
Abstract
Spondyloepimetaphyseal dysplasia with joint laxity, leptodactylic type (SEMDJL2), is a rare disorder due to a KIF22 gene mutation and characterized by postnatal short stature, midface hypoplasia and generalized ligamentous laxity. Radiologic hallmark includes severe involvement of the epiphyses and the slender appearance of the metacarpals and phalanges. The aim of the study was to evaluate radiologic findings of SEMDJL2 in a child followed from age 2 years 9 months to 11 years. Using whole-exome sequencing, we identified a single nucleotide de novo p.Pro148Leu mutation in the KIF22 gene. The child had midface hypoplasia, short stature, hip dislocation and generalized laxity of the joints in the first examination. Knee subluxation and bilateral severe genu valgum became prominent after 3.5 years of age. Short stature became evident gradually with increasing age, and height was 3.6 standard deviations below the mean for age. Small epiphyses with delayed maturation and metaphyseal vertical striations at the distal metaphysis of the femur were observed on initial radiographs. However, the slender metacarpals and proximal phalanges and progressive epiphyseal dysplasia with small and flattened epiphyses on both wrists and knees became more prominent after 7 years of age. In conclusion, we observed that typical radiologic findings became apparent after early childhood.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Joint laxity can be a cardinal feature in several skeletal dysplasia. Well-known examples include Larsen syndrome (MIM:150250), Desbuquois dysplasia (MIM:251450) and spondyloepimetaphyseal dysplasia with joint laxity (SEMDJL) type 1, or Beighton type (MIM:271640). SEMDJL type 2, or SEMDJL leptodactylic or Hall type, is an autosomal-dominant skeletal dysplasia that clinically manifests with postnatal short stature, midface hypoplasia and generalized joint laxity [1]. The skeletal hallmarks include mild spondylar dysplasia, progressive epiphyseal dysplasia with joint malalignment, metaphyseal striations, and slenderness of the short tubular bones. Several affected individuals were initially reported as examples of sponastrime dysplasia or its variant [2]. Later, however, Hall et al. [3] brought attention to an unusual constellation of clinical and radiologic manifestations of the disorder, and they clearly distinguished it from sponastrime dysplasia and SEMDJL1. It is known that a heterozygous mutation of KIF22 is responsible for SEMDJL2 [4, 5]. Herein we present the evolution of radiographic findings over time in a girl with lepto-SEMDJL who had a heterozygous missense mutation in the KIF22 gene.
Case report
Clinical findings
Our patient is an 11-year-old girl who was the first child of nonconsanguineous healthy parents. Her length at birth was normal. Hip dislocation on the left side was noticed at a routine neonatal examination. She had head control at 2 months, and she was able to sit and walk without support at 12 months and at 2 years 9 months, respectively. The girl’s history revealed that she was a hypotonic baby in infancy and she had recurrent respiratory tract infections and laryngomalacia until 2 years of age. Her length was 75 cm (−1.9 SD) at 18 months of age. At physical examination at the age of 2 years 9 months, her length was 83 cm (−2.7 SD). She had a high and broad forehead, bilateral epicanthal folds, flat mid-face, small nose, short neck, limitation of the elbow movements and the laxity of other joints. At 4.5 years of age, her height was 90 cm (−3.7 SD). She had difficulty walking, and a severe genu valgum deformity was detected in both knees, said to have developed especially in the right knee after 3.5 years of age. When she was at 6 years of age, corrective surgery for genu valgum deformity was performed three times, but genu valgum deformity occurred again. She was followed up until 11 years of age, at which point menarche occurred. She had developed severe scoliosis and her height was 117 cm (−3.5 SD).
Radiographic features
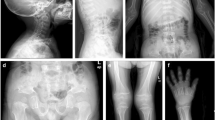
Marked delay of epiphyseal ossification and very small carpal bones were observed in the initial hand radiograph at 2 years 9 months of age. At 7 years of age, the girl’s hand radiographs revealed small and irregular carpal epiphyses. At 9 years of age, progressive epiphyseal dysplasia with small and flattened carpal epiphyses, gracile metacarpals and slender middle and proximal phalanges became prominent. At 11 years of age, prominent narrowing of epiphyseal plates and shortening of the distal ulna were detected. In the feet, mild slender metatarsal bones were observed on radiographs at 2.9 years of age, the gracile and slender metatarsals became prominent at 9 years of age. The upper limb radiographs showed elbow joint subluxation and cubitus valgus (Fig. 1).

