
Abstract
Angiosperm mitochondrial genes appear to have very low mutation rates, while non-gene regions expand, diverge, and rearrange quickly. One possible explanation for this disparity is that synonymous substitutions in plant mitochondrial genes are not truly neutral and selection keeps their occurrence low. If this were true, the explanation for the disparity in mutation rates in genes and non-genes needs to consider selection as well as mechanisms of DNA repair. Rps14 is co-transcribed with cob and rpl5 in most plant mitochondrial genomes, but in some genomes, rps14 has been duplicated to the nucleus leaving a pseudogene in the mitochondria. This provides an opportunity to compare neutral substitution rates in pseudogenes with synonymous substitution rates in the orthologs. Genes and pseudogenes of rps14 have been aligned among different species and the mutation rates have been calculated. Neutral substitution rates in pseudogenes and synonymous substitution rates in genes are significantly different, providing evidence that synonymous substitutions in plant mitochondrial genes are not completely neutral. The non-neutrality is not sufficient to completely explain the exceptionally low mutation rates in land plant mitochondrial genomes, but selective forces appear to play a small role.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Synonymous substitution rates in angiosperm mitochondrial genes are about 10-fold lower than in the nuclear genes (Drouin et al. 2008; Richardson et al. 2013; Wolfe et al. 1987) and approximately 100-fold lower than in animal mitochondria (Palmer and Herbon 1988). This low rate appears to be a derived trait in land plants (Smith 2015). Synonymous substitutions are often used to calculate mutation rates in genes under the assumption that they are selectively neutral (Nei et al. 2010). It might also be expected that mutations in non-coding or nonessential regions would also be neutral, and this could provide an interesting comparison to synonymous substitution rates. However, the non-gene regions of land plant mitochondrial genomes expand and rearrange so quickly, and to such an extent, that it is difficult to align the non-gene regions outside of very closely related species (Christensen 2013, 2014; Darracq et al. 2010; Kubo and Newton 2008; Mower et al. 2007; Palmer and Herbon 1988; Richardson et al. 2013; Sloan et al. 2012; Smith and Keeling 2015). If the mutation rate in plant mitochondrial genomes is truly low, then why do the non-gene regions diverge so quickly? One possible part of the explanation may be that synonymous substitutions in angiosperm mitochondria are not selectively neutral, and therefore underestimate the mutation rate. If so, the explanation for the paradox of low mutation rates in genes and high mutation rates in junk may need to be explained not just by DNA repair and maintenance mechanisms, but by a further understanding of the role of selection on synonymous substitutions.
This possibility has been addressed (Sloan and Taylor 2010) using patterns of codon usage in mitochondrial genes. Their study concluded that selection on synonymous sites was neutral or nearly neutral, and that selective effects on synonymous sites were too weak to explain the reduced substitution rates. They also identified a bias toward A-T bases and pyrimidines at synonymous sites, but this non-randomness is not fully understood. More recently, presumably neutral mutation rates in mitochondrial insertions of plastid DNA were measured, but were not able to be directly compared to homologous sequences under selection in mitochondria (Sloan and Wu 2014). Thus, the substitution rates of synonymous sites have never been directly compared to truly neutral substitution rates, such as the rates of homologous non-selected sequences. Such a comparison would provide a direct way of confirming that synonymous substitutions are truly neutral; however, the highly divergent nature of non-gene regions prevents proper alignment among lineages, and thus, there are very few opportunities for direct comparisons across diverse species.
Ribosomal protein small subunit 14 (rps14) is co-transcribed in many plant mitochondrial genomes (see Fig. 1) with ribosomal protein large subunit 5 (rpl5) and cytochrome b (cob) (Hoffmann et al. 1999; Quinones et al. 1996). In some lineages, a copy of rps14 has been relocated to the nucleus and the protein is imported by mitochondria. In these lineages, the mitochondrial copy of rps14 has become a pseudogene (Aubert et al. 1992; Figueroa et al. 1999; Ong and Palmer 2006). These pseudogenes accumulate frameshift mutations so are clearly non-functional and not under selection for protein coding capacity. Because both rps14 genes and pseudogenes are co-transcribed with and located between rpl5 and cob, large rearrangements of the area will be selected against, as cob would lose its promoter. These rps14 pseudogenes are thus a unique example of a non-coding sequence that can still be aligned to homologous coding sequences across very diverse lineages. Therefore, rps14 is a perfect candidate to measure neutral mutation rates. In lineages with functional rps14 genes, the synonymous substitution rate can be measured, while in lineages with ψrps14 pseudogenes, the total substitution rate is the neutral mutation rate. These rates can be compared to find out if synonymous substitutions in plant mitochondrial genes are selectively neutral.

