
Abstract
Dilated cardiomyopathy (DCM) is a myocardial disease characterized by bilateral or left ventricular cardiac dilation and systolic dysfunction that can lead to heart failure and sudden cardiac death in children. Many studies have focused on genetic variation in DCM-related genes in adult populations; however, the mutational landscape in pediatric DCM patients remains undetermined, especially in the Chinese population. We applied next-generation sequencing (NGS) technology to genetically analyze 46 pediatric DCM patients to reveal genotype–phenotype correlations. Our results indicated DCM-associated pathogenic mutations in 10 genes related to the structure or function of the sarcomere, desmosome, and cytoskeleton. We also identified 6 pathogenic mutations (5 novel) in the Titin (TTN) gene that resulted in truncated TTN variants in 6 (13%) out of 46 patients. Correlations between TTN mutations and clinical outcomes were assessed. Our data indicate that one-third of pediatric DCM cases are caused by genetic mutations. The role of TTN variants should not be underestimated in pediatric DCM and age-dependent pathogenic penetrance of these mutations should be considered for familial DCM cases. We argue that genetic testing of DCM cases is valuable for predicting disease severity, prognosis, and recurrence risk, and for screening first-degree relatives.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Dilated cardiomyopathy (DCM) is the most common myocardial disease, characterized by left or bilateral cardiac dilatation and systolic dysfunction in the absence of any other comorbid condition [1]. It has an estimated prevalence of 1:2500 in the general adult population, but its prevalence among children is less at 1 in 170,000 in the USA and 1 in 140,000 in Australia [2,3,4]. DCM can be inherited and can lead to arrhythmias, heart failure, and sudden cardiac death (SCD) [5]. About 30–50% of DCM cases are of familial origin, and sporadic DCM is more commonly observed in pediatric patients [6]. Male individuals are three times more susceptible to DCM than females [7, 8]. DCM is a genetically heterogeneous disease [9, 10] that produces varied patient phenotypes resulting from the interaction of underlying genetic susceptibility and environmental factors.
More than 60 genes related to structural or functional components of the cytoskeleton, desmosome, nuclear lamina, sarcomere, and mitochondria, or calcium-binding are associated with DCM pathogenesis in a Mendelian autosomal dominant pattern [6, 8, 11, 12]. Mutations in these genes exhibit variable expressivity and penetrance in DCM [13]. Identifying disease-causing genetic variants in probands may help asymptomatic family members to assess their risk of developing cardiomyopathy [14]. Genetic diagnosis techniques, including next-generation sequencing (NGS) technology, have been extensively used correlate genetic mutation with phenotypic presentation in numerous human diseases [15]. Previous studies have primarily focused on the relationship between genetic mutation and clinical phenotype in adult DCM patients. Truncations in Titin (TTN) are the most common cause of DCM, occurring in 25% of familial and 18% of sporadic cases, with these variants are over-represented in the A-band region of the TTN protein [16].
Presently, the genetic landscape of pediatric DCM remains undetermined. Therefore, in this study, we assembled a cohort of 46 pediatric DCM patients. We present their clinical phenotypes and results from NGS analysis, including whole-exome sequencing and targeted gene panel analysis in combination with cardiomyopathy-related gene-filtering to identify underlying pathogenic mutations. We demonstrate association between genetic variation, phenotypic presentation, and clinical outcomes in pediatric DCM patients from Shandong province, China. Furthermore, this study provides a comprehensive landscape of genetic variation in pediatric DCM patients by applying stringent American College of Medical Genetics and Genomics (ACMG) criteria for classification. This can support genetic testing and counseling of patients. Moreover, our investigation highlights a clinical-pathological correlation between truncating mutations in TTN, a crucial component of muscle fibers, and clinical outcomes in pediatric DCM patients of Chinese genetic background.
Materials and Methods
Subjects and Clinical Evaluation
Patients diagnosed with DCM were recruited at the pediatric department of Shandong Provincial Hospital between 2014 and 2020. Patients with specific etiologies, such as congenital heart disease, myocarditis, rheumatic heart disease, systemic hypertension, cardiotoxicity, ischemic heart disease, metabolic and syndromic diseases, were excluded. The study cohort consisted of 46 patients (n = 46; mean age 6.5 months; range: 1–156). All parents of the study patients gave their informed consent for the study according to the Declaration of Helsinki. Echocardiographic measurements were indexed to age and body surface area, and corresponding Z-scores were derived whenever applicable. DCM was diagnosed when the Z-score of the left ventricular end-diastolic (LVEDD) and/or end-systolic (LVESD) volume was above 2 standard deviations (SD) of body surface area (based on Detroit data) [17], with left ventricular ejection fraction (LVEF) < 45% or fractional shortening < 20% in the absence of any comorbidities [18].
Probands ≤ 13 years old and available family members were evaluated according to their medical history, and by physical examination, 12-lead electrocardiography, and transthoracic echocardiography. We recorded clinical parameters, such as age at onset of DCM, sex, family history of DCM or SCD, LVEF, and LVEDD. Positive family history was defined as cardiomyopathy or SCD reported to a clinical geneticist at the time of evaluation. Whenever possible, a positive family history was confirmed by obtaining clinical records. We set the DCM recovery parameter as LVEF ≥ 55%. All patients were followed up in outpatient clinics or by telephone interview until December 31, 2020 and the mean follow-up time was 12.5 months (range: 1–84). The latest echocardiographic data and outcomes were recorded. For the survival analysis, event-free survival was calculated from the date of onset to the date of heart failure-related death.
