
Abstract
Left ventricular hypertrabeculation/noncompaction (LVHT) is a cardiac abnormality of unknown etiology. Prenatal diagnosis of LVHT can be established by fetal echocardiography. A review of 106 published cases showed that 46 cases with prenatally diagnosed LVHT were alive 0.5–120 months after birth. Since the course of cases with prenatally LVHT after publication is unknown, we aimed to collect follow-up-information. Information regarding vital status, cardiac and extracardiac morbidity was gathered by contacting the authors of the 46 cases. Fourteen of the 28 authors answered and gave information about 18 cases (six females, seven males, five gender-unknown, age 18 months to 10 years, mean follow-up 60 months). No differences were found between the 18 cases with follow-up and the 28 cases without follow-up regarding age, gender, cardiac or extracardiac comorbidities, and interventions. Three of the 18 cases had died subsequently from heart failure, osteosarcoma, and enterocolitis, respectively. Mutations or chromosomal abnormalities were found in six of the seven examined patients, extracardiac abnormalities in nine patients. Three patients received a pacemaker because of complete AV block, and two patients underwent heart transplantation. Cardiac surgical or interventional procedures were carried out in four patients. None suffered from malignant arrhythmias or had a cardioverter–defibrillator implanted. Based on the limited information, there are indications that cases with fetal diagnosis of LVHT have a continuing morbidity and mortality, even if they receive appropriate care. Since fetal LVHT is frequently associated with genetic abnormalities, further research about survival and underlying genetic causes is needed.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Left ventricular hypertrabeculation/noncompaction (LVHT) is a cardiac abnormality of unknown etiology. LVHT is diagnosed in children as well as in adults and may be associated with normally sized and well contracting or dilated and poorly contracting left ventricles [1]. Patients with LVHT may have other cardiac features, neuromuscular abnormalities, or genetic defects [2]. Prenatal diagnosis of LVHT can be achieved by a standard fetal anatomic sonographic four-chamber view or by fetal echocardiography [3, 4]. A recent review of 106 published cases showed that only 46 (43 %) with prenatally diagnosed LVHT were reported to be alive 0.5–120 months (mean 28 months) after birth [5]. The remaining 60 cases were reported to have died due to termination of pregnancy (n = 18), intrauterine death (n = 8), within the first postnatal week (n = 25), later than in the first postnatal week (n = 3), or were lost to follow-up (n = 6) [5]. The surviving cases presented less frequently with fetal hydrops (13 vs. 62 %, p = 0.0004), complete heart block (27 vs. 78 %, p = 0.0076), more than three associated cardiac abnormalities (9 vs. 47 %, p = 0.0008), and more frequently with isolated LVHT (52 vs. 19 %, p = 0.009) than cases who died. Of the surviving patients, 16 received pharmacotherapy, three pacemakers, eight underwent surgical procedures, and four heart transplantation [5]. Since the course and outcome of these 46 patients after publication were unknown, we aimed to gather information about midterm follow-up of these previously published cases.
Patients and Methods
The corresponding authors of the 46 cases who were reported to be alive at the time of publication were contacted between March and May 2015. It was asked whether the patients were still alive. If so, it was asked by a questionnaire for their age, whether they had received any surgical or device therapy such as implantable cardioverters–defibrillator (ICD), pacemaker, assist device; developed heart failure, embolic events or arrhythmias, and for the type of pharmacotherapy prescribed. It was asked whether the extent or morphology of LVHT had changed during follow-up, whether the patient had extracardiac comorbidity, and whether there were any abnormalities in the intellectual development. We also searched information whether patients have been assessed by a neurologist; whether any manifestations of a neuromuscular disorder had been documented; whether there was CK elevation, muscle weakness, or easy fatigability; and whether a muscle biopsy had been carried out.
In case of death, the age and cause of death were requested. Further information regarding any surgical, device or interventional therapy between the publication and the death was requested and whether autopsy had been performed.
For surviving as well as death cases, it was asked whether any new information about the occurrence of LVHT or other cardiac or noncardiac diseases in the family of the patient had been obtained after the publication and whether follow-up data were published in the meantime.
