
Abstract
Inflammatory pseudotumor is a generic term used to designate a heterogeneous group of inflammatory mass-forming lesions histologically characterized by myofibroblastic proliferation with chronic inflammatory infiltrate. Inflammatory pseudotumor is multifactorial in etiology and generally benign, but it is often mistaken for malignancy given its aggressive appearance. It can occur throughout the body and is seen in all age groups. Inflammatory pseudotumor has been described in the literature by many organ-specific names, resulting in confusion. Recently within this generic category of inflammatory pseudotumor, inflammatory myofibroblastic tumor has emerged as a distinct entity and is now recognized as a fibroblastic/myofibroblastic neoplasm with intermediate biological potential and occurring mostly in children. We present interesting pediatric cases of inflammatory myofibroblastic tumors given this entity’s tendency to occur in children. Familiarity and knowledge of the imaging features of inflammatory pseudotumor can help in making an accurate diagnosis, thereby avoiding unnecessary radical surgery.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The umbrella term “inflammatory pseudotumor” is used to designate a group of mass-forming lesions that are histologically characterized by a proliferation of fibroblasts and myofibroblasts accompanied by a prominent chronic inflammatory infiltrate [1, 2]. Inflammatory pseudotumor is generally a benign process, but it is often mistaken for malignancy because of its mass-forming nature and frequently aggressive imaging appearance. In the literature, inflammatory pseudotumor has been described under various organ-specific names, reflecting its heterogeneous imaging appearance and variable histological characteristics and biological behavior [1, 3] (Table 1). Because various terms are used to describe the entity, accurate data regarding the incidence, anatomical distribution and patient demographics of inflammatory pseudotumors are difficult to obtain [2]. Inflammatory pseudotumor was originally described in the lung and orbit but has now been reported throughout the body [1]. It is seen in all age groups, with age distribution depending on the involved organ [2].
From among the inflammatory pseudotumors, inflammatory myofibroblastic tumor has recently emerged as a distinct entity with its own clinical, pathological and molecular features. Inflammatory myofibroblastic tumor is a true neoplastic entity and is categorized by the World Health Organization as a neoplastic fibroblastic/myofibroblastic tumor with intermediate biological potential. It occurs predominantly in the visceral soft tissue of children with a potential for local recurrence and rare distant metastasis [4]. Throughout this article we use inflammatory pseudotumor as an umbrella term inclusive of inflammatory myofibroblastic tumor because inflammatory myofibroblastic tumor cannot be differentiated from inflammatory pseudotumor on imaging, and tissue analysis is required to make the correct diagnosis. Our examples comprise mostly inflammatory myofibroblastic cases, though, and the specific pathological diagnosis of each is clarified in the figure captions.
Some inflammatory pseudotumors are found in association with immunoglobulin G4 (IgG4)-related disease, a systemic immune-mediated fibrosclerosing disease characterized by IgG4-positive plasma cell and T-cell infiltration of various tissues causing multicentric, mass-forming fibrous lesions, primarily in older adults. It is characterized by elevated serum titers of IgG4 and prototypical systemic manifestations, including orbital pseudotumor, autoimmune pancreatitis, retroperitoneal fibrosis, sclerosing mesenteritis, and sclerosing cholangitis [5]. Histologically, IgG4-related disease lesions have an infiltrative growth pattern with IgG4-positive plasma cells and a less cellular spindle cell and inflammatory infiltrate when compared to inflammatory myofibroblastic tumor [2].
Surgery is often the primary treatment for inflammatory pseudotumor, but radical surgery is usually unnecessary because of its overall benign nature. Some inflammatory pseudotumors have completely resolved after administration of steroids, anti-inflammatory agents, and antibiotics [6–12]. There have also been a few reports of inflammatory pseudotumor completely resolving after a period of observation without any treatment at all [8–10, 12]. Familiarity and knowledge of the imaging features of inflammatory pseudotumor can assist in making an accurate diagnosis, thereby avoiding unnecessary radical surgery.
The etiology of inflammatory pseudotumor is unknown, but various causes have been implicated. Some infectious organisms have been identified, including mycobacteria, mycoplasma and nocardia in pulmonary inflammatory pseudotumor, Epstein–Barr virus (EBV) in splenic and nodal inflammatory pseudotumor, and actinomycetes in hepatic inflammatory pseudotumor [1]. EBV and human herpesvirus-8 (HHV-8) have been detected in a variety of inflammatory pseudotumors, including lung, liver, spleen and bladder [13]. EBV-infected lymphocytes have been known to result in inflammatory pseudotumor or lymphomatoid granulomatosis [14]. Inflammatory pseudotumor may also be immune-related, as seen in the suggested relationship of orbital inflammatory pseudotumor to IgG4-related disease. Other inflammatory pseudotumors may be related to postoperative inflammatory reaction, such as central nervous system (CNS) inflammatory pseudotumor developing after long-standing ventriculoperitoneal shunt [15]. Finally, inflammatory myofibroblastic tumor, which had been grouped under inflammatory pseudotumor, is considered a true neoplasm with rare metastatic potential.
Histologically, inflammatory pseudotumors are characterized by a proliferation of fibroblasts and myofibroblasts associated with infiltration of inflammatory cells consisting of lymphocytes, plasma cells and histiocytes [16]. Inflammatory cytokines may stimulate the proliferation of fibroblasts. Fibroblasts are ubiquitous mesenchymal cells found in the stroma of all tissues; these fibroblasts can turn into myofibroblasts, which express smooth muscle actin and fibrogenic cytokines, resulting in fibrosis [17]. Regardless of the initial trigger, the common feature of all fibrotic diseases is the activation of myofibroblasts, key mediators of fibrotic tissue remodeling [17, 18].
By comparison, inflammatory myofibroblastic tumor is a distinct lesion composed of neoplastic myofibroblastic spindle cells accompanied by an inflammatory infiltrate of plasma cells, lymphocytes and eosinophils [16]. The neoplastic spindle cells are uniform and mostly myofibroblastic, with absence of nuclear hyperchromasia, low mitotic activity and rare atypical mitoses [2]. On immunohistochemistry, myofibroblastic cells stain positively for vimentin and smooth muscle actin [19]. Sixty percent of inflammatory myofibroblastic tumors harbor a rearrangement of the anaplastic lymphoma kinase (ALK) locus on chromosome 2p23, which causes abnormal tyrosine kinase receptor expression. The presence of ALK expression helps establish the diagnosis of inflammatory myofibroblastic tumor [20].
