Dendrobates spp.) and White’s tree frogs (Litoria caerulea). J Vet Diagn Invest 11:194–199
Hyatt A, Boyle DG, Olsen V, Boyle DB, Berger L, Obendorf D, Dalton A, Kriger K, Heros M, Hines H, Phillott R, Campbell R, Marantelli G, Gleason F, Coiling A (2007) Diagnostic assays and sampling protocols for the detection of Batrachochytrium dendrobatidis. Dis Aquat Organ 73:175–192
Shin J, Bataille A, Kosch TA, Waldman B (2014) Swabbing often fails to detect amphibian chytridiomycosis under conditions of low infection load. PLoS One 9, e111091
Reeder NMM, Pessier AP, Vredenburg VT (2012) A reservoir species for the emerging amphibian pathogen Batrachochytrium dendrobatidis thrives in a landscape decimated by disease. PLoS One 7, e33567
Goka K, Yokoyama JUN, Une Y, Kuroki T, Suzuki K, Nakahara M, Kobayashi A, Inaba S, Mizutani T, Hyatt AD (2009) Amphibian chytridiomycosis in Japan: distribution, haplotypes and possible route of entry into Japan. Mol Ecol 18:4757–4774
Bataille A, Fong JJ, Cha M, Wogan GOU, Baek HJ, Lee H, Min M-S, Waldman B (2013) Genetic evidence for a high diversity and wide distribution of endemic strains of the pathogenic chytrid fungus Batrachochytrium dendrobatidis in wild Asian amphibians. Mol Ecol 22:4196–4209
Bai C, Liu X, Fisher MC, Garner TWJ, Li Y (2012) Global and endemic Asian lineages of the emerging pathogenic fungus Batrachochytrium dendrobatidis widely infect amphibians in China. Divers Distrib 18:307–318
Farrer RA, Weinert LA, Bielby J, Garner TWJ, Balloux F, Clare F, Bosch J, Cunningham AA, Weldon C, du Preez LH, Anderson L, Pond SLK, Shahar-Golan R, Henk DA, Fisher MC (2011) Multiple emergences of genetically diverse amphibian-infecting chytrids include a globalized hypervirulent recombinant lineage. Proc Natl Acad Sci U S A 108:18732–18736
Martel A, Blooi M, Adriaensen C, Van Rooij P, Beukema W, Fisher MC, Farrer RA, Schmidt BR, Tobler U, Goka K, Lips KR, Muletz C, Zamudio KR, Bosch J, Lotters S, Wombwell E, Garner TWJ, Cunningham AA, Spitzen-van der Sluijs A, Salvidio S, Ducatelle R, Nishikawa K, Nguyen TT, Kolby JE, Van Bocxlaer I, Bossuyt F, Pasmans F (2014) Recent introduction of a chytrid fungus endangers Western Palearctic salamanders. Science 346:630–631
Masella A, Bartram A, Truszkowki J, Brown D, Neufeld J (2012) PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics 13:31
Schloss PD, Westcott SI, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541
Chun J, Lee H-J, Jung Y, Kim M, Kim S, Kim BK, Lim YW (2007) EzTaxon: a web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int J Syst Evol Microbiol 57:2259–2261
Unterseher M, Jumpponen A, Öpik M, Tedersoo L, Moora M, Dormann CF, Schnittler M (2011) Species abundance distribution and richness estimations in fungal metagenomics - lessons learned from community ecology. Mol Ecol 20:275–285
Zhou J, Wu L, Deng Y, Zhi X, Jiang Y-H, Tu Q, Xie J, Van Nostrand JD, He Z, Yang Y (2011) Reproducibility and quantitation of amplicon sequencing-based detection. ISME J 5:1303–1313
Faith DP, Baker AM (2006) Phylogenetic diversity (PD) and biodiversity conservation: some bioinformatics challenges. Evol Bioinform 2:121–128
Price MN, Dehal PS, Arkin AP (2009) FastTree: computing large minimum-evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26:1641–1650
Anderson MJ (2001) A new method for non-parametric multivariate analysis of variance. Austral Ecol 26:32–46
R Development Core Team (2011) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57:289–300
Thompson JD, Higgins DG, Gibson TJ (1994) Clustal W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Res 41:95–98
Kumar S, Dudley J, Nei M, Tamura K (2008) MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform 9:299–306
Jobb G (2011) TREEFINDER version of March 2011. Munich. Germany. Distributed by the author at www.treefinder.de
Barberio C, Delfino G, Mastromei G (1987) A low molecular weight protein with antimicrobial activity in the cutaneous ‘venom’ of the yellow-bellied toad (Bombina variegata pachypus). Toxicon 25:899–909
Mastromei G, Barberio C, Pistolesi S, Delfino G (1991) A bactericidal protein in Bombina variegata pachypus skin venom. Toxicon 29:321–328
Walke JB, Becker MH, Loftus SC, House LL, Cormier G, Jensen RV, Belden LK (2014) Amphibian skin may select for rare environmental microbes. ISME J 8:2207–2217
Shade A, Jones SE, Caporaso JG, Handelsman J, Knight R, Fierer N, Gilbert JA (2014) Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. mBio 5:e01371–14
Lam BA, Walke JB, Vredenburg VT, Harris RN (2010) Proportion of individuals with anti-Batrachochytrium dendrobatidis skin bacteria is associated with population persistence in the frog Rana muscosa. Biol Cons 143:529–531
Apprill A, Robbins J, Eren AM, Pack AA, Reveillaud J, Mattila D, Moore M, Niemeyer M, Moore KMT, Mincer TJ (2014) Humpback whale populations share a core skin bacterial community: towards a health index for marine mammals? PLoS One 9, e90785
Ruiz-Rodriguez M, Valdivia E, Soler JJ, Martin-Vivaldi M, Martin-Platero AM, Martinez-Bueno M (2009) Symbiotic bacteria living in the hoopoe’s uropygial gland prevent feather degradation. J Exp Biol 212:3621–3626
Lopanik NB (2014) Chemical defensive symbioses in the marine environment. Funct Ecol 28:328–340
Rainey SM, Shah P, Kohl A, Dietrich I (2014) Understanding the Wolbachia-mediated inhibition of arboviruses in mosquitoes: progress and challenges. J Gen Virol 95:517–530
Acknowledgments
We thank Jonathan Fong and Moonsuk Cha for assistance with fieldwork, and Dharmesh Singh for assistance with laboratory work.
Funding
This work was supported by the National Research Foundation of Korea (NRF) (grants 2012K1A2B1A03000496 to B.W., funded by the Ministry of Science, ICT and Future Planning, and 2014063422 to A.B.., funded by the Ministry of Education, government of the Republic of Korea).
Author information
Authors and Affiliations
Corresponding author
Additional information
Arnaud Bataille and Larisa Lee-Cruz contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 1479 kb)
Rights and permissions
About this article

Cite this article
Bataille, A., Lee-Cruz, L., Tripathi, B. et al. Microbiome Variation Across Amphibian Skin Regions: Implications for Chytridiomycosis Mitigation Efforts. Microb Ecol 71, 221–232 (2016). https://doi.org/10.1007/s00248-015-0653-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-015-0653-0