Radiographs of the hands (dorsi-palmar) and feet (dorsi-plantar) in a girl with spondyloepimetaphyseal dysplasia with joint laxity, leptodactylic type (SEMDJL2). a Hand radiograph shows delayed maturation of the carpal bones at age 2 years 9 months. Note progressive epiphyseal dysplasia with small, irregular and flattened carpal epiphyses, narrowed epiphyseal plates, gracile metacarpals and slender middle and proximal phalanges and the short distal ulna that were developed at age 7 (b), at 9 (c) and 11 (d) years. Radiography detected mild slender metatarsal bones of the feet at 2 years 9 months (e) and irregular and flattened of tarsal bones with slender metatarsals and proximal phalanges of the foot at 10 years of age (f). g Radiograph of the upper extremity at the age of 9 years shows cubitus valgus and subluxation of the elbow joints
The initial knee radiograph showed small, flat and irregular epiphyses and metaphyseal vertical striations at the distal femoral metaphysis at 2 years 9 months of age. Epiphyseal irregularity and flattening became prominent in knee radiographs with increasing age. Subluxation of both knees became apparent between 4 and 7 years of age. The patellae were not ossified until 9 years of age. Metaphyseal striations and irregularities in the knees (distal femur and proximal tibia) did not change throughout the follow-up period. However, epiphyseal irregularity and flattening and laterally displaced patellae became prominent at 11 years of age (Fig. 2).

Radiographs of the knees (anteroposterior) in the same girl as Fig. 1. a There are small and flat epiphyses and irregular metaphyses and vertical metaphyseal striations at age 2 years 9 months. Note the progressive epiphyseal irregularity and flattening and subluxation of the knee joints that become apparent only over time at age 4.5 (b), 7 (c), 9 (d) and 11 years (e). Lateral displacement of the patellae is visible at age 9 years (d)
Vertebral radiographs demonstrated mild irregularity and flattened vertebral endplate and mild lumbar scoliosis at 4 years 6 months of age. Although there was no prominent change in vertebral bodies, thoracolumbar scoliosis worsened in the radiographic reassessment of the spine at age 11 years (Fig. 3).

Radiograph of the spine (a–b anteroposterior, c–d lateral) in the same girl, who has spondyloepimetaphyseal dysplasia with joint laxity, leptodactylic type (SEMDJL2). At 4 years 6 months of age (a, c), note mildly flattened vertebral bodies and mild scoliosis. The corresponding radiographs at age 11 years (b, d) show worsening of severe scoliosis, but there is no change in vertebral bodies
Initial radiographs of the pelvis showed an irregular acetabular roof, small and flat capital femoral epiphyses as well as long and tapering femoral necks at age 2 years 9 months. Femoral epiphysis was smaller on the left than that on the right because of left hip dislocation. A dislocation of the right hip was also present. Flattening and dysplastic appearance of femoral epiphyses and gradual shortening of the femoral necks were noticed at 11 years of age (Fig. 4).

Anteroposterior radiographs of the pelvis in the same girl as the other figures. a There is an irregular acetabular roof, small and irregular capital femoral epiphyses, elongated femoral necks as well as dislocation on the left femoral head at age 2 years 9 months. b At 11 years of age the flattening and dysplastic appearance of the femoral epiphyses are more prominent, and there is shortening of the femoral necks
Molecular studies
Targeted capture of human whole-exome sequences using solution-based 2.1 M NimbleGen Exome Array (Roche, Basel, Switzerland) followed by Genome Analyzer IIx (Illumina, San Diego, CA) sequencing was performed as previously described [6]. We found a de novo mutation of the recurrent dominant p.Pro148Leu variant that was located at the exon 4 in KIF22 gene (uniprot:Q14807). This variation validated by direct Sanger sequencing in the proband was not present in the parents. The variant has been descibed in 11 patients from 7 families by Boyden et al. [5] and in two patients by Min et al. [4].
Discussion
Lepto-SEMDJL is characterized by a flat face, laryngotracheomalacia, infantile hypotonia, motor developmental delay, short stature, generalized joint laxity, limb deformity and normal intelligence. Craniofacial features comprise midfacial recession, frontal bossing, broad nasal bridge, small nose and anteverted nares [1]. Orthopedic problems include hip, elbow and knee dislocation and progressive genu valgus deformities [1, 7, 8]. It has been reported that short stature becomes evident in early childhood and final height has been reported as −6.9 SD in females, [7, 8].
The girl presented here had similar craniofacial features, laryngomalacia, motor developmental delay, dislocation of the left hip and generalized joint laxity except for the elbow joint. She had marked knee joint laxity and developed severe genu valgus deformity with increasing age. Surgical intervention was performed three times after 3.5 years of age, but genu valgum deformity recurred. Although her length at birth was normal (0 SD), her height was −2.7 SD and −3.6 SD at 2 years 9 months of age and at 11 years of age, respectively.
Similar radiologic findings of SEMDL-leptodactylic type were described by Hall et al. [1]. These findings include small, flat and irregular epiphyses with delayed maturation, short distal ulna, slender metacarpals/metatarsals, metaphyseal irregularities with vertical striations, mild flattened vertebral bodies, joint dislocation, genu valgum and mild thoracolumbar scoliosis. Nishimura et al. [7] found that mild platyspondyly was more prominent in infancy than in childhood and typical metaphyseal alterations gradually became less noticeable in adulthood, whereas a leptodactylic appearance of the hands became more prominent with increasing age. Boyden et al. [5] also reported that slender metacarpal bones and leptodactylic appearance became gradually apparent between 7 and 10 years of age.