Genetic Testing and Bioinformatics
Genomic DNA was extracted from peripheral blood samples using a QIAamp Blood Midi Kit (QIAGEN, Germany) according to the manufacturer’s instructions. Two target panels were designed for NGS-based genetic analysis. Twenty-five patients were analyzed by Sinopath genetic technology (Beijing, China), which included a panel of 175 cardiac genes (Panel 1) (Online Resource Table 1) and 16 patients were analyzed by Novocardio genetic technology Co. Ltd (Beijing, China), which included a panel of 101 cardiomyopathy-related genes (panel 2) (Online Resource Table 2). The genomic DNA of the remaining five patients was analyzed by whole-exome sequencing (trio-WES) on the Illumina platform.
After filtering out low-quality reads, Burrows–Wheeler Aligner (BWA-MEM v0.7.12) was used to align the clean reads to the reference genome (UCSC Genome Browser hg19) for sorting and duplicate marking. Insertion and/or deletion (InDel) sequence determination and base quality score calibration were carried out by local realignment using Genome Analysis Toolkit software (GATK v3.2) [19]. Single-nucleotide polymorphisms and InDel calling were performed by GATK’s Haplotype caller [20]. All determined variants were annotated by ANNOVAR and searched for in multiple databases, including 1000 genome, ESP6500, dbSNP, EXAC, and HGMD (Human Gene Mutation Database). Variant effects were predicted using SIFT, PolyPhen-2(PP2), MutationTaster (MT), and GERP++.
The pathogenicity of all variants was assessed in accordance with ACMG guidelines [21]. Sanger sequencing was then performed to confirm the presence of pathogenic or likely pathogenic variants and their parental origins.
Statistical Analysis
Statistical analysis was performed with SPSS (version 26.0) software. All data are expressed as the mean ± SD or, for non-parametric values, as the median and lower and upper quartiles. The difference in continuous variables was assessed by Student’s t-test and the Mann–Whitney U test was used when the distributions were asymmetrical. Characteristics of different groups, such as patients with or without disease-causing mutations, were compared using the chi-square test for categorical variables if appropriate; otherwise, Fisher’s exact test was used. The Kaplan–Meier method was used to calculate survival, and the log-rank test was used to compare survival curves between different patient groups. All statistical tests were two-sided, and P values < 0.05 were statistically significant.
Results
Clinical Characteristics
The clinical characteristics of 46 pediatric patients with DCM onset (26 female and 20 male) are presented in Tables 1 and 2. Almost all patients presented with a common respiratory syndrome-like shortness of breath and cough. Notably, heart rhythm disturbances [atrial tachycardia (1 patient), premature ventricular contractions with low voltage (2 patients), transient junctional rhythm (1 patient), premature ventricular contractions (7 patients), left anterior fascicular blocks (2 patients), left bundle branch block (1 patient), and atrial premature beats (3 patients)] were observed in 17 patients. It is important to note that 67.4% of patients (31/46) manifested the disease before 1 year of age with a median age at diagnosis of 6.5 months, and 84.8% (39/46) of patients were diagnosed before the child’s third birthday. At presentation, the mean LVEF was 28.5%, and the mean LVEDD Z-score was 7.02. The mean serum N-terminal pro-brain natriuretic peptide (NT-proBNP) level was 14,406 pg/ml, which is much higher than the reference range (< 125 pg/ml) (Table 3). At the median follow-up of 12.5 months, 54.3% of patients (25/46) had recovered from DCM and their LVEF was within the normal range. Ten out of 46 patients died because of severe DCM and the average time of death was 1–36 months (median 7 months) after the initial diagnosis. Importantly, 90% of deaths occurred within the first year after diagnosis, a critical time period reflected by the significant morbidity and mortality. Genetic analysis of DCM showed high heterogeneity in these patients with only three patients having a familial history of DCM. No significant differences were observed between the sexes for LVEF and LVEDD Z-scores, age of onset, serum NT-proBNP level at diagnosis, follow-up time, or death, or recovery outcome (all P > 0.05) (Table 2).
Genetic Characteristics of the Genotype-Positive Group
Based on the sequencing results, the cohort was divided into two groups: a genotype-positive group (16/46) and a genotype-negative group (30/46), as shown in Table 3. Considering the stringent selection criteria and reclassification of DCM according to the ACMG guidelines, the genetic analysis indicated that the 16 genotype-positive patients could be classified as either ‘pathogenic’ or ‘likely pathogenic’ mutant carriers. All carriers had only one pathogenic or likely pathogenic mutation in DCM-associated genes (Fig. 1). We identified 10 genes with disease-causing heterozygous mutations. These were genes with integral sarcomere functions: Titin (TTN) [(OMIM*604145), (n = 6, 37.5%)], Myosin heavy chain 7 (MYH7) [(OMIM*613426) (n = 1, 6.25%)], Troponin T2 (TNNT2) [(OMIM *601494) (n = 1, 6.25%)], Nexilin (NEXN) [(OMIM*613122) (n = 1, 6.25%)], Troponin I3 (TNNI3) [(OMIM*613286) (n = 1, 6.25%)]; cytoskeletal structure-related genes: Filamin-C (FLNC) [(OMIM*617047) (n = 1, 6.25%)], Vinculin (VCL) [(OMIM*611407) (n = 1, 6.25%)]; and other genes: RNA-binding motif Protein 20 (RBM20) [(OMIM*613172) (n = 2, 12.5%)], NK2 homeobox 5 (NKX2-5) [(OMIM:108900) (n = 1, 6.25%)], and PR domain containing 16 (PRDM16) [(OMIM*615373) (n = 1, 6.25%)]. Table 4 summarizes the main clinical features and the details of identified variants in DCM patients. Three patients had familial mutations in TTN, MYH7, and NEXN genes, and five de novo variants were identified in NKX2-5, TNNI3, PRDM16, and RBM20 (n = 2). Among the 16 mutations identified (5 missense, 5 nonsense, 4 frameshift, and 2 splice site), 2 were identified in a pair of monozygotic twin patients, and 10 (62.5%) were novel.