The answers of the authors were summarized. Group comparisons, except for age, were performed by the two-sided Fisher’s exact test. For age comparisons, the Tukey’s test was used because age was not normally distributed. The Kaplan–Meier estimator was used for indicating the survival function for died infants and for infants having received a heart transplant. For all statistical analyses, the statistical software “R” was used [6].
Results
Fourteen of the 28 corresponding authors answered. One author could not be reached because of missing e-mail address, and the remaining 13 authors did not answer.
We received detailed answers to our questions about 18 (39 %) cases from 11 authors [3, 7–16]. Three further corresponding authors answered, but did not provide information because they were no longer caring for the child [17], were retired subsequently [18–20], or follow-up data had been recently published [21, 22]. One corresponding author informed us that the patient’s mother has written a book about her son’s history after he had undergone cardiac transplantation because of heart failure due to LVHT (ISBN 1-880416-33-6-1) [17].
Altogether, data from 18 cases of which we received follow-up information were evaluated. No significant differences in the previously published data regarding age, gender, cardiac or extracardiac comorbidities, and interventions were found between the 18 cases in whom follow-up information was obtained and the 28 cases without follow-up information (Table 1).
The characteristics of the 18 cases that were included in the present study are reported in Table 2. There were six female, seven male patients, and the gender of the remaining five patients was unknown. The age of the surviving patients ranged between 18 months and 10 years, in five patients it was unknown. The duration of follow-up ranged from 2 to 119 months with a mean of 60 months and a median of 63 months.
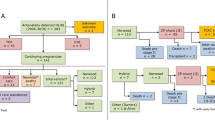
Three of the 18 cases (17 %) were no longer alive: One patient had died at the age of 5 years from heart failure, one had died at the age of 8 years due to metastatic osteosarcoma and sepsis, and one had died at the age of 8 weeks after bilateral branch pulmonary artery band placement with stent placement in the ductus arteriosus due to necrotizing enterocolitis [3, 14]. No autopsy was performed in these three cases. Of note, none of the patients had died from sudden cardiac death. For Kaplan–Meier estimates estimated for death and cardiac transplantation (Fig. 1), only data from 13/18 cases could be used [3, 7–15]. Data from the remaining 5/18 cases were missing [16].

Results of genetic analyses were reported for only seven patients, and mutations or chromosomal abnormalities were found in six of them (33 %) [7, 10, 15, 16]. Five of the patients had additional congenital cardiac abnormalities as listed in Table 2, and the remaining 13 patients had isolated LVHT. Three patients had received a pacemaker in the postnatal period because of complete AV block [3, 7, 9] and two further patients had undergone heart transplantation [3, 16]. One or more cardiac surgical or interventional procedures were carried out in four patients [3, 10, 11, 15]. None of the patients suffered from malignant arrhythmias, and none received ICDs. Extracardiac abnormalities were found in nine patients.
LVHT was reported to have changed over time in two cases: In one case, the appearance of the trabeculations slowly decreased over time [7], and in the other case, an increase in the spongiform crypt-like ventricular wall associated with reduced left ventricular function was observed [13]. Only four of the patients were reported to be without pharmacotherapy [9, 10, 12, 15]. The neurologic and intellectual development was reported as normal in seven cases [3, 7, 10, 11]. Mild mental disability was reported in three patients [8, 13, 15]. Seizures and an unclassified abdominal neurologic disorder were reported in one patient [8]. In none of the remaining patients, development of a neuromuscular disorder had been observed. A family history for LVHT, cardiomyopathy, or sudden death was found in five of the 18 cases [10, 13, 16].
Discussion
The results of this survey indicate that the prognosis of cases with LVHT diagnosed on fetal echocardiography is guarded, even if they receive appropriate care. Their life expectancy is limited by extracardiac and cardiac comorbidities, although the latter have been reported to occur more frequently in pediatric series of LVHT than in the presented cases with fetally diagnosed LVHT [23].
None of the 18 patients had died from sudden death or had received an ICD, only one patient had died from heart failure and two further patients underwent cardiac transplantation. This is in contrast to a recent publication about children with various cardiomyopathy phenotypes included in a long-term population-based study [24]. In that study, 29 patients with LVHT, aged at baseline 0.34 (0.1–1.3) years, had a cumulative incidence of 23 % of sudden cardiac death during a follow-up period of 6.8 (7–14) years, which was higher than in patients with dilated, hypertrophic, or restrictive cardiomyopathy [24]. Unfortunately, this paper did not describe under which circumstances sudden death occurred—whether it was triggered by physical exercise or occurred during rest. Since they excluded patients with congenital heart disease and progressive systemic, metabolic, or neuromuscular disease resulting in other severe organ involvement, the discrepancy between their findings and our cohort may be explained by different recruitment techniques and patients’ characteristics. Since we included all patients with different comorbidities, chance of poor outcome would be even higher.