Methods
Accession numbers of all sequences used are listed in Online Resource 1. In a few species, the synteny of cob with rpl5 and rps14 was disrupted, but it was still possible to identify rps14 or ψrps14 just downstream of rpl5. The ψrps14 pseudogenes were confirmed by the presence of internal stop codons or frameshifts. Four multiple alignments were used in this analysis: an alignment of the rps14 sequences in all species analyzed (Online Resource 2), an alignment of the concatenated sequences of atp4, rpl5, and cob in all species analyzed (Online Resource 3), an alignment of the functional rps14 sequences (Online Resource 4), and an alignment of the concatenated sequences of atp4, rpl5, and cob in only those species with a functional rps14 gene (Online Resource 5).
There is also RNA editing by pentatricopeptide repeat (PPR) proteins in the analyzed genes in several of these species (Uchida et al. 2011). A PPR protein binds to an mRNA and edits a cytosine to a uracil. These edits may change the amino acid encoded. A mutation at an edited site, or in the binding sequence of the PPR protein, may appear synonymous at the DNA level, but change the final protein, or may appear non-synonymous at the DNA level but leave the protein sequence unchanged. To avoid confounding the analysis, edited codons and the 18 upstream nucleotides representing potential PPR binding sites under selection have been deleted from analysis.
Two phylogenetic trees were constructed: one using the concatenated sequences of atp4, rpl5, and cob from all species analyzed, and one using the concatenated sequences of atp4, rpl5, and cob from only those species with a functional rps14 gene. The atp4 gene was chosen because it is independently transcribed (Forner et al. 2007). All alignments and phylogenetic trees were constructed with Mega5 (Tamura et al. 2011).
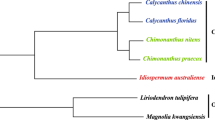
Analysis of functional rps14 genes was done using CodeML in PAML 4.8 implemented in PAMLX (Yang 2007). Branch lengths were calculated using synonymous substitutions, and the phylogenetic tree of the concatenated sequences of atp4, rpl5, and cob (Online Resource 6) was used to set the topology. This was done separately using the multiple alignment of the rps14 sequence including only species with functional rps14 genes (Fig. 2a) and the multiple alignment of the concatenated sequences of atp4, rpl5, and cob including only species with a functional rps14 gene (Fig. 2b). Taking the branch length of each terminal branch leading to a lineage on the rps14 tree and dividing it by the length of the same branch on the atp4, rpl5, and cob tree provides a ratio of the synonymous substitution rate of rps14 genes compared to the synonymous substitution rate of the other three genes.

Phylogenetic trees with terminal branch lengths calculated using PAML as described in methods. a Phylogenetic tree of functional rps14 genes using synonymous substitutions. b Phylogenetic tree of atp4, rpl5, and cob using synonymous substitutions and including only species with functional rps14 genes. c Phylogenetic tree of both rps14 genes and ψrps14 pseudogenes using total substitutions. Species with ψrps14 pseudogenes are labeled with a ψ. d Phylogenetic tree of atp4, rpl5, and cob using synonymous substitutions and including all species. For all trees, topology is based on an initial tree of atp4, rpl5, and cob sequences
Analysis of ψrps14 pseudogenes was done using BaseML in PAML 4.8 implemented in PAMLX (Yang 2007), branch lengths were calculated using total substitutions, and the phylogenetic tree of the concatenated sequences of atp4, rpl5, and cob (Online Resource 7) was used to set the topology. This was done using the multiple alignment of the rps14 sequence including all species (Fig. 2c). A phylogenetic tree using CodeML as described above was made using the multiple alignment of the concatenated sequences of atp4, rpl5, and cob including all species analyzed (Fig. 2d). Taking the branch length of each terminal branch leading to a lineage with an ψrps14 pseudogene on the rps14 tree and dividing it by the length of the same branch on the atp4, rpl5, and cob tree provides a ratio of the total substitution rate of the ψrps14 pseudogene compared to the synonymous substitution rate of the other three genes (Online Resource 8). Species with functional rps14 genes were included in these trees to avoid counting as much divergence before the pseudogenes became pseudogenes as possible. Indels were counted in all rps14 sequences. Indel rates per site were calculated.
Results
If synonymous substitutions in plant mitochondria are not neutral, then the synonymous substitution rate would erroneously underestimate the neutral mutation rate. In this event, we would expect rps14 genes to have a significantly lower synonymous substitution rate than the total substitution rate in an ψrps14 pseudogene. Alignments were done for ψrps14 of the chosen species as well as rps14 genes for the chosen species (Online Resources 2 and 4). Alignments were also done for the concatenated sequences of atp4, rpl5, and cob for all chosen species (Online Resources 3 and 5) in order to generate the trees shown in Fig. 2. Following alignments, we calculated both rates.
