
Abstract
Congenital genitourinary anomalies are among the most frequent types of birth defects in neonates. Some anomalies can be a significant cause of morbidity in infancy, while others remain asymptomatic even until adulthood and can be at times the only manifestation of a complex systemic disease. The spectrum of these anomalies results from the developmental insults that can occur at various embryologic stages, and an understanding of the formation of the genitourinary system is helpful in the evaluation and treatment of a child with a congenital genitourinary anomaly. Imaging plays an essential role in the diagnosis of congenital genitourinary anomalies and treatment planning. In this article, we highlight the embryologic and characteristic imaging features of various congenital genitourinary anomalies, demonstrate the utility of different imaging modalities in management, and review specific imaging modalities and protocols for image optimization.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Congenital anomalies of the genitourinary tract are among the most common organ system abnormalities in the neonate. Detection usually occurs in the antenatal or immediate postnatal period, with a significant proportion identified in older children with varying degrees of severity. Congenital abnormalities of the kidney and urinary tract are one of the leading causes of end-stage renal disease [1] and range from asymptomatic ectopic kidneys to fatal bilateral renal agenesis. The spectrum of congenital malformations of the genitalia is broad and beyond the scope of this paper. Disorders of sex development are covered in more detail in a separate article in this supplement [2]. For the purposes of this review, we focus on Müllerian duct anomalies, which are clinically important to diagnose because of the increased risk of infertility, endometriosis, and recurrent miscarriages later in life [3].
Congenital renal anomalies can be classified on an embryologic basis into abnormalities in the renal parenchymal development, aberrant embryonic migration, and abnormalities of the collecting system. Renal parenchymal abnormalities include multicystic dysplastic kidneys; renal hypoplasia; abnormalities in number, whether agenesis or supernumerary; and cystic renal diseases (Table 1). Aberrant embryonic migration encompasses abnormal location and fusion anomalies. Anomalies of the collecting system include duplex collecting systems and ureteropelvic junction (UPJ) obstruction. Congenital anomalies of the lower urinary tract can be classified into those involving the distal ureter, urachus, bladder and urethra. The spectrum of disease that occurs with congenital anomalies of the lower urinary tract is listed in Table 2.
In this manuscript, we review complex congenital genitourinary anomalies, emphasizing the imaging techniques that can aid in diagnosis. These malformations can coexist within the same case because of their common embryologic origin, which we also briefly summarize. We describe failures of normal development with each of the specific anomalies.
Embryology
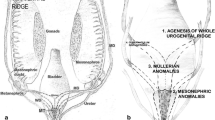
The urogenital system arises from intermediate mesoderm, which forms a urogenital ridge on either side of the aorta. The urogenital ridge develops into three sets of tubular nephric structures (from head to tail): the pronephros, the mesonephros and the metanephros (Fig. 1) [4]. The pronephros is the cranial-most set of tubes, and this mostly regresses [4, 5]. The mesonephros is located along the midsection of the embryo and develops into mesonephric tubules and the mesonephric duct (Wolffian duct) [4]. These tubules initially carry out some kidney function, but many of the tubules later regress. The mesonephric duct persists, however, and opens to the cloaca at the tail of the embryo. The metanephros gives rise to the definitive adult kidney [5].

Urogenital system development. Three sets of tubular nephric structures develop from the urogenital ridge: the pronephros, the mesonephros and the metanephros. The pronephros mostly regresses. The mesonephros develops into mesonephric tubules and the mesonephric duct (Wolffian duct). The metanephros gives rise to the definitive adult kidney
Before the 4th week of gestation, the cloaca, which is normally a transient structure during embryonic development, forms from the terminal portion of the hindgut by the confluence of the allantois and hindgut (Fig. 2) [4]. The cloaca is separated from the amniotic cavity by the cloacal membrane, which is the most distal portion of the cloaca [6]. Ureteral orifices form when caudal mesonephric ducts are absorbed into the developing bladder. The ureteral bud is an outgrowth of the mesonephric duct that further differentiates into ureters and the renal collecting system [4]. The genitals and the lower abdominal wall are formed by the genital tubercle.