Clinical presentation and imaging findings
Up to one-third of patients with inflammatory pseudotumor present with nonspecific constitutional signs and symptoms of fever, weight loss, microcytic anemia, failure to thrive, thrombocytosis, elevated sedimentation rate, polyclonal hypergammaglobulinemia, as well as symptoms from mass effect of the lesion itself [1]. These constitutional manifestations resolve after surgical resection and may reappear with recurrence [2]. Overproduction of interleukin-1, interleukin-6, and transforming growth factor β has been implicated as a cause of these systemic symptoms [1].
The most common locations of inflammatory pseudotumor are the abdominopelvic region, lung and retroperitoneum [2]. In one review of 275 cases of pediatric inflammatory pseudotumor, one-third of cases were pulmonary and two-thirds were extrapulmonary [21].
Inflammatory pseudotumors are characterized on imaging by their fibrous nature. Imaging features of fibrous lesions are nonspecific but demonstrate some common features. On US imaging, fibrous tissue is hypoechoic and relatively hypovascular [22]. On CT, fibrous tissue is iso-attenuating to muscle. On contrast-enhanced imaging, inflammatory pseudotumors show relative lack of enhancement in the parenchymal phase followed by characteristic delayed-phase enhancement as seen in fibrous tissue [22]. However, dual-phase CT examination is not recommended in children because of the overall increased radiation exposure.
Magnetic resonance imaging (MRI) is the modality of choice to image fibrous lesions. Features of fibrosis are hypo- to isointensity on T1-weighted images, marked hypointensity on T2-weighted images, and homogeneous or heterogeneous enhancement with more intense enhancement on delayed images [22]. In early stages of inflammatory pseudotumors, when inflammation predominates and fibrosis is not yet established, the T2 signal may be hyperintense. Diffusion-weighted imaging (DWI) varies depending on the degree of cellularity and acuity of the inflammation [22, 23]. Hypocellular lesions with edema and acute inflammation may not show diffusion restriction. Recent research in orbital lesions suggests that diffusion-weighted MRI may help distinguish inflammatory pseudotumor from malignancy, which tends to show lower apparent diffusion coefficient (ADC) values [24].
Differential diagnoses
Although its fibrous nature is a characteristic feature, inflammatory pseudotumor may still appear similar to any infiltrative, fibrous or hypercellular lesion, and thus requires histological confirmation by biopsy. Benign and malignant differential diagnoses for pediatric fibrous lesions are summarized in Table 2. Fibrous mass-forming lesions in the pediatric age group include fibroma, fibrosarcoma, fibromatoses, granulomatous inflammation, and malignancy with prominent inflammatory infiltrate. The fibromatoses are non-metastasizing benign fibroblastic/myofibroblastic proliferative lesions. Infantile myofibromatosis is one of the most common fibromatoses, mainly presenting in children younger than 2 years [19, 25, 26]. It can mimic inflammatory pseudotumor because of its fibrous nature, multicentricity and predilection for the pediatric population. Myofibromatosis can involve the lung, and this is a manifestation of multifocal systemic disease rather than metastatic disease. This is in contradistinction to inflammatory myofibroblastic tumor, which can metastasize to lung [27].
Patient age is important for narrowing the differential diagnosis for suspected inflammatory pseudotumor. Inflammatory myofibroblastic tumor, particularly with visceral involvement, can occur in any age group but is most frequent in the first two decades of life, with a mean age at diagnosis of 10 years old [4]. Infantile myofibromatosis occurs in infants and young children. IgG4-related disease tends to occur in older adults, with a mean age of presentation of 60–65 years [5], though it rarely occurs in the pediatric population [28]. When a putative diagnosis of inflammatory myofibroblastic tumor is made in an older adult, an alternative diagnosis such as IgG4-related disease should be considered [2].
Presentations of inflammatory pseudotumor vary by organ location and are reviewed in this article to help familiarize radiologists with this heterogeneous group of mass-forming lesions. Table 3 summarizes imaging findings of inflammatory pseudotumor based on location, important differential diagnoses, current treatment and key pearls.
Head and neck
Orbital inflammatory pseudotumor
Orbital inflammatory pseudotumor is nonspecific orbital inflammation of unknown etiology. Diagnosis is usually made by clinical history and exclusion [1]. Patients often present with acute painful proptosis, swelling and ophthalmoplegia rather than the chronic pain seen in most neoplasms [1]. Orbital inflammatory pseudotumor can develop at any age but more commonly affects older adults [7]. Pediatric cases comprise 6–16% of all orbital inflammatory pseudotumor cases [29]. Clinical symptoms in orbital inflammatory pseudotumor reflect the degree of inflammatory response and location of inflammation. In acute forms patients may present with sudden onset of painful exophthalmos [29]. In chronic fibrosclerosing forms of inflammatory pseudotumor, patients may exhibit symptoms from fixation of orbital structures or lesion mass effect, such as vision loss and diplopia [29].
On CT, inflammation involving different parts of the orbit may be seen, including dacryoadenitis (enlarged lacrimal gland), episcleritis or perineuritis (diffuse thickening along the ocular wall or optic nerve, respectively), and myositis (enlarged extraocular muscles) [1]. Orbital inflammatory pseudotumor may present as focal or diffuse inflammation, or as a localized mass [12]. Rarely it demonstrates aggressive features such as erosive bony changes or intracranial extension [1]. Tolosa–Hunt syndrome is a variant of orbital inflammatory pseudotumor, which extends through the orbital apex to involve the cavernous sinus. The middle cranial fossa and cavernous sinus are the two most common intracranial sites of involvement by orbital pseudotumor [30].
On MRI, orbital inflammatory pseudotumor may demonstrate low T1 signal compared to muscle; relatively low T2 signal, depending on the degree of fibrosis, with more chronic/fibrotic lesions having lower T2 signal intensity; and usually marked contrast enhancement [29, 30] (Fig. 1). Diffusion-weighted imaging is helpful to differentiate orbital inflammatory pseudotumor from orbital lymphoma, which has significantly lower ADC values [24].

Orbital inflammatory pseudotumor in a 19-year-old woman with a 1-week history of right eye pain, swelling and diplopia. Axial T2-weighted (a) and axial T1-weighted post-gadolinium (b) MR images demonstrate a thickened right lateral rectus muscle with slightly hyperintense T2 signal (arrow) and avid enhancement (arrow) compared to the uninvolved extraocular muscles. The high T2 signal may indicate a more acute inflammation
The main differential diagnoses include infection, sarcoidosis, Wegener granulomatosis, connective tissue disease, lymphoma and metastatic disease, especially from neuroblastoma and rhabdomyosarcoma [29]. In adults, orbital inflammatory pseudotumor is typically unilateral, and bilateral involvement may suggest underlying systemic disease such as IgG4-related disease [31]. However in children, orbital inflammatory pseudotumor can manifest bilaterally without systemic disease in more than half of patients [7].