Distribution of pathogenic or likely pathogenic variants in the DCM cohort. Distribution of pediatric DCM patients based on mutations in genes related to either sarcomere, cytoskeletal structure, or other cellular functions. Thirty 30 out of 46 patients had no underlying genetic mutation related to DCM pathogenesis. The majority of mutation-positive patients exhibited pathogenic or likely pathogenic mutations in sarcomeric genes
In total, we identified six (five novel) TTN truncation variants, including three nonsense, one frameshift, and two splice-site variants, in 13% of patients (Table 4). Furthermore, we mapped the identified variants to protein domains of TTN (Online Resource Fig. 1a). Consistent with previous reports, this showed that four variants were in the I-band region, and the remaining two were in the A-band and M-band regions, respectively.
Genotype and Clinical Phenotype Analyses
The severity of patient phenotype at presentation was assessed by LVEDD Z-score, LVEF, and serum NT-proBNP levels. There were no significant differences in distribution based on sex (P = 0.202), age (P = 0.23), LVEF (P = 0.935), LVEDD Z-score (P = 0.42), or serum NT-proBNP levels (P = 0.115) between genotype-positive and genotype-negative groups. More cardiac arrhythmia was observed in genotype-positive patients (P = 0.01). The genotype-positive group probands exhibited lower phenotypic severity (echocardiographic parameters) than genotype-negative group probands, with the same LVEF of 28.5% and LVEDD Z-score of 6.4 versus 7.08, respectively. We also observed a trend that the onset age (median 9.5 months) in the genotype-positive group was slightly higher than that in the genotype-negative group (median 6 months). Nine genotype-positive patients developed DCM before 12 months of age, and the age distribution was between 1 month and 13 years.
Despite better echocardiographic parameters and seemingly better phenotypes at presentation in genotype-positive patients, there was no significant difference in outcome (the number of deceased patients, P = 0.145 and LVEF recovery patients, P = 0.665) between the two groups during the 12.5 months (median) follow-up time (P = 0.972). Heart failure-related death occurred in 37.5% (6/16) of genotype-positive patients compared with only 13% (4/30) of genotype-negative patients. No significant differences in survival (P = 0.093) were documented. Cardiac-related death occurred in patients with truncating mutations in TTN (c.50065C>T, c.98421_98422insGG, c.37454–2A>T), NKX2-5 (c.242delA), TNNT2 (c.422G>A), and TNNI3 (c.544G>A). Notably, five out of six disease-causing mutations belong to sarcomeric genes (TT n = 3, MYH7, TNNT2, and TNNI3).
A significant proportion of the patients exhibited marked improvement and better prognostic outcomes in response to heart failure treatment. In both groups, almost 50% of pediatric patients recovered (LVEF above 50%).
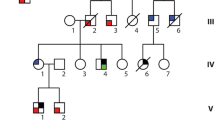
The clinical and genetic characteristics of four probands with four truncating TTN mutations and of twin brothers with RBM20 mutations are summarized in Table 4 and in Online Resource Figs. 1–3. We identified the functional domains of the TTN protein and showed that the DCM-linked pathogenic or likely pathogenic mutations reside mostly in the I-band domain; other mutations were scattered in the A-band and M-band. The TTN protein sequence is highly conserved among different vertebrates and the residues corresponding to mutation sites are shown in the various TTN proteins in Online Resource Fig. 1. The pedigree tree and Sanger sequencing which confirmed the variants are shown in Online Resource Fig. 2.
Discussion
DCM is a common pediatric heart disease that can lead to poor clinical outcomes and heart transplantation [22, 23]. In this cohort, with a mean LVEF of 28.5% and a mean LVEDD Z-score of 7.02, DCM presented as a severe disease and 67.4% of patients (31/46) manifested DCM before 1 year of age. DCM affects men more commonly than women. In a large heart disease cohort, male sex was an independent predictor of mortality, and women with heart failure had better transplant‐free survival compared with men [24, 25]. Another study also showed better prognosis for women with DCM [25]. However, in the present study, no significant sex difference was observed for LVEF, LVEDD Z-scores, age at onset, serum NT-proBNP level, either at diagnosis or follow-up, or for the outcome of death, or recovery (all P > 0.05). This may be because our patient sample size was relatively small, although ethnic differences should also be considered.
The prognosis of pediatric DCM is usually bad with a high mortality rate [5]. In this cohort, although 21.7% (10/46) of patients died after medical therapy without heart transplantation, half of the patients recovered reflecting major advances in medical technology. A limitation of our and similar studies is that patients who agree to participate may not completely reflect the recovery ratio in the general population. However, this positive prognosis is worthy of further study and we believe that a well-balanced, large population-based study is warranted.