Embryology of early the lower urinary tract. The allantois (A) incorporates into the distal portion of the hindgut (H) to form the cloaca (C) before the 4th week of gestation. Ureteral orifices form from caudal mesonephric ducts (M) that are absorbed into the developing bladder. The ureteral bud (B) differentiates into ureters and the renal collecting system. The genitals and the lower abdominal wall form from the genital tubercle (G). CM cloacal membrane
By the end of the 7th week of gestation, the urorectal septum separates the cloaca into dorsal and ventral parts and develops into the perineal body (Fig. 3) [7]. There is further differentiation of the urogenital sinus into vesical (cranial), pelvic (middle) and phallic (caudal) divisions (Fig. 3) [6]. The vesical division forms the bladder dome, ventral bladder wall and urachus. The pelvic division forms the urethra, and the phallic division the genitals. The bladder trigone is formed by caudal ends of the mesonephric ducts distal to the ureteral bud [6]. By week 16, the urachus involutes to become the median umbilical ligament extending from the apex of the bladder to the umbilicus. The developing genitals are normally cephalad to the opening of the urogenital sinus (Fig. 3) [7].

Development of the lower urinary tract (a), urogenital sinus and rectum (b) and urinary bladder (c). a The urorectal septum separates the cloaca into dorsal (D) and ventral (V) parts and develops into the perineal body. A allantois, M mesonephric ducts. b The formation of the perineal body later divides the cloaca into the urogenital sinus anteriorly and rectum posteriorly. c The urogenital sinus further differentiates into cranial and vesical (Ve), middle and pelvic (P), and caudal and phallic (Ph) divisions. The urachus (U) involutes and becomes the median umbilical ligament by week 16. Developing genitals (G) are normally positioned cephalad to the opening of urogenital sinus
Mesodermal folds lateral to the cloacal membrane enlarge and form the paired primordia of the genital tubercles before the 4th week of gestation [6]. By the 5th week of gestation, the definitive genital tubercle forms by rapid enlargement, medial migration and fusion of these primordia [6]. The abdominal wall musculature and anterior pelvic bones are formed by additional mesodermal tissue that accompanies the genital tubercle, which is located in the lower developing abdominal wall.
All embryos develop female internal genitalia unless a functioning testis is present [8]. In the presence of androgens, male genitalia develops (Fig. 4) [6]. Sertoli cells in the fetal testes produce Müllerian-inhibiting factor, a glycoprotein that inhibits development of the paramesonephric (Müllerian) ducts [6]. Leydig cells in the fetal testes produce testosterone, which stabilizes the mesonephric (Wolffian) ducts and promotes further development of vasa deferentia, epididymides and seminal vesicles. In females, the mesonephric (Wolffian) ducts atrophy and the paramesonephric (Müllerian) ducts persist and develop into the fallopian tubes, uterus and upper vagina (Fig. 4). Because the formation of the urinary tract and genitalia occur intimately, renal anomalies are commonly seen with genital anomalies [9].