Mild forms of orbital inflammation can be treated with watchful waiting [12]. Most cases (more than 75%) of orbital inflammatory pseudotumor are treated with steroids and show dramatic, rapid improvement within 2–3 days [6, 12]. About half of cases resolve completely with steroids, but the recurrence rate can still be high [7, 32]. Antibiotics and non-steroidal anti-inflammatory drugs (NSAIDs) are alternative treatments [1]. In cases of refractory orbital inflammatory pseudotumor, cytotoxic chemotherapy and low-dose radiation therapy may be useful [1, 33].
Brain, head and neck sinonasal inflammatory pseudotumor
Inflammatory pseudotumor involving the central nervous system (CNS) is extremely rare. Clinically, people may present with seizure or headache and systemic inflammatory symptoms such as fever. Several etiologies for CNS inflammatory pseudotumor have been implicated, including genetic (ALK 2p23 gene translocation in inflammatory myofibroblastic tumor), post-infectious (isolation of viral DNA [EBV, HHV-8] [16]), and post-traumatic (CNS inflammatory pseudotumor arising from chronic inflammation at the site of ventriculoperitoneal shunts [15]). On CT, inflammatory pseudotumor involving the brain most commonly presents as an enhancing extra-axial meningeal-based mass, but it can also be intraparenchymal, mixed intraparenchymal/meningeal or intraventricular [34, 35]. On MRI, brain inflammatory pseudotumor demonstrates isointense T1 signal intensity, markedly low T2 signal intensity, avid enhancement and moderately restricted diffusion [15, 36]. The main differential diagnoses for brain inflammatory pseudotumor include entities causing mass lesions of the dura: meningiomas, sarcoidosis, tuberculosis and non-Langerhans cell histiocytosis [36]. Meningiomas and sarcoidosis are unusual in children. Treatment of CNS inflammatory pseudotumor is total surgical resection, with removal of the mass or inciting factor such as a ventriculoperitoneal shunt. Recurrence is rare but has been reported [16].
Head and neck inflammatory pseudotumor may involve the temporal bone, skull base, upper cervical spine, neck soft tissues, infratemporal fossa, nasopharynx, parapharyngeal space, larynx, clivus, maxillary sinuses, pterygopalatine fossa, sella and pituitary gland and major salivary glands [1, 3, 37–39]. Patients present with a variety of symptoms based on the region involved. Inflammatory pseudotumor should be suspected when multiple groups of muscles, bones and salivary glands are simultaneously affected (Fig. 2). Ultimately, inflammatory pseudotumor can be difficult to differentiate from malignancy or aggressive infection on imaging alone, and thus biopsy is necessary to confirm the diagnosis [23, 38].

Central nervous system inflammatory pseudotumor involving the pituitary gland, cavernous sinus, pineal gland, orbits and skull base in an 8-year-old boy who presented with growth delay, polyuria, polydipsia and right eye esotropia. Axial T2-weighted (a), T1 post-gadolinium (b), and positron emission tomography (c) images demonstrate a T2-hypointense, avidly enhancing, hypermetabolic sellar/suprasellar mass involving the cavernous sinus (arrow). d Sagittal T1-W post-gadolinium MR image depicts enhancing pituitary and pineal masses (arrows). e Sagittal T1-W post-gadolinium MR image shows an enlarged, enhancing left superior rectus muscle and maxillary sinus lesion (arrows). f Axial bone window CT image demonstrates diffuse skull base bony sclerosis involving the orbits and greater sphenoid wing. g Maxillary sinus wall biopsy was performed, and high-powered photomicrograph (original magnification ×400; hematoxylin–eosin stain) reveals a sclerosing inflammatory process with mixed B- and T-lymphocyte infiltration (per immunohistochemistry) and abundant macrophages (arrow). No Langerhans histiocytes were present
On MRI, the low T2 signal of skull-based inflammatory pseudotumor can help differentiate it from other malignant primary skull-based tumors such as chordoma, chondrosarcoma and nasopharyngeal carcinoma, which are hyperintense on T2-weighted imaging [32]. Recent reports suggest that MRI characteristics may predict therapeutic response, with lesions showing high T2 signal intensity and strong enhancement having a relative abundance of free water and mobile protons, indicating more acute inflammation that will respond well to steroids [23]. Response to high-dose steroid therapy is thought to roughly parallel the acuity of the inflammatory process, with acute lesions responding to a high dose of steroids [23]. Inflammatory pseudotumor with low T2 signal may correlate with more fibrosis and poorer response to steroids, thus necessitating surgical resection [23]. Some authors also suggest that lesions involving the temporal bone or petrous apex with bony destruction and aggressive erosion of inner ear structures are best treated by excision, with radiation and steroid therapy reserved for inoperable tumors [32]. Recently, chemotherapy with ALK-inhibitors such as crizotinib have been used to treat some head and neck ALK-positive inflammatory myofibroblastic tumors with varying degrees of success [39].
Thorax
Lung
Lung inflammatory pseudotumor is the most common primary pediatric lung mass, constituting 50% of benign pulmonary masses [40]. In a large single institutional review over 90 years, Yu et al. [41] reported that carcinoid, inflammatory pseudotumor, and pleuropulmonary blastoma were the three leading primary pediatric lung tumors. In a review of 60 patients with lung inflammatory pseudotumor, the mean age at diagnosis was 28 years, with a range of 17 months to 61 years [42]. Common presenting symptoms include cough, fever, shortness of breath, and hemoptysis, and 20% have a history of antecedent infection [42]. Historically, lung inflammatory pseudotumor had various names, including plasma cell granuloma, fibrous histiocytoma and now inflammatory myofibroblastic tumor [1].
Inflammatory pseudotumor of the lung typically appears as a solitary, peripheral, sharply circumscribed and lobulated mass, predominantly in the lower lobes [3, 42] (Fig. 3). Less commonly, it presents as an endobronchial mass with obstructive atelectasis in 10% of cases (Fig. 4) or as multiple lesions in 5% of cases [42]. On CT, they may have heterogeneous attenuation and enhancement. On MRI, they are usually iso- or slightly hyperintense on T1-W images and hyperintense on T2-W images compared to skeletal muscle, which is in contrast to inflammatory pseudotumors in other locations, which are usually hypointense on T2 [42]. Intralesional calcification is seen in 15% of cases and can range from amorphous, mixed or a fine fleck-like pattern to heavy calcification [42] (Fig. 5). Calcification occurs more frequently in children than adults [42]. Cavitation and lymphadenopathy are rare [1]. Lung inflammatory pseudotumor may be FDG-avid on PET/CT [3] (Figs. 4 and 5). In one retrospective review of FDG PET/CT findings of inflammatory myofibroblastic tumors, these lesions showed varied low to high FDG uptake, which may reflect tumor cellularity, biological behavior, composition/proportion of inflammatory cells, and extent of inflammatory cell activity [43]. Thus, the authors suggested that the role of FDG PET/CT may not be to distinguish inflammatory myofibroblastic tumor from other pulmonary parenchymal masses, but rather to help identify the primary tumor and evaluate for local recurrence and distant metastases [43].