Genetic factors play an important role in DCM pathogenesis. However, genetic and prognostic understanding are still a challenge for DCM therapy. Hence, we sought to reveal genetic variations that are prevalent in pediatric patients with DCM. Among our 46 pediatric patients with DCM, 16 (34.8%) carried at least one pathogenic or likely pathogenic mutation in a disease-causing gene. Consistent with previous studies, mutations were most frequently detected in sarcomere genes in all 46 DCM patients [26]. The prevalence of pathogenic mutations seemed to be similar to those in recently published cohorts [16, 27]. A recent multicenter study in North America found 35% of familial DCM and 9% of idiopathic DCM patients carried pathogenic or likely pathogenic mutants; however, only two truncating TTN mutations were identified [28]. These high mutation rates cannot be ignored and further confirmed the value of genetic testing in DCM. There was no difference in clinical presentation or prognosis between genotype-positive and genotype-negative groups, except for arrhythmia.
Mutations are frequently identified in child-onset DCM, especially in sarcomeric genes. Importantly, we found TTN-related mutations in 10 out of 16 genotype-positive patients. The truncated TTN carrier rate was 0.6–1.2% in the USA, and the odds ratio for DCM in patients of European ancestry was 10.8–18.7% [29]. The truncated TTN carrier rate in the general Chinese population is unknown. Truncating TTN mutations account for 12–27% of all adult DCM cases, indicating the importance of diagnostic sequencing [30,31,32,33,34]. However, truncating TTN mutations have been rarely identified in pediatric patients [35,36,37] and recent studies in pediatric DCM have shown similar results [14]. Notably, genetic analysis of a 66-patient cohort of severe childhood cardiomyopathy, including 37 DCM cases, did not identify any truncating TTN mutations [32]. Likewise, another study involving 30 Chinese pediatric patients with sporadic DCM pathology did not find any pathogenic truncating TTN mutations [33]. However, in another study only one pathogenic truncating TTN variant was identified in a 16-year-old boy among 36 pediatric DCM patients [34]. A study of 70 pediatric probands, including 56 DCM patients who underwent genetic evaluation, showed that 16 carried pathogenic mutations but only three TTN mutations were identified [36]. Our results are clearly different from these findings; we identified six different truncating TTN variants in 6 (13%) of 46 pediatric patients.
Arrhythmias were more common in the genotype-positive group (10/16) (P = 0.01). Consistently, many studies have identified early and life-threatening arrhythmias in DCM associated with gene mutations, especially truncating TTN mutations and LMNA mutations, although the mechanism for this association remains incompletely understood [38, 39]. The types of arrhythmia also vary, including atrial or ventricular arrhythmia. Endomyocardial interstitial fibrosis may be a factor of arrhythmia in TTN-DCM [39] and the correlation between arrhythmia and DCM genotype should be investigated further. The prognosis of DCM patients with truncating TTN mutations is different and inconsistent in adults with moderate to severe outcomes. Notably, DCM patients with TTN mutations can have a good response to treatment [35]. A cohort of 70 patients with end-stage DCM showed recovery after left ventricle assist device implantation [36]. Another study showed that TTN mutation-positive patients frequently present severe cardiomyopathy and a worse 5-year prognosis [4].
We also observed different clinical outcomes in TTN genotype-positive patients. Three patients with TTN mutations (c.18230–1G>A, c.43298T>G, and c.105541A>T) recovered after medical treatment with no further symptoms, while three other patients harboring TTN mutations (c.50065C>T, c.98421_98422insGG, and c.37454-2A>T) showed no response to treatment and died from heart failure, indicating clinical heterogeneity. These conflicting prognoses indicate that apart from the identified genetic factors, post-transcriptional, environmental, hormonal, and other factors may also modify the rate of disease progression. Therefore, it might be difficult to predict clinical outcomes in pediatric DCM cases based on truncating TTN mutations alone.
The penetrance of truncating TTN mutations can reach up to 100% by the age of 70 years [40], which leads to discordant segregation with phenotype, and the same variant has also been detected in unaffected relatives. In some pedigrees, especially for familial DCM, clinical follow-up with aging should be performed for unaffected relatives. The age at onset of girl P13 (3 months) with a truncating TTN mutation (c.105541A>T, p.K35181X) was obviously different from that of her mother (23 years) and her grandfather (36 years), indicating the possible involvement of other factors.
We also identified a de novo pathogenic RBM20 variant (p.E916K) in twin patients (P6 and P7) with pediatric DCM. RBM20 mutations have been associated with cardiomyopathy [41]. Notably, the RBM20 variant c.2746G>A (p.E916K) identified in this study was located in exon 11, a known hot spot for cardiomyopathy-associated mutations (Online Resource Fig. 2). However, the mechanistic relationship of RBM20 mutations with DCM onset is still unclear.
A de novo mutation in PRDM16 in an 8-month-old girl (P9) was associated with pediatric DCM. Previously identified mutations in this gene are mostly missense mutations; however, a nonsense mutation leading to functional loss of PRDM16 was detected in this case. The role of PRDM16 mutations could be very important, warranting further studies to investigate their pathogenicity.
This study suffers from the following limitations: (1) it was a single-center, retrospective, and a small cohort study; (2) follow-up time was not long enough to estimate long-term outcomes; (3) clinical assessment was highly recommended for family members of the probands, but only a minority of the relatives were willing to participate in this evaluation; therefore, gene mutation carriers in a family could be underestimated in our cohort; (4) bioinformatic prediction can give useful information about the pathogenicity of mutants associated with DCM; however, it cannot reveal the real pathobiology of mutations in cardiac myocytes; (5) this study lacked analysis of endomyocardial biopsies.