Embryonic development of males and females. Differential development of the male and female ducts is directed by the gonads. Wolffian ducts regress in females in the absence of testosterone, leaving Müllerian ducts to form female reproductive organs. In males, testosterone supports the growth of the Wolffian ducts, which differentiate into male urogenital tract derivatives
Imaging techniques and protocols
In addition to the more traditional methods to image the genitourinary tract in children, which include US, fluoroscopy and in some cases nuclear imaging, MRI and contrast-enhanced voiding urosonography (ceVUS) can aid in diagnosis and management. CT can also be helpful in certain situations.
Ultrasound and fluoroscopy are commonly performed first in the initial evaluation of the genitourinary system at most institutions. Contrast-enhanced VUS can be a helpful adjunct, combining both anatomical and functional imaging of the genitourinary system with no radiation exposure. However, these primary imaging modalities might be inadequate for the full characterization of certain genitourinary anomalies. MRI can help by providing morphological and functional information on genitourinary structures without ionizing radiation or limitations of patient body habitus or overlying bowel gas. In addition, MRI has good spatial, temporal and contrast resolution that is ideally suited for evaluating the genitourinary tract [10].
There are two primary MRI techniques for evaluating the pediatric genitourinary system: an MRI pelvis protocol for assessing urogenital anatomy, and MR urography for specific morphological and functional evaluation of the kidneys and upper urinary tract. MRI examinations can be performed at either 1.5-tesla (T) or 3.0-T magnet strength using multi-channel phased-array surface coils. Imaging performed at 3.0 T provides increased signal, which can be used to improve spatial resolution in younger children, albeit with the trade-off of increasing artifacts and decreasing homogeneous fat suppression [11].
The primary aim of the standard MRI pelvis protocol is to clearly define the anatomy of the genitourinary system and assess abnormalities (Table 3). Common indications include evaluation of Müllerian duct anomalies or ambiguous genitalia, or identification of the ovaries/testes. The coverage area should be focused on the pelvis, especially the perineum to include the external genitalia, with survey images of the kidneys. All sequences (aside from the diffusion-weighted images) should be performed with high resolution and a minimum in-plane resolution of 1 × 1-mm pixels and 3- to 4-mm slice thickness.
The most common indications for pediatric MR urography are to evaluate complex renal and urinary tract anatomy, suspected urinary tract obstruction, and pre- and postoperative assessment. A typical MR urography protocol should include sequences for MR hydrography and post-contrast MR urography (Table 4). MR hydrography leverages the long T2 relaxation time of urine to create high-contrast-resolution images of the urinary tract. This technique excels at evaluating a dilated or obstructed upper urinary tract without contrast material, although it is less ideal when the urinary tract is not dilated. Post-contrast MR urography images the urinary tract following the administration of an intravenous gadolinium-based contrast agent (GBCA), typically gadobutrol at our institution. This technique provides gross evaluation of renal perfusion and excretion as well as better visualization of nondilated ureters and their insertion. Additionally, when using a GBCA, quantitative renal function data such as renal blood flow, glomerular filtration rate, and split renal function can be assessed [12]. The field-of-view should include the entire urinary tract from the kidneys through the perineum to assess for ectopic ureter insertion. All sequences should be performed with high resolution and a minimum in-plane resolution of 1 × 1-mm pixels and 3- to 4-mm slice thickness.
Spectrum of imaging findings
Renal agenesis
Renal development begins in the 5th week of gestation [13]. Renal agenesis results from the abnormal interaction between the ureteric bud of the mesonephric duct and metanephric mesenchyme [14]. If bilateral, renal agenesis leads to the classic triad of oligohydramnios, lung hypoplasia, and dysmorphic facies known as Potter syndrome. It is a uniformly fatal condition [14]. If unilateral, however, children are typically asymptomatic with a normal life expectancy [14].
On imaging, there is complete absence of the kidney with no ectopia or fusion anomaly. A linear configuration of the ipsilateral adrenal gland can often be an indicator of renal agenesis [15]. Compensatory hypertrophy of the remaining kidney might also be seen.
Contralateral renal abnormalities are seen in up to 25% of cases of unilateral renal agenesis, in which vesicoureteral reflux is most common [16, 17]. Other contralateral anomalies that occur include ureteropelvic junction obstruction, megaureter and collecting system duplication [17, 18].
Unilateral renal agenesis is also frequently associated with genital anomalies, including seminal vesicle cysts in males and Müllerian duct anomalies in females (discussed later in the article) [17]. Seminal vesicle cysts can be congenital or acquired and are thought to be caused by an obstruction at the junction of the seminal vesicle and ejaculatory duct [17]. Failure of the distal part of the mesonephric duct to develop before the 7th week of gestation causes atresia of the ejaculatory ducts, resulting in distension of the seminal vesicles from insufficient drainage and further leading to the formation of a cyst [17]. Patients usually become symptomatic in young adulthood because of the accumulation of secretions.
Ejaculatory duct obstruction and seminal vesicle cysts can be seen on US as anechoic structures in the pelvis (Fig. 5), sometimes with low-level echoes indicating proteinaceous material or hemorrhage. MRI can aid in the detailed assessment and characterization of the genital anatomy and shows a periprostatic cystic mass ipsilateral to the renal agenesis, without enhancement (Fig. 5). Associated findings include an ectopic ureteral insertion into the seminal vesicle, ejaculatory duct, prostatic urethra or vas deferens; or agenesis of the vas deferens (Fig. 6) [19, 20]. If a child presents with unilateral agenesis of the vas deferens, a renal US is recommended to assess for renal agenesis.

Ejaculatory duct obstruction in a 15-year-old boy with fever and abdominal pain. a Transverse US demonstrates avascular, tubular cystic structures (asterisks) filled with debris, compatible with ejaculatory duct obstruction and seminal vesicle cysts. b Coronal T2-weighted MRI shows left renal agenesis and seminal vesicle cysts (arrow)