Terminal branch lengths for the genes were calculated using PAML 4.8 (Yang 2007), and are shown in Fig. 2 and Online Resource 8. For rps14 genes, the normalized neutral mutation rate is calculated by dividing the terminal branch length of the rps14 tree by the terminal branch length of the atp4, rpl5, and cob tree, both calculated using synonymous substitutions per synonymous site. For ψrps14 pseudogenes, the normalized neutral mutation rate is calculated by dividing the terminal branch length of the rps14 tree (calculated using total substitutions per site) by the terminal branch length of the atp4, rpl5, and cob tree (calculated using synonymous substitutions per synonymous site).
The neutral mutation rates normalized with the atp4, rpl5 and cob genes are shown in Table 1 and Fig. 3. The average normalized neutral mutation rate of the functional rps14 genes is 0.276, and the average normalized neutral mutation rate of the ψrps14 pseudogenes is 1.32. Using a Student’s t test, these rates are significantly different (p = 0.0099). One species, Citrullus lanatus, had branch lengths of zero for both ψrps14 and atp4, rpl5, cob, and was excluded from analysis. Despite having no lineage specific substitutions when compared to neighboring species, C. lanatus differed by several indels.

Comparison of the neutral mutation rate of species with functional rps14 genes and species with ψrps14 pseudogenes. Rates are from Table 1 and Online Resource 6. Mean ± standard error are shown
In addition to substitutions, we also measured indel rates. Indels should be strongly selected against in functional genes, but neutral in pseudogenes. The ψrps14 pseudogenes had an average indel rate of 0.011 indels per site. The rps14 genes had an average indel rate of 0 indels per site. These rates are significantly different (p = 0.00043), as expected.
Discussion
Because there is no selective pressure on a non-functional pseudogene, substitutions will be neutral. The availability of both genes and alignable pseudogenes of rps14 allowed us to measure the neutral substitution rate directly and compare it to the synonymous substitution rate, often used as a proxy for the neutral rate. The normalized synonymous substitution rate of the rps14 genes is significantly different from the neutral substitution rate of the ψrps14 pseudogenes (Fig. 3; Table 1). Therefore, it can be inferred that the number of observable synonymous substitutions in plant mitochondria is lower than we would expect in the absence of any selection.
One possible explanation for the apparent selection on synonymous substitutions is RNA stability and translation efficiency. If synonymous substitutions affect the stability of mitochondrial RNA or the association with the translation machinery, then there will be selective pressure to repair them even without a difference in the encoded protein. Another possibility is that mutational processes may be responsible for the A-T and pyrimidine biases in codon usage observed by Sloan and Taylor (2010), as well as the A-T bias in mutations of neutral insertions of plastid DNA in mitochondrial genomes (Sloan and Wu 2014). In other systems, it has been estimated that the rate of cytosine deamination which causes G-C to A-T transitions is at least 50-fold higher than deamination reactions that could cause A-T to G-C transitions (Friedberg et al. 2006). The oxidation of guanine to 8-oxo-guanine, which can result in G-C to T-A transversions, appears to occur in plant mitochondria as well (Christensen 2013; Markkanen et al. 2012; van Loon et al. 2010). These two processes may skew the overall mutational spectrum toward an A-T bias, resulting in the non-randomness at synonymous sites previously observed (Sloan and Taylor 2010; Sloan and Wu 2014).
Another possible explanation for the apparent selection on synonymous substitutions is that synonymous substitutions might be repaired simultaneously with non-synonymous substitutions via gene conversion if gene conversion tracts are long enough. In genes, the selective pressure on deleterious mutations is very high, so repaired mutations should be frequent. In the pseudogene, there will not be selection to repair mutations, so nearby neutral mutations will not be repaired as a result of a selective sweep.
The low mutation rate in land plant mitochondrial genes compared to non-genes does not appear to be due to differences in repair processes available, but is likely due to differences in selection on the repaired products (Christensen 2013, 2014). Gross rearrangements or even small indels would be strongly selected against in gene sequences, while they would not be selected against in non-genes, including pseudogenes. These events appear to be common on evolutionary timescales, explaining the large divergence of non-coding sequences.
This study is the first direct comparison of plant mitochondrial synonymous substitution rates with a neutral substitution rate in homologous pseudogenes. Although we have found that synonymous substitutions are not completely neutral, we still concur with the conclusion of Sloan and Taylor (2010) that the non-neutrality is not sufficient to explain the large disparity between the low mutation rates in genes and the much higher mutation, rearrangement, and expansion rates of the non-coding sequences in plant mitochondria.