We followed the evolution of the radiographic findings in a girl from age 2 years 9 months to 11 years. Although a delay of bone maturation was the only finding in initial hand and knee radiographs, the radiographic hallmarks of the disease such as progressive epiphyseal dysplasia with small and flattened epiphyses in wrists and knees and typical slender metacarpal and metatarsal bones were prominent after the age of 7 years. Vertical metaphyseal striations at the knee joint, which were present at 2.5 years of age, did not progress with age (Figs. 1 and 2).
In a study by Kim et al. [8] all seven patients had a genu valgum or genu valgus deformity of varying severity; three children required recurrent corrective surgery for these deformities. Genu valgus deformity recurred in our patient despite corrective surgery, which was performed three times. Our patient also had late ossification of the patellae and patellar subluxations like the patients reported by Kim et al. [8]. Severe thoracolumbar scoliosis developed late in childhood rather than in younger ages in other reported patients [1, 8]; this feature was seen at 11 years of age in our patient (Fig. 3). In the report by Hall et al. [1], femoral necks were slender and tapered at 3–4 years but they were found to be short in an adult patient. Similarly, in our patient femoral necks were found to be slender and tapered in early childhood and they became shortened with advancing age. Premature osteoarthritis has been reported in this disorder [1, 8]. Our patient had been complaining of hip pain for a year at her last follow-up. However the pathological changes of osteoarthritis were not evident in our patient at that time (Fig. 4).
We describe a de novo case of the recurrent p.Pro148Leu variant in KIF22 gene in a girl. Min et al. [4] also found the recurrent heterozygous missense mutations p.Pro148Leu [c.443C>T] and p.Arg149Gln [c.446G>A]) in six patients previously included in the study by Kim et al. [8] and in two new patients with lepto-SEMDJL. Min et al. [4] also suggested that KIF22 could be involved in ciliogenesis, a process of major importance for skeletal development. KIF22 mRNA expression was detected in bone, cartilage, joint capsule, ligament, skin and primary cultured chondrocytes harvested from human donors. Heterozygous missense mutations in KIF22 gene also have been identified in a total of 32 cases with sporadic and familial leptodactylic SEMDJL [5]. In all cases, the mutations affected two adjacent amino acids located in a region exhibiting a specific protein function. It was concluded that the specific mutations of KIF22 affecting amino acids 148 or 149 are pathogenic for lepto-SEMDJL. When we compared the patient presented here with the patients reported by Kim et al. [8], the clinical and radiologic findings were similar in the patients with the specific mutations of KIF22 affecting both 148 and 149 amino acids. These heterozygous missense mutations lead to lepto-SEMDJL, demonstrating a clear genotype–phenotype correlation.
In conclusion, we report a longitudinal evolution of radiographic findings with advancing age in a girl with a molecular diagnosis of SEMDJL2. Because typical radiologic findings of this disorder are evident only after early childhood, patients could be underdiagnosed or misdiagnosed and treated as having a growth hormone deficiency. Therefore SEMDJL2 should be considered in the presence of short stature with joint laxity and midface hypoplasia in the early childhood period.
References
Hall CM, Elcioglu NH, Shaw DG (1998) A distinct form of spondyloepimetaphyseal dysplasia with multiple dislocations. J Med Genet 35:566–572
Nishimura G, Mikawa M, Fukushima M (1998) Another observation of Langer-type sponastrime dysplasia variant. Am J Med Genet 80:288–290
Hall CM, Elcioglu NH, MacDermot KD et al (2002) Spondyloepimetaphyseal dysplasia with multiple dislocations (Hall type): three further cases and evidence of autosomal dominant inheritance. J Med Genet 39:666–670
Min BJ, Kim N, Chung T et al (2011) Whole-exome sequencing identifies mutations of KIF22 in spondyloepimetaphyseal dysplasia with joint laxity, leptodactylic type. Am J Hum Genet 89:760–766
Boyden ED, Campos-Xavier AB, Kalamajski S et al (2011) Recurrent dominant mutations affecting two adjacent residues in the motor domain of the monomeric kinesin KIF22 result in skeletal dysplasia and joint laxity. Am J Hum Genet 89:767–772
Bilguvar K, Ak O, Louvi A et al (2010) Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature 467:207–210
Nishimura G, Honma T, Shiihara T et al (2003) Spondyloepimetaphyseal dysplasia with joint laxity leptodactylic form: clinical course and phenotypic variations in four patients. Am J Med Genet 117A:147–153
Kim OH, Cho TJ, Song HR (2009) A distinct form of spondyloepimetaphyseal dysplasia with joint laxity (SEMDJL)-leptodactylic type: radiological characteristics in seven new patients. Skelet Radiol 38:803–811
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Tüysüz, B., Yılmaz, S., Erener-Ercan, T. et al. Spondyloepimetaphyseal dysplasia with joint laxity, leptodactylic type: longitudinal observation of radiographic findings in a child heterozygous for a KIF22 mutation. Pediatr Radiol 45, 771–776 (2015). https://doi.org/10.1007/s00247-014-3159-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00247-014-3159-x