In conclusion, DCM is a genetically heterogeneous disease in children and adults. Using genetic testing (NGS analysis), we detected that more than one-third of DCM cases were caused by mutations in genes related to the structure or function of the sarcomere, desmosome, or cytoskeleton. We also assessed genotype–phenotype correlations in pediatric DCM patients. We discovered six truncating TTN mutations (five of which were novel) that correlated with severe disease phenotypes. Furthermore, we identified 16 mutations in 10 genes in 16 patients that were likely to be associated with DCM pathogenesis. Most pediatric patients were diagnosed with DCM before 1 year of age. Also, most deaths occurred within the first year of life after diagnosis. Death occurred in patients harboring mutations in TTN (3 patients), NKX2-5 (1 patient), and TNNT2 (1 patient). Hence, this study advances the genetic understanding of pediatric DCM and highlights certain mutations with severe clinical courses. Further studies are needed to define the mechanisms by which pathogenic TTN variants affect outcomes in pediatric and adult patients with DCM. DCM may have a molecular cause that can be identified through genetic testing.
Data Availability
The data and materials are available upon request.
Code Availability
N/A.
Abbreviations
- ACMG:
-
American College of Medical Genetics and Genomics
- ALT:
-
Alanine transaminase
- AST:
-
Aspartate transaminase
- CT:
-
Computed tomography
- DCM:
-
Dilated cardiomyopathy
- FLNC :
-
Filamin C
- HGMD:
-
Human Gene Mutation Database
- LVEDD:
-
Left ventricular end-diastolic diameter
- LVEF:
-
Left ventricular ejection fraction
- LVNC:
-
Left ventricular noncompaction
- MYH7 :
-
β-Myosin heavy chain
- NEXN :
-
Nexilin F-actin binding protein
- NKX2-5 :
-
NK2 homeobox 5
- NGS:
-
Next generation sequencing
- NT-proBNP:
-
N-terminal pro-brain-natriuretic peptide
- PRDM16 :
-
Positive regulatory domain16
- RBM20 :
-
RNA-binding protein20
- TNNI3 :
-
Troponin I3
- TNNT2 :
-
Troponin T2
- TTN :
-
Titin
- VCL :
-
Vinculin
- WES:
-
Whole-exome sequencing
References
Taylor MR, Carniel E, Mestroni L (2006) Cardiomyopathy, familial dilated. Orphanet J Rare Dis 1:27. https://doi.org/10.1186/1750-1172-1-27
Puggia I, Merlo M, Barbati G, Rowland TJ, Stolfo D, Gigli M, Ramani F, Di Lenarda A, Mestroni L, Sinagra G (2016) Natural history of dilated cardiomyopathy in children. J Am Heart Assoc 5(7):12. https://doi.org/10.1161/jaha.116.003450
Nugent AW, Daubeney PE, Chondros P, Carlin JB, Cheung M, Wilkinson LC, Davis AM, Kahler SG, Chow CW, Wilkinson JL, Weintraub RG (2003) The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med 348(17):1639–1646. https://doi.org/10.1056/NEJMoa021737
Wilkinson JD, Sleeper LA, Alvarez JA, Bublik N, Lipshultz SE (2008) The pediatric cardiomyopathy registry: 1995–2007. Prog Pediatr Cardiol 25(1):31–36. https://doi.org/10.1016/j.ppedcard.2007.11.006
Dellefave L, McNally EM (2010) The genetics of dilated cardiomyopathy. Curr Opin Cardiol 25(3):198–204. https://doi.org/10.1097/HCO.0b013e328337ba52
Akinrinade O, Ollila L, Vattulainen S, Tallila J, Gentile M, Salmenperä P, Koillinen H, Kaartinen M, Nieminen MS, Myllykangas S, Alastalo TP, Koskenvuo JW, Heliö T (2015) Genetics and genotype-phenotype correlations in Finnish patients with dilated cardiomyopathy. Eur Heart J 36(34):2327–2337. https://doi.org/10.1093/eurheartj/ehv253
Merlo M, Pivetta A, Pinamonti B, Stolfo D, Zecchin M, Barbati G, Di Lenarda A, Sinagra G (2014) Long-term prognostic impact of therapeutic strategies in patients with idiopathic dilated cardiomyopathy: changing mortality over the last 30 years. Eur J Heart Fail 16(3):317–324. https://doi.org/10.1002/ejhf.16
Jefferies JL, Towbin JA (2010) Dilated cardiomyopathy. Lancet (London, England) 375(9716):752–762. https://doi.org/10.1016/s0140-6736(09)62023-7
Long PA, Larsen BT, Evans JM, Olson TM (2015) Exome sequencing identifies pathogenic and modifier mutations in a child with sporadic dilated cardiomyopathy. J Am Heart Assoc 4(12):12. https://doi.org/10.1161/jaha.115.002443
Long PA, Zimmermann MT, Kim M, Evans JM, Xu X, Olson TM (2016) De novo RRAGC mutation activates mTORC1 signaling in syndromic fetal dilated cardiomyopathy. Hum Genet 135(8):909–917. https://doi.org/10.1007/s00439-016-1685-3
Simpson S, Edwards J, Ferguson-Mignan TF, Cobb M, Mongan NP, Rutland CS (2015) Genetics of human and canine dilated cardiomyopathy. Int J Genomics 2015:204823. https://doi.org/10.1155/2015/204823