LVHT is reported to be associated with a variety of genetic and neuromuscular disorders [2]. Likewise in our survey in six of the 18 patients (33 %), chromosomal abnormalities or mutations were detected, as listed in Table 2, and in three further patients, the family history suggested a genetic disease. Since genetic testing was not carried out in all patients, it can be speculated that a larger proportion of cases had a genetic cause.
Limitations of the study are that information could not be obtained in 61 % of the previously published cases, the small number of included patients, the lack of detailed information about associated morbidity, the small number of patients who underwent neurologic investigations, the lack of application of formal tests about intellectual and neurologic development, and that results of genetic testing were only reported in 39 %.
From our findings, we conclude that cases with fetally diagnosed LVHT have a continuing morbidity and mortality, even if they receive appropriate care which starts perinatally and is continued during childhood. Sudden cardiac death, however, appears to be less frequent than in other pediatric LVHT series. LVHT diagnosed by fetal echocardiography is frequently associated with genetic abnormalities, and there is a need for research about the relationship between survival and underlying genetic etiology. Due to the rare occurrence of fetal LVHT, a prospective multicenter study would be an appropriate tool to investigate the natural history and appropriate therapies of these patients.
References
Gati S, Rajani R, Carr-White GS, Chambers JB (2014) Adult left ventricular noncompaction: reappraisal of current diagnostic imaging modalities. JACC Cardiovasc Imaging 7:1266–1275. doi:10.1016/j.jcmg.2014.09.005
Finsterer J (2009) Cardiogenetics, neurogenetics, and pathogenetics of left ventricular hypertrabeculation/noncompaction. Pediatr Cardiol 30:659–681. doi:10.1007/s00246-008-9359-0
Arunamata A, Punn R, Cuneo B, Bharati S, Silverman NH (2012) Echocardiographic diagnosis and prognosis of fetal left ventricular noncompaction. J Am Soc Echocardiogr 25:112–120. doi:10.1016/j.echo.2011.09.019
International Society of Ultrasound in Obstetrics and Gynecology, Carvalho JS, Allan LD, Chaoui R, Copel JA, DeVore GR, Hecher K, Lee W, Munoz H, Paladini D, Tutschek B, Yagel S (2013) ISUOG Practice Guidelines (updated): sonographic screening examination of the fetal heart. Ultrasound Obstet Gynecol 41:348–359. doi:10.1002/uog.12403
Stöllberger C, Wegner C, Finsterer J (2015) Fetal ventricular hypertrabeculation/noncompaction: clinical presentation, genetics, associated cardiac and extracardiac abnormalities and outcome. Pediatr Cardiol 36:1319–1326. doi:10.1007/s00246-015-1200-y
R Core Team R (2014) A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Acherman RJ, Evans WN, Schwartz JK, Dombrowski M, Rollins RC, Castillo W, Haltore S, Berthody DP (2008) Right ventricular noncompaction associated with long QT in a fetus with right ventricular hypertrophy and cardiac arrhythmias. Prenat Diagn 28:551–553. doi:10.1002/pd.2014
Betrián Blasco P, Albert Brotóns DC, Menduña QF, Noguer FR, García GG (2010) Severe foetal hypertrophic cardiomyopathy evolving to left ventricular non-compaction. Eur J Echocardiogr 11:E36. doi:10.1093/ejechocard/jeq089
Hamela-Olkowska A, Dangel J, Miszczak-Knecht M (2009) Successful outcome of a pregnancy with an extremely low fetal heart rate (34 bpm) due to isolated complete heart block—case report. Ginekol Polska 80:708–711