Solitary right kidney in a 1-month-old boy. a Sagittal color Doppler sonographic image demonstrates a complex, avascular cystic structure (arrow) posterior to the bladder and separate from the bladder and rectum. b Magnetic resonance fistulogram three-dimensional (3-D) reformatted image from a sagittal projection shows that the retrovesicular cyst correlates to a prostatic utricle cyst, with a fistula between the terminal rectum and the utricle cyst. Ectopic insertion of the bilateral ductus deferens and left seminal vesicle into the utricle cyst was also seen (not shown). The right seminal vesicle was not identified and thought to be secondary to agenesis
Ureteropelvic junction obstruction
Congenital ureteropelvic junction (UPJ) obstruction is one of the most common causes of prenatal hydronephrosis and the most common cause of postnatal hydronephrosis [17]. It is more common in males and has a left-side predominance [17, 21]. It is often associated with other urinary anomalies [17], including contralateral multicystic dysplastic kidney (MCDK), which requires prompt intervention because MCDK is nonfunctional and UPJ obstruction can compromise the remaining renal function. Prognosis is usually excellent if renal function has not been compromised by longstanding, high-grade obstruction [22].
The etiology of UPJ obstruction is debatable and includes intrinsic causes from inadequate canalization during 10–12 weeks of gestation or extrinsic obstructions secondary to bands, kinks or aberrant vessels [21]. These children can be asymptomatic or present with recurrent urinary tract infections, stone formation or palpable flank mass.
Antenatal and postnatal US are diagnostic and show a disproportionately dilated renal pelvis with dilatation of the calices and non-dilated ureters. Nuclear mercaptoacetyltriglycine (MAG3) renal scan can then be used to grade the degree of obstruction and differential function and to determine whether surgical intervention is required. MR urography might be a viable single-test alternative, optimizing anatomical and functional assessment, especially if proximal ureteral kinking or abnormal kidney rotation is suspected (Fig. 7). Evaluation for crossing vessels as a cause is important for appropriate surgical correction. Following successful surgery, pelvocaliectasis can persist for years on imaging. Postoperative imaging assessment for appropriate renal growth, changes in the degree of dilation, and morphologic parenchymal changes that might indicate worsening obstruction can help measure surgical success [23, 24].

Ureteropelvic junction (UPJ) obstruction in a 6-month-old boy with a history of prenatal hydronephrosis. Coronal maximum-intensity MR urogram demonstrates marked right hydronephrosis with no visualization of the right ureter, compatible with UPJ obstruction (arrow)
Duplex collecting system
A duplex collecting system is one of the most common congenital renal tract abnormalities [17], characterized by an incomplete fusion of upper and lower pole moieties resulting in a variety of complete or incomplete duplications of the collecting system. Duplication occurs when two separate ureteric buds arise from a single Wolffian duct. Based on the degree of fusion, it can present as a bifid renal pelvis or partial ureteric duplication and lead to a Y-shape ureter, incomplete ureteric duplication with ureters joining near or in the bladder wall causing a V-shape ureter, or complete ureteric duplication with separate urinary bladder ureteric orifices. Duplex collecting systems are often asymptomatic, clinically insignificant and incidentally detected.
Contrast-enhanced VUS can aid in the simultaneous visualization of the refluxing lower pole moiety ureter and obstructed upper pole moiety ureter in contrast to fluoroscopy alone (Fig. 8). An MR urogram can help in demonstrating the detailed anatomy of the collecting system, level of fusion and status of ureteric orifices (Fig. 9). In complete duplex collecting systems, the ectopic upper pole moiety ureter can insert into the prostatic urethra in males or distal to the external sphincter or in the wall of the vagina resulting in incontinence in females. Also, a ureterocele can prolapse into the urethra and lead to bladder outlet obstruction.

History of urinary tract infections and outside report of left vesicoureteral reflux in a 4-month-old boy. a Sagittal gray-scale sonographic image demonstrates dilation of the left upper pole moiety ureter (arrows) of a duplicated left kidney to the level of the urinary bladder, which contains a large ureterocele (asterisk). b Dual-display sagittal gray-scale and contrast-enhanced voiding urosonography (ceVUS) image of the left kidney demonstrates reflux of contrast agent into the lower pole moiety (arrow) with hydronephrosis of the adjacent obstructed upper pole moiety (asterisks). c Transverse ceVUS dual-display image obtained more inferiorly demonstrates reflux into the dilated lower pole moiety ureter (arrow) and hydroureter of the obstructed upper pole moiety (asterisks)