References
Aubert D, Bisanz-Seyer C, Herzog M (1992) Mitochondrial rps14 is a transcribed and edited pseudogene in Arabidopsis thaliana. Plant Mol Biol 20:1169
Christensen AC (2013) Plant mitochondrial genome evolution can be explained by DNA repair mechanisms. Genome Biol Evol 5:1079
Christensen AC (2014) Genes and junk in plant mitochondria-repair mechanisms and selection. Genome Biol Evol 6:1448
Darracq A, Varre JS, Touzet P (2010) A scenario of mitochondrial genome evolution in maize based on rearrangement events. BMC Genomics 11:233
Drouin G, Daoud H, Xia J (2008) Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol Phylogenet Evol 49:827
Figueroa P, Gomez I, Carmona R, Holuigue L, Araya A, Jordana X (1999) The gene for mitochondrial ribosomal protein S14 has been transferred to the nucleus in Arabidopsis thaliana. Mol Gen Genet 262:139
Forner J, Weber B, Thuss S, Wildum S, Binder S (2007) Mapping of mitochondrial mRNA termini in Arabidopsis thaliana: t-elements contribute to 5′ and 3′ end formation. Nucleic Acids Res 35:3676
Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T (2006) DNA repair and mutagenesis. ASM Press, Washington, DC
Hoffmann M, Dombrowski S, Guha C, Binder S (1999) Cotranscription of the rpl5-rps14-cob gene cluster in pea mitochondria. Mol Gen Genet 261:537
Kubo T, Newton KJ (2008) Angiosperm mitochondrial genomes and mutations. Mitochondrion 8:5
Markkanen E, Hubscher U, van Loon B (2012) Regulation of oxidative DNA damage repair: the adenine:8-oxo-guanine problem. Cell Cycle 11:1070
Mower JP, Touzet P, Gummow JS, Delph LF, Palmer JD (2007) Extensive variation in synonymous substitution rates in mitochondrial genes of seed plants. BMC Evol Biol 7:135
Nei M, Suzuki Y, Nozawa M (2010) The neutral theory of molecular evolution in the genomic era. Annu Rev Genomics Hum Genet 11:265
Ong HC, Palmer JD (2006) Pervasive survival of expressed mitochondrial rps14 pseudogenes in grasses and their relatives for 80 million years following three functional transfers to the nucleus. BMC Evol Biol 6:55
Palmer JD, Herbon LA (1988) Plant mitochondrial DNA evolves rapidly in structure, but slowly in sequence. J Mol Evol 28:87
Quinones V, Zanlungo S, Moenne A, Gomez I, Holuigue L, Litvak S, Jordana X (1996) The rpl5-rps14-cob gene arrangement in Solanum tuberosum: rps14 is a transcribed and unedited pseudogene. Plant Mol Biol 31:937
Richardson AO, Rice DW, Young GJ, Alverson AJ, Palmer JD (2013) The “fossilized” mitochondrial genome of Liriodendron tulipifera: ancestral gene content and order, ancestral editing sites, and extraordinarily low mutation rate. BMC Biol 11:29
Sloan DB, Taylor DR (2010) Testing for selection on synonymous sites in plant mitochondrial DNA: the role of codon bias and RNA editing. J Mol Evol 70:479
Sloan DB, Wu Z (2014) History of plastid DNA insertions reveals weak deletion and AT mutation biases in angiosperm mitochondrial genomes. Genome Biol Evol 6:3210
Sloan DB, Alverson AJ, Chuckalovcak JP, Wu M, McCauley DE, Palmer JD, Taylor DR (2012) Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol 10:e1001241
Smith DR (2015) Mutation rates in plastid genomes: they are lower than you might think. Genome Biol Evol 7:1227
Smith DR, Keeling PJ (2015) Mitochondrial and plastid genome architecture: reoccurring themes, but significant differences at the extremes. Proc Natl Acad Sci USA 112:10177
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731
Uchida M, Ohtani S, Ichinose M, Sugita C, Sugita M (2011) The PPR-DYW proteins are required for RNA editing of rps14, cox1 and nad5 transcripts in Physcomitrella patens mitochondria. FEBS Lett 585:2367
van Loon B, Markkanen E, Hubscher U (2010) Oxygen as a friend and enemy: how to combat the mutational potential of 8-oxo-guanine. DNA Repair (Amst) 9:604
Wolfe K, Li W, Sharp P (1987) Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast and nuclear DNAs. Proc Natl Acad Sci USA 84:9054
Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586
Acknowledgments
We are grateful to Etsuko Moriyama and Julien Gradnigo for advice and help with software. Comments by an anonymous reviewer were especially helpful in ensuring that the analysis was accurate. Julien Gradnigo was very helpful with the use of PAML. Chad Brassil gave us helpful advice on the palatability of Spirodela polyrhiza. This research was supported in part by the National Science Foundation (MCB-1104677 and MCB-1413152 to ACC).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wynn, E.L., Christensen, A.C. Are Synonymous Substitutions in Flowering Plant Mitochondria Neutral?. J Mol Evol 81, 131–135 (2015). https://doi.org/10.1007/s00239-015-9704-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-015-9704-x