Hoedemaekers YM, Cohen-Overbeek TE, Frohn-Mulder IM, Dooijes D, Majoor-Krakauer DF (2013) Prenatal ultrasound diagnosis of MYH7 non-compaction cardiomyopathy. Ultrasound Obstet Gynecol 41:336–339. doi:10.1002/uog.12279
Jacobs K, Giacobbe L, Aguilera M, Ramin K, Sivanandam S (2014) A case of fetal diagnosis of noncompaction cardiomyopathy and coarctation of the aorta. AJP Rep 4:45–48. doi:10.1055/s-0034-1371750
Pipitone S, Curcio P (2015) Fetal myocardial calcifications and non-compaction: a shared etiology? Ultrasound Obstet Gynecol 45:232–233. doi:10.1002/uog.14626
Sleurs E, De Catte L, Benatar A (2005) Prenatal diagnosis of isolated ventricular noncompaction of the myocardium. J Ultrasound Med 24:1325–1329
Tomar M, Radhakrishnan S (2009) Biventricular noncompaction: a rare cause of fetal distress and tricuspid regurgitation. Images Paediatr Cardiol 11:1–5
Wang JC, Dang L, Mondal TK, Khan A (2007) Prenatally diagnosed mosaic trisomy 22 in a fetus with left ventricular non-compaction cardiomyopathy. Am J Med Genet 143A:2744–2746
Weber R, Kantor P, Chitayat D, Friedberg MK, Golding F, Mertens L, Nield LE, Ryan G, Seed M, Yoo SJ, Manlhiot C, Jaeggi E (2014) Spectrum and outcome of primary cardiomyopathies diagnosed during fetal life. JACC Heart Fail 2:403–411. doi:10.1016/j.jchf.2014.02.010
Bleyl SB, Mumford BR, Brown-Harrison MC, Pagotto LT, Carey JC, Pysher TJ, Ward K, Chin TK (1997) Xq28-linked noncompaction of the left ventricular myocardium: prenatal diagnosis and pathologic analysis of affected individuals. Am J Med Genet 72:257–265
Bader RS, Punn R, Silverman NH (2014) Evaluation of risk factors for prediction of outcome in fetal spectrum of atrioventricular septal defects. Congenit Heart Dis 9:286–293. doi:10.1111/chd.12136
Friedberg MK, Ursell PC, Silverman NH (2005) Isomerism of the left atrial appendage associated with ventricular noncompaction. Am J Cardiol 96:985–990
Kohl T, Villegas M, Silverman N (1995) Isolated noncompaction of ventricular myocardium—detection during fetal life. Cardiol Young 5:187–189
Dhar R, Reardon W, McMahon CJ (2014) Biventricular non-compaction hypertrophic cardiomyopathy in association with congenital complete heart block and type I mitochondrial complex deficiency. Cardiol Young 15:1–3. doi:10.1017/S1047951114001279
Dhar R, Reardon W, McMahon CJ (2015) Response to commentary: biventricular non-compaction hypertrophic cardiomyopathy in association with congenital complete heart block and type I mitochondrial complex deficiency. Cardiol Young 25:1024–1025. doi:10.1017/S1047951115000323
Punn R, Silverman NH (2010) Cardiac segmental analysis in left ventricular noncompaction: experience in a pediatric population. J Am Soc Echocardiogr 23:46–53. doi:10.1016/j.echo.2009.09.003
Bharucha T, Lee KJ, Daubeney PE, Nugent AW, Turner C, Sholler GF, Robertson T, Justo R, Ramsay J, Carlin JB, Colan SD, King I, Weintraub RG, Davis AM, NACCS (National Australian Childhood Cardiomyopathy Study) Investigators (2015) Sudden death in childhood cardiomyopathy: results from a long-term national population-based study. J Am Coll Cardiol 65:2302–2310. doi:10.1016/j.jacc.2015.03.552
Acknowledgments
We thank for providing information about the patients Ruben J. Acherman, Las Vegas, USA/Pedro Betrián Blasco, Barcelona, Spain/Katarzyna Bieganowska, Warszawa, Poland/Salvatore Pipitone, Palermo, Italy/Munesh Tomar, Gurgaon, India.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Stöllberger, C., Wegner, C., Benatar, A. et al. Postnatal Outcome of Fetal Left Ventricular Hypertrabeculation/Noncompaction. Pediatr Cardiol 37, 919–924 (2016). https://doi.org/10.1007/s00246-016-1369-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00246-016-1369-8