Pérez-Serra A, Toro R, Sarquella-Brugada G, de Gonzalo-Calvo D, Cesar S, Carro E, Llorente-Cortes V, Iglesias A, Brugada J, Brugada R, Campuzano O (2016) Genetic basis of dilated cardiomyopathy. Int J Cardiol 224:461–472. https://doi.org/10.1016/j.ijcard.2016.09.068
Watkins H, Ashrafian H, Redwood C (2011) Inherited cardiomyopathies. N Engl J Med 364(17):1643–1656. https://doi.org/10.1056/NEJMra0902923
Jensen MK, Havndrup O, Christiansen M, Andersen PS, Diness B, Axelsson A, Skovby F, Køber L, Bundgaard H (2013) Penetrance of hypertrophic cardiomyopathy in children and adolescents: a 12-year follow-up study of clinical screening and predictive genetic testing. Circulation 127(1):48–54. https://doi.org/10.1161/circulationaha.111.090514
Precone V, Del Monaco V, Esposito MV, De Palma FD, Ruocco A, Salvatore F, D’Argenio V (2015) Cracking the code of human diseases using next-generation sequencing: applications. Chall Perspect 2015:161648. https://doi.org/10.1155/2015/161648
Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJ, Cook SA, Mestroni L, Seidman JG, Seidman CE (2012) Truncations of titin causing dilated cardiomyopathy. N Engl J Med 366(7):619–628. https://doi.org/10.1056/NEJMoa1110186
Pettersen MD, Du W, Skeens ME, Humes RA (2008) Regression equations for calculation of z scores of cardiac structures in a large cohort of healthy infants, children, and adolescents: an echocardiographic study. J Am Soc Echocardiogr 21(8):922–934. https://doi.org/10.1016/j.echo.2008.02.006
Daubeney PE, Nugent AW, Chondros P, Carlin JB, Colan SD, Cheung M, Davis AM, Chow CW, Weintraub RG (2006) Clinical features and outcomes of childhood dilated cardiomyopathy: results from a national population-based study. Circulation 114(24):2671–2678. https://doi.org/10.1161/circulationaha.106.635128
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20(9):1297–1303. https://doi.org/10.1101/gr.107524.110
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43(5):491–498. https://doi.org/10.1038/ng.806
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424. https://doi.org/10.1038/gim.2015.30
Stehlik J, Edwards LB, Kucheryavaya AY, Benden C, Christie JD, Dobbels F, Kirk R, Rahmel AO, Hertz MI (2011) The registry of the international society for heart and lung transplantation: twenty-eighth adult heart transplant report-2011. J Heart Lung Transplant 30(10):1078–1094. https://doi.org/10.1016/j.healun.2011.08.003
McNally EM, Golbus JR, Puckelwartz MJ (2013) Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Investig 123(1):19–26. https://doi.org/10.1172/JCI62862
Martinez-Selles M, Doughty RN, Poppe K, Whalley GA, Earle N, Tribouilloy C, McMurray JJ, Swedberg K, Kober L, Berry C, Squire I (2012) Meta-Analysis Global Group In Chronic Heart, Gender and survival in patients with heart failure: interactions with diabetes and aetiology. Results from the MAGGIC individual patient meta-analysis. Eur J Heart Fail 14(5):473–479. https://doi.org/10.1172/jci62862
Halliday BP, Gulati A, Ali A, Newsome S, Lota A, Tayal U, Vassiliou VS, Arzanauskaite M, Izgi C, Krishnathasan K, Singhal A, Chiew K, Gregson J, Frenneaux MP, Cook SA, Pennell DJ, Collins P, Cleland JGF, Prasad SK (2018) Sex- and age-based differences in the natural history and outcome of dilated cardiomyopathy. Eur J Heart Fail 20(10):1392–1400. https://doi.org/10.1002/ejhf.1216
Zhao Y, Feng Y, Zhang YM, Ding XX, Song YZ, Zhang AM, Liu L, Zhang H, Ding JH, Xia XS (2015) Targeted next-generation sequencing of candidate genes reveals novel mutations in patients with dilated cardiomyopathy. Int J Mol Med 36(6):1479–1486. https://doi.org/10.3892/ijmm.2015.2361
Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, Böhm M, Duboc D, Gimeno J, de Groote P, Imazio M, Heymans S, Klingel K, Komajda M, Limongelli G, Linhart A, Mogensen J, Moon J, Pieper PG, Seferovic PM, Schueler S, Zamorano JL, Caforio AL, Charron P (2016) Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J 37(23):1850–1858. https://doi.org/10.1093/eurheartj/ehv727
Ware SM, Wilkinson JD, Tariq M, Schubert JA, Sridhar A, Colan SD, Shi L, Canter CE, Hsu DT, Webber SA, Dodd DA, Everitt MD, Kantor PF, Addonizio LJ, Jefferies JL, Rossano JW, Pahl E, Rusconi P, Chung WK, Lee T, Towbin JA, Lal AK, Bhatnagar S, Aronow B, Dexheimer PJ, Martin LJ, Miller EM, Sleeper LA, Razoky H, Czachor J, Lipshultz SE (2021) Pediatric Cardiomyopathy Registry study, genetic causes of cardiomyopathy in children: first results from the pediatric cardiomyopathy genes study. J Am Heart Assoc 10(9):7731. https://doi.org/10.1161/JAHA.120.017731