Hydronephrosis in a 2-month-old boy. Coronal T2-weighted MR urogram demonstrates a duplicated left renal collecting system, with hydronephrosis of both upper pole and lower pole moieties
Prune belly syndrome
Prune belly syndrome is defined by the coexistence of three major findings: bilateral cryptorchidism, urinary tract abnormalities, and absence or deficiency of abdominal wall musculature, which can be associated with a number of other anomalies including respiratory, gastrointestinal, musculoskeletal and cardiovascular anomalies [25]. Occurring almost exclusively in males, the etiology for this syndrome is unknown. One theory proposes that a mesenchymal insult to the fetus at about 6 weeks of gestation produces deficient abdominal muscular development [26].
Urinary tract findings are characterized by tortuous and dilated ureters, a dilated prostatic urethra and renal dysmorphism. The bladder is often enlarged and elongated without trabeculation. There is bladder wall thickening caused by replacement of normal smooth muscle with connective tissue, which can similarly result in ureteral dilatation. However, ureteral dilatation can also be secondary to vesicoureteral reflux, which is present in the majority of cases [26].
On imaging, bilateral hydronephrosis with dilated, tortuous ureters is commonly visualized (Fig. 10). There can also be renal dysplasia with cystic changes, and a urachal anomaly might be seen, most frequently a urachal diverticulum [26].

Prune belly syndrome in a 4-day-old boy. Coronal MR urogram demonstrates severe bilateral hydronephrosis and markedly dilated and tortuous ureters (arrows)
Cloacal malformation and urogenital sinus
Cloacal malformation is a persistence of the early embryonic state in which the urinary, genital and gastrointestinal (GI) tracts all drain through a common perineal opening, resulting from failure of the urorectal septum to join the cloacal membrane during the 4th to 6th weeks of embryonic life (Fig. 11) [26]. Occurring only in phenotypical girls, this rare anomaly presents with a normal abdominal wall and a single perineal opening through which urine, genital secretions, and feces or meconium are excreted (Fig. 12). These children commonly present with associated findings including lower urinary tract abnormalities, genital abnormalities and abnormalities of the bony pelvis and lower spinal cord [26]. Although fluoroscopy is typically used to evaluate the cloaca, which is essential for diagnosis, a contrast genitosonogram can be performed as an alternative to a more traditional fluoroscopic genitogram (Fig. 13). The kidneys can be imaged by US, and anomalies of the lower spinal cord can be evaluated by MRI.

Flow chart shows one arm in the development of the genitourinary system. Disruptions in the normal embryologic development (denoted by the negative signs) at distinct time points result in the different congenital anomalies: cloacal malformation, urogenital sinus anomaly and bladder exstrophy–epispadias–cloacal exstrophy complex

Physical exam findings in a 3-month-old girl with cloacal malformation. Clinical photograph shows a single perineal opening (arrow) through which urine, genital secretions, and feces or meconium are excreted

Transperineal contrast-enhanced genitosonography in a 3-month-old girl with cloacal malformation. Catheters were placed in the bladder (B), vagina (V) and rectum (R) under cystoscopy, and these structures were opacified with a US contrast agent. Note the low confluence near the skin surface of the urethra (arrow), vagina and rectum. Case courtesy of Dr. Jeanne Chow, Department of Radiology, Boston Children’s Hospital
If there is persistence of the embryonic state in which the urinary and genital tracts drain through a common perineal opening but the GI tract drains separately, a urogenital sinus anomaly occurs (Fig. 11). Like cloacal malformation, urogenital sinus occurs only in phenotypical girls. However, in contrast to cloacal malformation, this anomaly presents with two perineal orifices: the anterior orifice for the bladder and vagina, and the posterior anal orifice. A contrast-enhanced genitosonogram can also be performed to delineate anatomy (Fig. 14). This imaging modality is covered in more detail in a separate article in this supplement [2].

Contrast-enhanced genitosonogram in a 2-month-old girl with urogenital sinus. The anterior perineal opening was catheterized, and a US contrast agent was injected. There is an elongated urethra (U) with confluence (arrow) of the urethra and the vagina (V) through a common perineal opening. The rectum is not opacified because it drains through a separate opening. B bladder
Bladder exstrophy–epispadias–cloacal exstrophy complex
In bladder exstrophy–epispadias complex, the normal arrangement of the genital tubercle fusing cephalad to the distal aspect of the urogenital sinus is reversed, leading to direct contact of the urogenital sinus and anterior body wall [27]. As a result, the pelvic portion of the urogenital sinus, which forms the urethra, lies cephalad to the genital tubercle, which forms the body of the genitals. Thus, epispadias forms with the urethra on the dorsal aspect of the penis.
Bladder exstrophy involves a migration failure of the lateral mesodermal folds of the infraumbilical anterior abdominal wall and rupture of the resulting enlarged, persistent cloacal membrane before the 8th week of gestation (Fig. 11) [27, 28]. A wide communication between the posterior bladder wall and the outside environment results (Fig. 15).