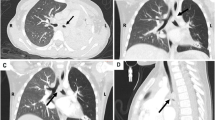
Pulmonary inflammatory pseudotumor in an 8-year-old girl with an isolated right upper lobe nodule, which grew over time. a Axial CT shows a solitary, peripheral, well-circumscribed right upper lobe nodule (arrow). b The nodule (arrow) is hyperintense on T2-weighted MR image. Pathology of right upper lobe wedge resection (not shown) showed inflammatory myofibroblastic tumor

Tracheobronchial inflammatory pseudotumor in a 15-year-old boy who presented with gradually worsening sharp chest pain. a Axial CT post-contrast image demonstrates an oblong lesion (arrow) in the proximal right mainstem bronchus with post-obstructive collapse of the right lower lobe. b The endobronchial lesion (arrow) is hypermetabolic on coronal FDG PET/CT image. c Mainstem bronchus biopsy photomicrograph (original magnification ×400; hematoxylin–eosin stain) shows inflammatory myofibroblastic tumor with proliferating spindle cells (white arrow) and an inflammatory infiltrate consisting predominantly of lymphocytes with plasma cells (black arrows) and numerous macrophages (black arrowhead). FDG [F-18]2-fluoro-2-deoxyglucose, PET positron emission tomography

Pulmonary inflammatory pseudotumor in a 6-year-old girl who presented with a lung mass. Axial CT (a) and corresponding fused axial FDG PET/CT (b) show a heterogeneously enhancing, lobulated right lower lobe lung mass with coarse central calcification (arrows). Right lower lobectomy and mediastinal mass excision revealed inflammatory myofibroblastic tumor. c Histology. Low-powered photomicrograph (original magnification ×200; hematoxylin–eosin stain) shows compact fascicles of myofibroblastic spindle cells (white arrow) with a background inflammatory infiltrate of lymphocytes and plasma cells (black arrow). d Diffusely positive anaplastic lymphoma kinase (ALK) cytoplasmic staining, represented by areas of brown pigmented staining on low-powered photomicrograph (original magnification ×200; ALK stain), indicates ALK protein overexpression caused by gene fusion involving ALK at chromosome 2p23. FDG [F-18]2-fluoro-2-deoxyglucose, PET positron emission tomography
The differential diagnoses vary depending on the imaging presentation. Key differential diagnoses for calcified intraparenchymal mass lesions include hamartoma and calcified granuloma [42]. For solid masses, benign congenital lesions such as sequestration and congenital pulmonary airway malformation, as well as metastases, should be considered. Another unique entity to be included in the pediatric differential diagnosis is pleuropulmonary blastoma, a large pleural-based malignant mass that may contain cysts. Rarely, inflammatory pseudotumor can have aggressive features such as bony destruction and vascular or mediastinal invasion, and in such cases inflammatory pseudotumor may be difficult to differentiate from sarcoma or lymphoma [42]. For endobronchial lesions, key differentials include carcinoid and metastatic disease [42].
Although lung inflammatory pseudotumor is generally recognized as a benign lesion, aggressive behavior with rapid growth, local invasion, and recurrences have been reported [44]. Conversely, there are rare reports of regression, either spontaneously or following steroid treatment [8]. Given these varied outcomes, it is no wonder that various therapeutic approaches have been proposed, from steroids and antibiotics to radical surgery and chemotherapy [44]. Overall, prognosis has been found to be excellent after complete surgical resection, with low incidence of recurrence [44]. In general, initial treatment is complete surgical resection for tumors, with either wedge resection or lobectomy. For non-surgical cases such as those with extensive mediastinal and central vasculature involvement, steroids, chemotherapy and radiotherapy have been used with varying degrees of success [1, 3]. Consideration of inflammatory pseudotumor is important to avoid a premature decision to pursue aggressive treatment.
Cardiac
Cardiac inflammatory pseudotumor is rare and most commonly affects children and young adults. The most common sites of involvement are the right atrium and right ventricle [45]. On MRI, cardiac inflammatory pseudotumor looks similar to other tumors, with slightly high T1 signal and moderately high T2 signal compared to myocardium and with heterogeneous enhancement [45]. The majority of primary pediatric cardiac tumors are benign, with the two most common primary tumors being rhabdomyoma and fibroma, which are associated with tuberous sclerosis and Gorlin syndrome, respectively [46]. Calcifying fibrous pseudotumor involving the heart is a rare manifestation of possibly a late sclerosing stage of inflammatory myofibroblastic tumor (Fig. 6) [47]. Several papers have reported calcifying fibrous pseudotumors and inflammatory myofibroblastic tumors arising in locations near to each other and hypothesized that they may be related entities [47]. They are distinct from fibrous mesothelioma and solitary fibrous tumor, two other rare fibrous tumors of the pericardium [47]. Histologically, calcifying fibrous pseudotumor is characterized by a dense collagenized fibrous mass with psammomatous and dystrophic calcifications and variable lymphoplasmacytic infiltrate. It is negative for ALK but positive for CD34 and smooth muscle actin stains. It is generally benign and local resection is curative, although local recurrences have been reported [47]. In cases where there is aggressive local growth, surgical excision may be impossible, necessitating cardiac transplantation for cure (Fig. 6).

Cardiac calcifying fibrous pseudotumor in a 17-year-old girl. a Coronal T2-weighted MR image shows a left ventricular wall mass originating from the free wall of the left ventricle with extremely low T2 signal intensity. b CT angiography oblique plane image further demonstrates that the left coronary artery and its branches are deeply imbedded in the tumor (black arrow). Prominent tumoral calcification (white arrow) is also seen. The encasement of the left anterior descending artery precluded surgical resection. c, d Histology. Myocardial mass biopsy via thoracotomy with cardioparesis revealed a densely collagenized mass with psammomatous calcifications (arrow in c) and foci of lymphoplasmacellular infiltrates (arrow in d), both on low-powered photomicrographs (original magnification ×100; hematoxylin–eosin stain) (Adapted with permission from [47])
Abdomen
Three quarters of inflammatory pseudotumor cases occur in the abdomen, with frequent sites of involvement including the mesentery, omentum, liver, retroperitoneum, pelvis and abdominal soft tissue [19]. Constitutional symptoms of fever, weight loss, malaise and elevated inflammatory laboratory markers are seen in 19% of patients [20]. Other clinical symptoms include abdominal pain, nausea, vomiting and compressive symptoms depending on the size and site of origin. Imaging appearances vary from an ill-defined infiltrating process to a well-circumscribed soft-tissue mass. The variation most likely corresponds to the differing degrees of inflammatory and fibrotic components in the lesion [22]. In general hepatic, splenic, retroperitoneal and genitourinary lesions are well-circumscribed, while those involving the gastrointestinal tract and biliary tree are ill-defined and infiltrative. Mesenteric lesions may be either infiltrative or mass-like [22].