Haggerty CM, Damrauer SM, Levin MG, Birtwell D, Carey DJ, Golden AM, Hartzel DN, Hu Y, Judy R, Kelly MA, Kember RL, Lester Kirchner H, Leader JB, Liang L, McDermott-Roe C, Babu A, Morley M, Nealy Z, Person TN, Pulenthiran A, Small A, Smelser DT, Stahl RC, Sturm AC, Williams H, Baras A, Margulies KB, Cappola TP, Dewey FE, Verma A, Zhang X, Correa A, Hall ME, Wilson JG, Ritchie MD, Rader DJ, Murray MF, Fornwalt BK, Arany Z (2019) Genomics-first evaluation of heart disease associated with titin-truncating variants. Circulation 140(1):42–54. https://doi.org/10.1161/CIRCULATIONAHA.119.039573
Fatkin D, Huttner IG (2017) Titin-truncating mutations in dilated cardiomyopathy: the long and short of it. Curr Opin Cardiol 32(3):232–238. https://doi.org/10.1097/hco.0000000000000382
Ellepola CD, Knight LM, Fischbach P, Deshpande SR (2018) Genetic testing in pediatric cardiomyopathy. Pediatr Cardiol 39(3):491–500. https://doi.org/10.1007/s00246-017-1779-2
Vasilescu C, Ojala TH, Brilhante V, Ojanen S, Hinterding HM, Palin E, Alastalo TP, Koskenvuo J, Hiippala A, Jokinen E, Jahnukainen T, Lohi J, Pihkala J, Tyni TA, Carroll CJ, Suomalainen A (2018) Genetic basis of severe childhood-onset cardiomyopathies. J Am Coll Cardiol 72(19):2324–2338. https://doi.org/10.1016/j.jacc.2018.08.2171
Dai G, Pu Z, Cheng X, Yin J, Chen J, Xu T, Zhang H, Li Z, Chen X, Chen J, Qin Y, Yang S (2019) Whole-exome sequencing reveals novel genetic variation for dilated cardiomyopathy in pediatric Chinese patients. Pediatr Cardiol 40(5):950–957. https://doi.org/10.1007/s00246-019-02096-1
Zaklyazminskaya E, Mikhailov V, Bukaeva A, Kotlukova N, Povolotskaya I, Kaimonov V, Dombrovskaya A, Dzemeshkevich S (2019) Low mutation rate in the TTN gene in paediatric patients with dilated cardiomyopathy—a pilot study. Sci Rep 9(1):16409. https://doi.org/10.1038/s41598-019-52911-1
Jansweijer JA, Nieuwhof K, Russo F, Hoorntje ET, Jongbloed JD, Lekanne Deprez RH, Postma AV, Bronk M, van Rijsingen IA, de Haij S, Biagini E, van Haelst PL, van Wijngaarden J, van den Berg MP, Wilde AA, Mannens MM, de Boer RA, van Spaendonck-Zwarts KY, van Tintelen JP, Pinto YM (2017) Truncating titin mutations are associated with a mild and treatable form of dilated cardiomyopathy. Eur J Heart Fail 19(4):512–521. https://doi.org/10.1002/ejhf.673
Felkin LE, Walsh R, Ware JS, Yacoub MH, Birks EJ, Barton PJ, Cook SA (2016) Recovery of cardiac function in cardiomyopathy caused by titin truncation. JAMA Cardiol 1(2):234–235. https://doi.org/10.1001/jamacardio.2016.0208
Anderson JL, Christensen GB, Escobar H, Horne BD, Knight S, Jacobs V, Afshar K, Hebl VB, Muhlestein JB, Knowlton KU, Carlquist JF, Nadauld LD (2020) Discovery of TITIN gene truncating variant mutations and 5-year outcomes in patients with nonischemic dilated cardiomyopathy. Am J Cardiol 137:97–102. https://doi.org/10.1016/j.amjcard.2020.09.026
Tobita T, Nomura S, Fujita T, Morita H, Asano Y, Onoue K, Ito M, Imai Y, Suzuki A, Ko T, Satoh M, Fujita K, Naito AT, Furutani Y, Toko H, Harada M, Amiya E, Hatano M, Takimoto E, Shiga T, Nakanishi T, Sakata Y, Ono M, Saito Y, Takashima S, Hagiwara N, Aburatani H, Komuro I (2018) Genetic basis of cardiomyopathy and the genotypes involved in prognosis and left ventricular reverse remodeling. Sci Rep 8(1):1998. https://doi.org/10.1038/s41598-018-20114-9
Verdonschot JAJ, Hazebroek MR, Derks KWJ, Barandiaran Aizpurua A, Merken JJ, Wang P, Bierau J, van den Wijngaard A, Schalla SM, Abdul Hamid MA, van Bilsen M, van Empel VPM, Knackstedt C, Brunner-La Rocca HP, Brunner HG, Krapels IPC, Heymans SRB (2018) Titin cardiomyopathy leads to altered mitochondrial energetics, increased fibrosis and long-term life-threatening arrhythmias. Eur Heart J 39(10):864–873. https://doi.org/10.1093/eurheartj/ehx808
Kayvanpour E, Sedaghat-Hamedani F, Amr A, Lai A, Haas J, Holzer DB, Frese KS, Keller A, Jensen K, Katus HA, Meder B (2017) Genotype-phenotype associations in dilated cardiomyopathy: meta-analysis on more than 8000 individuals. Clin Res Cardiol 106(2):127–139. https://doi.org/10.1007/s00392-016-1033-6
Watanabe T, Kimura A, Kuroyanagi H (2018) Alternative splicing regulator RBM20 and cardiomyopathy. Front Mol Biosci 5:105. https://doi.org/10.3389/fmolb.2018.00105