Cloacal exstrophy, epispadias and imperforate anus in a 25-day-old boy. a Clinical photograph shows cloacal exstrophy with a large lower anterior abdominal wall defect, bifid scrotum (white arrows), bilateral ureters emptying into herniated bladder plates (asterisks), and bowel (black arrow). b Anteroposterior abdominal radiograph demonstrates splayed pubic and ischial bones (arrows). c Axial post-contrast T1 fat saturated excretory phase MRI shows a markedly distended rectal pouch containing debris (solid arrow) and the right ureter (dotted arrow) coursing anterolaterally with its orifice to the right of midline at the herniated bladder plate (not shown)
Cloacal exstrophy must be differentiated from cloacal malformation. Although both of these anomalies contain the word “cloaca,” which is Latin for sewer, they differ greatly in their embryogenesis and clinical features. In contrast to cloacal malformation, exstrophy of the cloaca is seen in both boys and girls and involves a failure of the lower abdominal wall to close. Superficially, the anomaly resembles bladder exstrophy, but the defect is larger [28].
Imaging helps to distinguish bladder exstrophy from cloacal exstrophy and variants [28]. Radiographs can show diastasis of the pubic symphysis, commonly seen in bladder exstrophy (Fig. 15) [28, 29]. Spinal anomalies, ranging from segmental spinal dysgenesis to open spinal dysraphisms, are more common in cloacal exstrophy. Thus, both radiographs and spinal US can help to define the type of spinal dysraphism [30]. MRI is useful for detailed assessment of the complex anatomy involved prior to closure and reconstruction (Fig. 15) [28].
Müllerian duct anomalies
In the female embryo, estrogens stimulate development of Müllerian ducts and differentiation of female external genitalia. The Müllerian ducts are paired laterally, longitudinal embryologic structures that undergo fusion and resorption to give rise to the uterus, fallopian tubes, cervix and upper two-thirds of the vagina (Fig. 16). Interruption in the normal development at one of the steps of formation, fusion and resorption can result in Müllerian duct anomalies (Fig. 17) [3, 31]. If neither Müllerian duct forms, complete Müllerian agenesis occurs. If only one of the bilateral Müllerian ducts forms, a unicornuate uterus results. Sometimes, there is partial development of one side, resulting in a rudimentary horn that may or may not communicate with the completely developed side. Incomplete fusion of the formed ducts results in a didelphys uterus, with partial fusion resulting in a bicornuate uterus. Arrest at any point during the normal septal resorption phase can result in varying septal lengths, ranging in an arcuate, partial or complete septate uterus, which is the most common Müllerian duct anomaly [3, 32].

Müllerian duct development and differentiation of female external genitalia. The Müllerian ducts start as laterally situated structures (1), which course medially at their caudal aspect. The ducts abut the urogenital sinus, inducing the formation of a solid vaginal plate. As the ovaries descend from the gonadal ridge, the caudal Müllerian ducts fuse (2), forming the corpus of the uterus, cervix and upper vagina, and upper portions of ducts form fallopian tubes. Initially, a septum is present because of the fused segments of distal paired ducts, but later the septum degenerates and the vaginal plate becomes canalized (3). The result is paired fallopian tubes that communicate with the abdominal cavity cranially and uterine cavity caudally, uterus and cervix with a single central cavity, and vagina that is derived both from Müllerian ducts and urogenital sinus (4)

Müllerian duct anomalies. Abnormalities can develop from problems with formation, fusion or septal resorption. Adapted from [31]
There is a high associated risk of infertility, endometriosis and miscarriage in women with Müllerian duct anomalies [33]. Commonly associated renal anomalies include renal agenesis, ectopia, hypoplasia, fusion, malrotation and duplication [34, 35]. Other abnormalities that can occur with Müllerian duct anomalies include vertebral body anomalies, cardiac anomalies and syndromes such as Klippel–Feil syndrome and Bardet–Biedl syndrome [36, 37] (Fig. 18).

Genetically confirmed Bardet–Biedl syndrome in a 3-day-old girl. a Sagittal T2-weighted MRI shows a large fluid-filled structure posterior to the bladder and anterior to the rectum, compatible with hydrocolpos (asterisk) from vaginal atresia. b Coronal T2-weighted MRI shows right hydronephrosis and multiple cysts in both kidneys (arrows) in a pattern seen better on renal US (not shown) to be suggestive of medullary cystic renal dysplasia. In addition, the girl had post-axial polydactyly
Ultrasound is the preferred imaging modality in the initial workup of Müllerian duct anomalies in younger patients. However, MRI provides the best anatomical detail for assessing both the uterine cavity and external fundal contour [38].
The Mayer–Rokitansky–Küster–Hauser syndrome is characterized by complete agenesis of the Müllerian ducts, leading to aplasia of the uterus and the upper two-thirds of the vagina. Because ovarian function is normal, these girls usually present during adolescence with primary amenorrhea in the presence of normal pubertal development and secondary sexual characteristics [39]. Renal agenesis and ectopia are the most common associated congenital anomalies [40]. MRI allows for accurate visualization of normal ovaries bilaterally and absence of the uterus and upper two-thirds of the vagina (Fig. 19).