Liver
Liver inflammatory pseudotumors have been infrequently reported but are now recognized with increasing frequency, especially in Asian countries [10]. They have been reported in pediatric patients ranging from age 3 months to 15 years [9]. Clinically, patients present with abdominal pain, vomiting, fever and signs of chronic inflammation including microcytic anemia and elevated C-reactive protein [10]. Serum alpha-fetoprotein is normal in liver inflammatory pseudotumor, which can help to differentiate it from hepatoblastoma or hepatocellular carcinoma [9]. The etiology of liver inflammatory pseudotumor is unknown, but the inflammatory pattern of pathology and the clinical and laboratory data suggest an inflammatory response to an underlying infection [10]. No definite microorganism has been identified, though EBV has been recovered from some lesions [10, 48].
Liver inflammatory pseudotumor usually presents as a single, well-circumscribed mass (Fig. 7) but can also present as multiple lesions [49]. Hepatic hilar lesions, which comprise one-third of cases, may cause biliary or portal vein obstruction and portal hypertension [9, 50]. On US most of these lesions are iso- or hypoechoic but can have heterogeneous echogenicity [9]. On CT the lesions hypoattenuate relative to surrounding liver and may have calcifications. On contrast CT, inflammatory pseudotumors are non-encapsulated but well-demarcated masses, with most showing peripheral enhancement [3, 51] and others showing delayed intense enhancement of the fibrous component. Cholangiography may show strictures or dilatation of intra- and extra-hepatic bile ducts. On MRI, these lesions are usually T1 hypointense but may have varying T2 appearance from hypointense to hyperintense signal relative to the surrounding liver [3, 49].

Liver inflammatory pseudotumor in a 5-year-old girl presenting with an abdominal mass. a Coronal post-contrast CT image shows a large, heterogeneously enhancing, infiltrative left liver lobe mass with an exophytic component (arrow). The mass obstructs the biliary tree at the hepatic hilum, resulting in intrahepatic biliary dilatation. b Axial T2-weighted, (c) axial T1-weighted and (d) axial T1-weighted post-gadolinium images demonstrate that the mass (arrows) has a mildly hyperintense T2 rim and center but is otherwise isointense to background liver, is hypointense on T1, and avidly enhances. e Needle biopsy and left lateral liver segmental resection revealed inflammatory myofibroblastic tumor with spindle cells (white arrow) and mixed inflammatory infiltrate (black arrow) on low-powered photomicrograph (original magnification ×200; hematoxylin–eosin stain). f The spindle cells are strongly reactive for smooth muscle actin on immunohistochemistry on low-powered photomicrograph (original magnification ×200; smooth muscle actin stain)
When presenting as a focal mass, these tumors can mimic metastasis, abscess or focal nodular hyperplasia. Infiltrative lesions at the porta hepatis can mimic lymphoma, rhabdomyosarcoma or cholangiocarcinoma [50].
Typical treatment is complete surgical resection, with good prognosis [9]. Hilar lesions with biliary obstruction may necessitate biliary drainage. Unresectable lesions may require liver transplantation. However, there have been isolated case reports of complete or near-complete resolution of liver inflammatory pseudotumor with antibiotics, NSAIDs, steroids and even no treatment at all [9, 10]. The natural history of liver inflammatory pseudotumor is unpredictable, varying from rapidly progressive to spontaneous regression without treatment [9]. Thus an initial trial of conservative therapy of hepatic inflammatory pseudotumor is reasonable when histology is confirmed by biopsy. Surgical resection is indicated if the lesion persists or progresses, or if there is local invasion into vital structures [9].
Gastrointestinal
Gastrointestinal inflammatory pseudotumors are uncommon, with occasional cases involving the stomach or ileocecal region, often occurring in young girls [1]. Children present with abdominal pain, upper gastrointestinal hemorrhage, abdominal mass and laboratory findings of iron-deficiency anemia and elevated inflammatory markers [52].
Imaging features include ulceration, wall infiltration, and extramural extension, similar to findings in malignant disease [3]. Inflammatory pseudotumor involving the stomach and duodenum may show diffuse, infiltrative wall thickening with homogeneous low attenuation on CT [50]. Gastric inflammatory pseudotumor may show high metabolic activity on FDG PET and can be helpful in reassessing location of recurrence on follow-up exams [43, 53] (Fig. 8).

Gastric inflammatory pseudotumor in a 7-year-old girl with recurrent fevers and left upper abdominal mass. a Post-contrast coronal CT image demonstrates an infiltrative, enhancing exophytic gastric mass (arrow) extending into the mesentery. The girl underwent laparotomy and resection of the mass involving the greater curvature of the stomach wall and surrounding mesentery. Pathology revealed gastric inflammatory myofibroblastic tumor. b Initial preoperative coronal fused FDG PET/CT image shows hypermetabolic activity in the mass (arrow). c Follow-up fused FDG PET/CT 7 months after initial surgery revealed tumor recurrence in multiple locations, including the surgical bed and throughout the omentum (arrows). d High-powered photomicrograph (original magnification ×400; hematoxylin–eosin stain) shows uniform spindle cell nuclei (white arrow) and an inflammatory background composed of lymphocytes and plasma cells (black arrow). Immunohistochemistry of spindle cells was positive for anaplastic lymphoma kinase, vimentin and smooth muscle actin
Complete surgical resection is the treatment of choice for all pediatric gastric inflammatory pseudotumors, with an overall good prognosis [52, 54]. However there is a risk of recurrence (reported 1 in 16) [52], as was the case with our patient who had recurrence and several new intraperitoneal sites of disease (Fig. 8). The high recurrence rate may be a result of factors precluding complete resection, such as adherence to vital structures or multifocality [55]. Recurrent tumors can be large and locally invasive [54]. Adjuvant chemotherapy may help in these cases, though its use has been rarely reported in the pediatric population [55]. This is likely partly because of the inability of imaging to reliably distinguish inflammatory pseudotumor from malignancy prior to surgery.