Miszalski-Jamka K, Jefferies JL, Mazur W, Głowacki J, Hu J, Lazar M, Gibbs RA, Liczko J, Kłyś J, Venner E, Muzny DM, Rycaj J, Białkowski J, Kluczewska E, Kalarus Z, Jhangiani S, Al-Khalidi H, Kukulski T, Lupski JR, Craigen WJ, Bainbridge MN (2017) Novel genetic triggers and genotype-phenotype correlations in patients with left ventricular noncompaction, circulation. Cardiovasc Genet 10(4):20. https://doi.org/10.1161/circgenetics.117.001763
Parikh VN, Caleshu C, Reuter C, Lazzeroni LC, Ingles J, Garcia J, McCaleb K, Adesiyun T, Sedaghat-Hamedani F, Kumar S, Graw S, Gigli M, Stolfo D, Dal Ferro M, Ing AY, Nussbaum R, Funke B, Wheeler MT, Hershberger RE, Cook S, Steinmetz LM, Lakdawala NK, Taylor MRG, Mestroni L, Merlo M, Sinagra G, Semsarian C, Meder B, Judge DP, Ashley E (2019) Regional variation in RBM20 causes a highly penetrant arrhythmogenic cardiomyopathy. Circ Heart Fail 12(3):e005371. https://doi.org/10.1161/CIRCHEARTFAILURE.118.005371
Hoedemaekers YM, Cohen-Overbeek TE, Frohn-Mulder IM, Dooijes D, Majoor-Krakauer DF (2013) Prenatal ultrasound diagnosis of MYH7 non-compaction cardiomyopathy. Ultrasound Obstet Gynecol 41(3):336–339
Genomics of complex disorders I (2008) Genome Med 2(3–4) 303–330. https://doi.org/10.1007/s11568-009-9104-7
Cuenca S, Ruiz-Cano MJ, Gimeno-Blanes JR, Jurado A, Salas C, Gomez-Diaz I, Padron-Barthe L, Grillo JJ, Vilches C, Segovia J, Pascual-Figal D, Lara-Pezzi E, Monserrat L, Alonso-Pulpon L, Garcia-Pavia P (2016) Genetic basis of familial dilated cardiomyopathy patients undergoing heart transplantation. Transplantation 35(5):625–635. https://doi.org/10.1016/j.healun.2015.12.014
Lakdawala NK, Funke BH, Baxter S, Cirino AL, Roberts AE, Judge DP, Johnson N, Mendelsohn NJ, Morel C, Care M, Chung WK, Jones C, Psychogios A, Duffy E, Rehm HL, White E, Seidman JG, Seidman CE, Ho CY (2012) Genetic testing for dilated cardiomyopathy in clinical practice. J Cardiac Fail 18(4):296–303. https://doi.org/10.1016/j.cardfail.2012.01.013
Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, Mazzarotto F, Blair E, Seller A, Taylor JC, Minikel EV, Exome Aggregation C, MacArthur DG, Farrall M, Cook SA, Watkins H (2017) Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genetics Med 19(2):192–203. https://doi.org/10.1038/gim.2016.90
Meng L, Pammi M, Saronwala A, Magoulas P, Ghazi AR, Vetrini F, Zhang J, He W, Dharmadhikari AV, Qu C, Ward P, Braxton A, Narayanan S, Ge X, Tokita MJ, Santiago-Sim T, Dai H, Chiang T, Smith H, Azamian MS, Robak L, Bostwick BL, Schaaf CP, Potocki L, Scaglia F, Bacino CA, Hanchard NA, Wangler MF, Scott D, Brown C, Hu J, Belmont JW, Burrage LC, Graham BH, Sutton VR, Craigen WJ, Plon SE, Lupski JR, Beaudet AL, Gibbs RA, Muzny DM, Miller MJ, Wang X, Leduc MS, Xiao R, Liu P, Shaw C, Walkiewicz M, Bi W, Xia F, Lee B, Eng CM, Yang Y, Lalani SR (2017) Use of exome sequencing for infants in intensive care units: ascertainment of severe single-gene disorders and effect on medical management. JAMA Pediatr 171(12):e173438. https://doi.org/10.1001/jamapediatrics.2017.3438
Funding
This study was funded by the Natural Science Foundation of China (Grant Number: 81873498), the Natural Science Foundation of Shandong Province (Grant Number: ZR2019MH015), the Jinan Science and Technology Development Plan (Grant Number 201805020), and Special Expert of Taishan Scholars (Grant Number: TS201511099).
Author information
Authors and Affiliations
Contributions
All authors contributed to study conception and design. YW and BH collected patient data and prepared the manuscript. YF, XY, JL, and JW contributed to the clinical evaluation of patients and revision of the manuscript. YY, HY, LZ, and JZ analyzed and interpreted the genetic data and surveyed the literature relevant to the mutations. All authors reviewed the results and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Consent to Participate
N/A.
Consent for Publication
All authors and study participants declare their unconditional consent toward publication of this manuscript and its associated data in a peer-reviewed journal.
Ethical Approval
Our study received ethics approval (NSFC: NO.2018-115) from the ethics committee of Shandong Provincial Hospital, Cheeloo College of Medicine, Shandong University.
Informed Consent
Informed consent was obtained from the parents or carers of all participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, Y., Han, B., Fan, Y. et al. Next-Generation Sequencing Reveals Novel Genetic Variants for Dilated Cardiomyopathy in Pediatric Chinese Patients. Pediatr Cardiol 43, 110–120 (2022). https://doi.org/10.1007/s00246-021-02698-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00246-021-02698-8