Mayer–Rokitansky–Küster–Hauser syndrome in a 17-year-old girl who presented with primary amenorrhea. a, b Sagittal T2-weighted MR images demonstrate normal right (a) and left (b) ovaries (arrows). c Sagittal T2-weighted MRI shows complete absence of the uterus and upper two-thirds of the vagina. The girl was also found to have left renal agenesis (not shown). Findings were compatible with Mayer–Rokitansky–Küster–Hauser syndrome. Case courtesy of Dr. Yen-Ying Wu, Department of Radiology, Loma Linda University Children’s Hospital
Conclusion
Congenital genitourinary anomalies are commonly encountered in routine imaging. When an infant is suspected of having an underlying genitourinary structural anomaly, screening US and fluoroscopy are commonly the initial diagnostic studies performed. However, ceVUS can be a viable imaging alternative to fluoroscopy where available. MR urography plays an important role in displaying dilated collecting systems, ectopic ureters and ureteroceles and has an advantage over US in that it can demonstrate ectopic extravesical ureteric insertions, thereby providing a global view of the malformation. MRI is also currently the imaging modality of choice for the diagnosis of Müllerian duct anomalies because of its reliability and accuracy in classifying these anomalies.
Because the formation of the urinary tract and genitalia occur intimately, it is important to remember that renal anomalies are commonly seen with genital anomalies. An understanding of the embryologic failure, associated findings, and potential complications is important for timely diagnosis and optimal patient outcome.
References
Isert S, Müller D, Thumfart J (2020) Factors associated with the development of chronic kidney disease in children with congenital anomalies of the kidney and urinary tract. Front Pediatr 8:298
Hryhorczuk A, Phelps A, Yu R, Chow J (2021) A radiologist’s role in assessing differences of sex development. Pediatr Radiol. https://doi.org/10.1007/s00247-021-05147-z
Behr SC, Courtier JL, Qayyum A (2012) Imaging of Müllerian duct anomalies. Radiographics 32:E233–250
Rehman S, Ahmed D (2021) Embryology, kidney, bladder, and ureter. StatPearls. https://www.ncbi.nlm.nih.gov/books/NBK547747/. Accessed 26 Mar 2021
Blake J, Rosenblum ND (2014) Renal branching morphogenesis: morphogenetic and signaling mechanisms. Semin Cell Dev Biol 36:2–12
No authors listed (2015) Urogenital development. Duke Medicine website. https://embryology.oit.duke.edu/urogenital/urogenital.html. Accessed 30 Jun 2021
Levin TL, Han B, Little BP (2007) Congenital anomalies of the male urethra. Pediatr Radiol 37:851–862
Mullen RD, Behringer RR (2014) Molecular genetics of Müllerian duct formation, regression and differentiation. Sex Dev 8:281–296
Massé J, Watrin T, Laurent A et al (2009) The developing female genital tract: from genetics to epigenetics. Int J Dev Biol 53:411–424
Epelman M, Daneman A, Donnelly LF et al (2014) Neonatal imaging evaluation of common prenatally diagnosed genitourinary abnormalities. Semin Ultrasound CT MRI 35:528–554
Morin CE, McBee MP, Trout AT et al (2018) Use of MR urography in pediatric patients. Curr Urol Rep 19:1–11
Dickerson EC, Dillman JR, Smith EA et al (2015) Pediatric MR urography: indications, techniques, and approach to review. Radiographics 35:1208–1230
Rosenblum S, Pal A, Reidy K (2017) Renal development in the fetus and premature infant. Semin Fetal Neonatal Med 22:58–66
Ramanathan S, Kumar D, Khanna M et al (2016) Multi-modality imaging review of congenital abnormalities of kidney and upper urinary tract. World J Radiol 8:132
Hoffman CK, Filly RA, Callen PW (1992) The “lying down” adrenal sign: a sonographic indicator of renal agenesis or ectopia in fetuses and neonates. J Ultrasound Med 11:533–536
Shapiro E, Goldfarb DA, Ritchey ML (2003) The congenital and acquired solitary kidney. Rev Urol 5:2–8