Mesentery
Mesenteric inflammatory pseudotumor manifests in one of two forms: well-circumscribed mass or infiltrative ill-defined lesion extending into adjacent bowel loops [22, 56] (Fig. 9). On US imaging mesenteric inflammatory pseudotumor can be either well-defined or infiltrative with mixed echotexture. By Doppler US, internal blood flow is seen. On contrast-enhanced CT, a variety of enhancement patterns can be seen, including delayed enhancement of the fibrotic component. MRI shows low signal intensity on T1- and T2-weighted images and absence of diffusion restriction, though this depends on the cellularity of the lesion [22].

Mesenteric/peritoneal inflammatory pseudotumor in an 8-year-old girl presenting with an abdominal mass, poor appetite and weight loss over 5 months, following an episode of flu-like illness. a–c Contrast-enhanced CT images in coronal (a) and axial (b, c) planes through the lower abdomen (b) and pelvis (c) reveal a large, infiltrative enhancing mesenteric/peritoneal mass (arrows) with surrounding ascites. Biopsy of the mass showed a proliferation of spindled tumor cells in a collagenous background with variable positivity for smooth muscle actin and cytoplasmic anaplastic lymphoma kinase (ALK), consistent with inflammatory myofibroblastic tumor. Peritoneal fluid was also positive for similar ALK-positive spindle tumor cells
The differential diagnosis for mesenteric masses includes benign tumors such as desmoid, gastrointestinal stromal tumor, and sclerosing mesenteritis as well as malignant neoplasms such as lymphoma, metastatic carcinoid, soft-tissue sarcomas, and embryonal rhabdomyosarcoma, especially if the mass is near the bladder [56]. Inflammatory pseudotumors presenting as multifocal omental or mesenteric masses can mimic peritoneal carcinomatosis (Fig. 9) [57].
Diagnosis of mesenteric inflammatory pseudotumor requires laparoscopy or laparotomy for tissue sampling because percutaneous biopsy is often not helpful. Complete surgical resection is the treatment of choice and is curative in most people [56]. Adjuvant therapy with corticosteroids, anti-inflammatory agents and chemotherapy in cases of incomplete resection have been used but without great success. Recurrence rates of inflammatory pseudotumor in the mesentery and retroperitoneum have been reported between 15% and 37%, with recurrence usually found within 1 year [20].
Genitourinary
Genitourinary tract inflammatory pseudotumor is rare and most commonly arises from the urinary bladder. In the past, bladder inflammatory pseudotumor was called low-grade fibrosarcoma [2], reflecting the difficulty in distinguishing inflammatory pseudotumor from a sarcoma with extensive fibrotic proliferation based on clinical, radiologic and histological features. Bladder inflammatory pseudotumor is rare in children and typically occurs in young adults (mean age of 28 years) with male predominance [1]. The most common symptom is asymptomatic gross hematuria from exophytic and ulcerated lesions, sometimes significant enough to cause anemia. Other symptoms are dysuria and urinary frequency. The majority of cases occur de novo, rather than following recurrent infection or surgical instrumentation [58].
On CT, bladder inflammatory pseudotumor may present as a polypoid enhancing intraluminal mass or submucosal mass and can sometimes extend into the perivesical fat [58]. On MRI, they may show intermediate T1 signal and varying T2 signal ranging from hypointense to hyperintense [59, 60].
An important differential diagnosis in children is rhabdomyosarcoma, which is treated by total cystectomy with pelvic lymphadenectomy. When bladder inflammatory pseudotumor is suspected, careful biopsy of the lesion is warranted to exclude rhabdomyosarcoma [61]. By doing so radical surgery can be avoided because bladder inflammatory pseudotumor can be treated with conservative surgery such as partial cystectomy or local transurethral resection, with excellent outcomes [61]. There are no reported cases of recurrence or metastases from pediatric bladder inflammatory pseudotumor [61].
Renal inflammatory pseudotumor is very rare and usually presents clinically with flank pain and hematuria. It may present as a hypoechoic or heterogeneously echogenic mass on US imaging, a well-circumscribed, hypoattenuating, mildly enhancing mass on CT, and a hypovascular mass on MRI [11, 62]. Treatment is usually nephrectomy or partial nephrectomy because of the inability to differentiate it preoperatively from Wilms tumor or renal cell carcinoma. Regression of renal inflammatory pseudotumor with steroids has been reported [11].
Prognosis
Although inflammatory pseudotumors occasionally behave aggressively, in general its nature is relatively benign. Many of the most noticeable effects of inflammatory pseudotumor are from mass effect or fibrosis of involved organs, such as nerve compression or obstruction of bile ducts or bowel. In general, the prognosis is much better than that of the tumors for which they may be mistaken.
Inflammatory myofibroblastic tumor is a neoplasm with intermediate biological potential, with a tendency for local recurrence and a small risk of distant metastasis [2]. The recurrence rate of inflammatory myofibroblastic tumor varies by anatomical site, occurring in only 2% of pulmonary lesions versus 25% of extrapulmonary tumors [20]. Distant metastases are rare, occurring in less than 5% of cases [4]. In a study of 59 cases with atypical histology or clinical aggressiveness over a mean clinical follow-up of 3.6 years, slightly higher percentages of local recurrence and distant metastasis were reported, at 56% and 10%, respectively [20]. The mortality rate from disease was 10% in this study [20]. Recurrences are most common with incomplete resection or multifocal intra-abdominal tumors [2]. The most common sites of metastases are lung, brain, liver and bone, in order of decreasing frequency [2]. Metastatic disease is usually identified at presentation or within a year of diagnosis, but occasionally patients develop metastases as many as 9 years after excision [20]. Some data suggest that ALK-negative inflammatory myofibroblastic tumors have a higher likelihood of metastasis, but there is still no clear-cut relationship between ALK expression and prognosis for inflammatory myofibroblastic tumor. However, the presence of targeted agents such as the ALK-inhibitor crizotinib could impact long-term prognosis of people with ALK-positive inflammatory myofibroblastic tumor [2, 20, 63].
Treatment
Familiarity and knowledge of the imaging features of inflammatory pseudotumors can help in accurate diagnosis and avoidance of unnecessary radical surgery. Because inflammatory pseudotumors have a relatively benign clinical course, if inflammatory pseudotumor is diagnosed via biopsy, a trial of initial conservative treatment with non-surgical or limited surgical intervention may be attempted. Initial treatment using observation alone, steroids, antibiotics, anti-inflammatory agents and other conservative treatments may be attempted in certain cases, because there have been rare case reports of complete resolution with conservative treatment of orbital, pulmonary, hepatic and renal inflammatory pseudotumors [6–12]. The primary treatment for orbital inflammatory pseudotumor is steroids. The radiologic suggestion of the lesion and histological confirmation are important to avoid premature decisions to perform radical surgery or invasive procedures.