Houat AP, Guimarães CTS, Takahashi MS et al (2021) Congenital anomalies of the upper urinary tract: a comprehensive review. Radiographics 41:462–486
Westland R, Schreuder MF, Ket JCF, van Wijk JAE (2013) Unilateral renal agenesis: a systematic review on associated anomalies and renal injury. Nephrol Dial Transplant 28:1844–1855
Kim B, Kawashima A, Ryu JA et al (2009) Imaging of the seminal vesicle and vas deferens. Radiographics 29:1105–1121
Arora SS, Breiman RS, Webb EM et al (2007) CT and MRI of congenital anomalies of the seminal vesicles. AJR Am J Roentgenol 189:130–135
Williams B, Tareen B, Resnick MI (2007) Pathophysiology and treatment of ureteropelvic junction obstruction. Curr Urol Rep 8:111–117
Amling CL, O’Hara SM, Wiener JS et al (1996) Renal ultrasound changes after pyeloplasty in children with ureteropelvic junction obstruction: long-term outcome in 47 renal units. J Urol 156:2020–2024
Cost NG, Prieto JC, Wilcox DT (2010) Screening ultrasound in follow-up after pediatric pyeloplasty. Urology 76:175–179
Heinlen JE, Manatt CS, Bright BC et al (2009) Operative versus nonoperative management of ureteropelvic junction obstruction in children. Urology 73:521–525
Jennings RW (2000) Prune belly syndrome. Semin Pediatr Surg 9:115–120
Berrocal T, López-Pereira P, Arjonilla A, Gutiérrez J (2002) Anomalies of the distal ureter, bladder, and urethra in children: embryologic, radiologic, and pathologic features. Radiographics 22:1139–1164
Kulkarni B, Chaudhari N (2008) Embryogenesis of bladder exstrophy: a new hypothesis. J Indian Assoc Pediatr Surg 13:57–60
Dunn EA, Kasprenski M, Facciola J et al (2019) Anatomy of classic bladder exstrophy: MRI findings and surgical correlation. Curr Urol Rep 20:48
Pierre K, Borer J, Phelps A, Chow JS (2014) Bladder exstrophy: current management and postoperative imaging. Pediatr Radiol 44:768–786
Ebert A-K, Reutter H, Ludwig M, Rösch WH (2009) The exstrophy–epispadias complex. Orphanet J Rare Dis 4:23
No authors listed (1988) The American Fertility Society classifications of adnexal adhesions, distal tubal occlusion, tubal occlusion secondary to tubal ligation, tubal pregnancies, Müllerian anomalies and intrauterine adhesions. Fertil Steril 49:944–955
Fajardo RS, DeAngelis GA (2021) Septate uterus. Applied Radiology website. https://www.appliedradiology.com/articles/septate-uterus. Accessed 13 Jul 2021
Wold ASD, Pham N, Arici A (2006) Anatomic factors in recurrent pregnancy loss. Semin Reprod Med 24:25–32
Troiano RN, McCarthy SM (2004) Müllerian duct anomalies: imaging and clinical issues. Radiology 233:19–34
Li S, Qayyum A, Coakley FV, Hricak H (2000) Association of renal agenesis and Mullerian duct anomalies. J Comput Assist Tomogr 24:829–834
Gell JS (2003) Müllerian anomalies. Semin Reprod Med 21:375–388
Kimberley N, Hutson JM, Southwell BR, Grover SR (2012) Vaginal agenesis, the hymen, and associated anomalies. J Pediatr Adolesc Gynecol 25:54–58
Mueller GC, Hussain HK, Smith YR et al (2007) Müllerian duct anomalies: comparison of MRI diagnosis and clinical diagnosis. AJR Am J Roentgenol 189:1294–1302
Oppelt P, Renner SP, Kellermann A et al (2006) Clinical aspects of Mayer–Rokitansky–Kuester–Hauser syndrome: recommendations for clinical diagnosis and staging. Hum Reprod 21:792–797
Pittock ST, Babovic-Vuksanovic D, Lteif A (2005) Mayer–Rokitansky–Küster–Hauser anomaly and its associated malformations. Am J Med Genet Part A 135A:314–316
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
None
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Acharya, P.T., Ponrartana, S., Lai, L. et al. Imaging of congenital genitourinary anomalies. Pediatr Radiol 52, 726–739 (2022). https://doi.org/10.1007/s00247-021-05217-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00247-021-05217-2