If inflammatory pseudotumors involve vital structures such as the CNS, if there is a high likelihood of recurrence as seen in visceral lesions, or if there is poor response to conservative treatment, complete surgical resection is the treatment of choice. Some lesions showing hypointense T2 signal may be more fibrotic and have a poorer response to steroids, necessitating surgical resection [23]. For unresectable tumors, chemotherapy (cyclosporine, methotrexate, azathioprine and cyclophosphamide) or radiation therapy are alternative treatment options [1, 33]. A newer therapeutic option for inflammatory myofibroblastic tumor is crizotinib, an orally available small-molecule inhibitor of ALK tyrosine kinase that has been used to treat ALK-translocated inflammatory myofibroblastic tumors with some success [63, 64].
Summary
Inflammatory pseudotumors are a heterogeneous group of benign, mass-forming lesions characterized by a proliferation of fibroblasts and myofibroblasts accompanied by a prominent chronic inflammatory infiltrate. Within the family of inflammatory pseudotumors, inflammatory myofibroblastic tumors have emerged as a distinct, low-grade neoplastic entity associated with ALK-gene translocation that occurs predominantly in children. Inflammatory pseudotumors may exhibit characteristic hypointense T2 signal on MRI because of their fibrous nature. Ultimately imaging characteristics cannot make a specific diagnosis and biopsy is required. Familiarity and knowledge of the imaging features of inflammatory pseudotumor can help radiologists consider this entity in the differential diagnosis of pediatric soft-tissue masses, thereby avoiding unnecessary radical surgery.
References
Narla LD, Newman B, Spottswood SS et al (2003) Inflammatory pseudotumor. Radiographics 23:719–729
Gleason BC, Hornick JL (2008) Inflammatory myofibroblastic tumours: where are we now? J Clin Pathol 61:428–437
Patnana M, Sevrukov AB, Elsayes KM et al (2012) Inflammatory pseudotumor: the great mimicker. AJR Am J Roentgenol 198:W217–W227
Fletcher CDM, Unni KK, Mertens F et al (2002) Pathology and genetics of tumours of soft tissue and bone. IARC Press, Lyon
Vlachou PA, Khalili K, Jang HJ et al (2011) IgG4-related sclerosing disease: autoimmune pancreatitis and extrapancreatic manifestations. Radiographics 31:1379–1402
Chaudhry IA, Shamsi FA, Arat YO et al (2008) Orbital pseudotumor: distinct diagnostic features and management. Middle East Afr J Ophthalmol 15:17–27
Mottow LS, Jakobiec FA (1978) Idiopathic inflammatory orbital pseudotumor in childhood. I. Clinical characteristics. Arch Ophthalmol 96:1410–1417
Maurya V, Aditya Gupta U, Dewan RK et al (2013) Spontaneous resolution of an inflammatory pseudotumour of the lung subsequent to wedge biopsy. Arch Bronconeumol 49:31–34
Nagarajan S, Jayabose S, McBride W et al (2013) Inflammatory myofibroblastic tumor of the liver in children. J Pediatr Gastroenterol Nutr 57:277–280
Koea JB, Broadhurst GW, Rodgers MS et al (2003) Inflammatory pseudotumor of the liver: demographics, diagnosis, and the case for nonoperative management. J Am Coll Surg 196:226–235
Tarhan F, Gül AE, Karadayi N et al (2004) Inflammatory pseudotumor of the kidney: a case report. Int Urol Nephrol 36:137–140
Swamy BN, McCluskey P, Nemet A et al (2007) Idiopathic orbital inflammatory syndrome: clinical features and treatment outcomes. Br J Ophthalmol 91:1667–1670
Mergan F, Jaubert F, Sauvat F et al (2005) Inflammatory myofibroblastic tumor in children: clinical review with anaplastic lymphoma kinase, Epstein-Barr virus, and human herpesvirus 8 detection analysis. J Pediatr Surg 40:1581–1586
Moritani T, Aihara T, Oguma E et al (2001) Spectrum of Epstein-Barr virus infection in Japanese children: a pictorial essay. Clin Imaging 25:1–8
De Oliveira RS, Amato MC, Brassesco MS et al (2009) Clinical and cytogenetic analysis of an intracranial inflammatory myofibroblastic tumor induced by a ventriculoperitoneal shunt. J Neurosurg Pediatr 4:372–377
Swain RS, Tihan T, Horvai AE et al (2008) Inflammatory myofibroblastic tumor of the central nervous system and its relationship to inflammatory pseudotumor. Hum Pathol 39:410–419
Phan SH (2008) Biology of fibroblasts and myofibroblasts. Proc Am Thorac Soc 5:334–337
Wynn TA, Ramalingam TR (2012) Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 18:1028–1040
Coffin CM, Alaggio R (2012) Fibroblastic and myofibroblastic tumors in children and adolescents. Pediatr Dev Pathol 15:127–180
Coffin CM, Hornick JL, Fletcher CD (2007) Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol 31:509–520
Janik JS, Janik JP, Lovell MA et al (2003) Recurrent inflammatory pseudotumors in children. J Pediatr Surg 38:1491–1495
George V, Tammisetti VS, Surabhi VR et al (2013) Chronic fibrosing conditions in abdominal imaging. Radiographics 33:1053–1080
Han MH, Chi JG, Kim MS et al (1996) Fibrosing inflammatory pseudotumors involving the skull base: MR and CT manifestations with histopathologic comparison. AJNR Am J Neuroradiol 17:515–521
Sepahdari AR, Aakalu VK, Setabutr P et al (2010) Indeterminate orbital masses: restricted diffusion at MR imaging with echo-planar diffusion-weighted imaging predicts malignancy. Radiology 256:554–564
Laffan EE, Ngan BY, Navarro OM (2009) Pediatric soft-tissue tumors and pseudotumors: MR imaging features with pathologic correlation: part 2. Tumors of fibroblastic/myofibroblastic, so-called fibrohistiocytic, muscular, lymphomatous, neurogenic, hair matrix, and uncertain origin. Radiographics 29:e36
Kaçar A, Paker I, Orhan D et al (2012) Childhood fibroblastic and myofibroblastic tumors: a multicenter documentation and review of the literature. Turk Patoloji Derg 28:24–30
Dishop MK, Kuruvilla S (2008) Primary and metastatic lung tumors in the pediatric population: a review and 25-year experience at a large children’s hospital. Arch Pathol Lab Med 132:1079–1103
Mannion M, Cron RQ (2011) Successful treatment of pediatric IgG4 related systemic disease with mycophenolate mofetil: case report and a review of the pediatric autoimmune pancreatitis literature. Pediatr Rheumatol Online J 9:1
Weber AL, Romo LV, Sabates NR (1999) Pseudotumor of the orbit. Clinical, pathologic, and radiologic evaluation. Radiol Clin N Am 37:151–168
Lee EJ, Jung SL, Kim BS et al (2005) MR imaging of orbital inflammatory pseudotumors with extraorbital extension. Korean J Radiol 6:82–88
Fujita A, Sakai O, Chapman MN et al (2012) IgG4-related disease of the head and neck: CT and MR imaging manifestations. Radiographics 32:1945–1958
Strasnick B, Vaughan A (2008) Inflammatory pseudotumor of the temporal bone: a case series. Skull Base 18:49–52
Kovach SJ, Fischer AC, Katzman PJ et al (2006) Inflammatory myofibroblastic tumors. J Surg Oncol 94:385–391
Weber MA, Viehoever A, Stieltjes B et al (2005) Intracerebral manifestation of an atypical monoclonal plasma cell hyperplasia depicted by MR perfusion and diffusion tensor imaging and MR spectroscopy. AJR Am J Roentgenol 185:784–787
Häusler M, Schaade L, Ramaekers VT et al (2003) Inflammatory pseudotumors of the central nervous system: report of 3 cases and a literature review. Hum Pathol 34:253–262
Whitehead MT, Grimm J, Nelson MD (2012) Case 185: inflammatory myofibroblastic tumor. Radiology 264:912–916
Wong S, Lam WY, Wong WK et al (2007) Hypophysitis presented as inflammatory pseudotumor in immunoglobulin G4-related systemic disease. Hum Pathol 38:1720–1723
Lee JH, Kim K, Chung SW et al (2001) A case report of inflammatory pseudotumor involving the clivus: CT and MR findings. Korean J Radiol 2:231–234
Devaney KO, Lafeir DJ, Triantafyllou A et al (2012) Inflammatory myofibroblastic tumors of the head and neck: evaluation of clinicopathologic and prognostic features. Eur Arch Otorhinolaryngol 269:2461–2465
Cohen MC, Kaschula RO (1992) Primary pulmonary tumors in childhood: a review of 31 years’ experience and the literature. Pediatr Pulmonol 14:222–232
Yu DC, Grabowski MJ, Kozakewich HP et al (2010) Primary lung tumors in children and adolescents: a 90-year experience. J Pediatr Surg 45:1090–1095
Agrons GA, Rosado-de-Christenson ML, Kirejczyk WM et al (1998) Pulmonary inflammatory pseudotumor: radiologic features. Radiology 206:511–518
Dong A, Wang Y, Dong H et al (2014) Inflammatory myofibroblastic tumor: FDG PET/CT findings with pathologic correlation. Clin Nucl Med 39:113–121
Messineo A, Mognato G, D’Amore ES et al (1998) Inflammatory pseudotumors of the lung in children: conservative or aggressive approach? Med Pediatr Oncol 31:100–104
Das Narla L, Siddiqi NH, Hingsbergen EA (2001) Inflammatory pseudotumor of the right atrium. Pediatr Radiol 31:351–353
Becker AE (2000) Primary heart tumors in the pediatric age group: a review of salient pathologic features relevant for clinicians. Pediatr Cardiol 21:317–323
Kirby PA, Sato Y, Tannous R et al (2006) Calcifying fibrous pseudotumor of the myocardium. Pediatr Dev Pathol 9:384–387
Arber DA, Kamel OW, van de Rijn M et al (1995) Frequent presence of the Epstein-Barr virus in inflammatory pseudotumor. Hum Pathol 26:1093–1098
Sakai M, Ikeda H, Suzuki N et al (2001) Inflammatory pseudotumor of the liver: case report and review of the literature. J Pediatr Surg 36:663–666
Kim SJ, Kim WS, Cheon JE et al (2009) Inflammatory myofibroblastic tumors of the abdomen as mimickers of malignancy: imaging features in nine children. AJR Am J Roentgenol 193:1419–1424
Fukuya T, Honda H, Matsumata T et al (1994) Diagnosis of inflammatory pseudotumor of the liver: value of CT. AJR Am J Roentgenol 163:1087–1091
Estêvão-Costa J, Correia-Pinto J, Rodrigues FC et al (1998) Gastric inflammatory myofibroblastic proliferation in children. Pediatr Surg Int 13:95–99
Murphy JJ, Tawfeeq M, Chang B et al (2008) Early experience with PET/CT scan in the evaluation of pediatric abdominal neoplasms. J Pediatr Surg 43:2186–2192
Sanders BM, West KW, Gingalewski C et al (2001) Inflammatory pseudotumor of the alimentary tract: clinical and surgical experience. J Pediatr Surg 36:169–173
Dishop MK, Warner BW, Dehner LP et al (2003) Successful treatment of inflammatory myofibroblastic tumor with malignant transformation by surgical resection and chemotherapy. J Pediatr Hematol Oncol 25:153–158
Levy AD, Rimola J, Mehrotra AK et al (2006) From the archives of the AFIP: benign fibrous tumors and tumorlike lesions of the mesentery: radiologic-pathologic correlation. Radiographics 26:245–264
Singhal M, Ramanathan S, Das A et al (2011) Omental inflammatory myofibroblastic tumour mimicking peritoneal carcinomatosis. Cancer Imaging 11:19–22
Miki SC, Kwatra A, Kawashima A et al (1997) Pseudosarcomatous fibromyxoid tumor of the bladder: biphasic contrast-enhanced helical CT findings. J Comput Assist Tomogr 21:271–273
Fujiwara T, Sugimura K, Imaoka I et al (1999) Inflammatory pseudotumor of the bladder: MR findings. J Comput Assist Tomogr 23:558–561
Sugita R, Saito M, Miura M et al (1999) Inflammatory pseudotumour of the bladder: CT and MRI findings. Br J Radiol 72:809–811
Houben CH, Chan A, Lee KH et al (2007) Inflammatory myofibroblastic tumor of the bladder in children: what can be expected? Pediatr Surg Int 23:815–819
Boo YJ, Kim J, Kim JH et al (2006) Inflammatory myofibroblastic tumor of the kidney in a child: report of a case. Surg Today 36:710–713
Mossé YP, Lim MS, Voss SD et al (2013) Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children’s Oncology Group phase 1 consortium study. Lancet Oncol 14:472–480
Butrynski JE, D’Adamo DR, Hornick JL et al (2010) Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med 363:1727–1733
Acknowledgments
This report was presented as an educational exhibit at the Radiological Society of North America 2013 meeting.
Conflicts of interest
Dr. M.B. McCarville received financial support from GE Healthcare.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Lai, L.M., McCarville, M.B., Kirby, P. et al. Shedding light on inflammatory pseudotumor in children: spotlight on inflammatory myofibroblastic tumor. Pediatr Radiol 45, 1738–1752 (2015). https://doi.org/10.1007/s00247-015-3360-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00247-015-3360-6