
Abstract
Purpose
We reviewed the genotypes and the imaging appearances of cochleae in CHARGE patients from two large tertiary centres and analysed the observed cochlear anomalies, providing detailed anatomical description and a grading system. The goal was to gain insight into the spectrum of cochlear anomalies in CHARGE syndrome, and thus, in the role of the CHD7 gene in otic vesicle development.
Methods
We retrospectively reviewed CT and/or MR imaging of CHARGE patients referred to our institutions between 2005 and 2022. Cochlear morphology was analysed and, when abnormal, divided into 3 groups in order of progressive severity. Other radiological findings in the temporal bone were also recorded. Comparison with the existing classification system of cochlear malformation was also attempted.
Results
Cochlear morphology in our CHARGE cohort ranged from normal to extreme hypoplasia. The most common phenotype was cochlear hypoplasia in which the basal turn was relatively preserved, and the upper turns were underdeveloped. All patients in the cohort had absent or markedly hypoplastic semicircular canals and small, misshapen vestibules. Aside from a stenotic cochlear aperture (fossette) being associated with a hypoplastic or absent cochlear nerve, there was no consistent relationship between cochlear nerve status (normal, hypoplasia, or aplasia) and cochlear morphology.
Conclusion
Cochlear morphology in CHARGE syndrome is variable. Whenever the cochlea was abnormal, it was almost invariably hypoplastic. This may shed light on the role of CHD7 in cochlear development. Accurate morphological description of the cochlea contributes to proper clinical diagnosis and is important for planning surgical treatment options.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
CHARGE (coloboma, heart defects, choanal atresia, growth restriction, genital abnormalities and ear abnormalities) syndrome is a rare congenital disorder which is associated with substantial morbidity, and, rarely, mortality [1, 2]. Considering aetiology, there is a strong genetic component, with 90–95% of cases meeting Blake’s or Verloes’ diagnostic criteria, related to a mutation in the CHD7 gene [3,4,5]. This gene encodes a protein involved in chromatin remodelling, which is pivotal in many developmental pathways [6, 7]. Disruption of this protein leads to a particular disturbance of neural crest development, which is believed to underpin the most significant manifestations of the syndrome [8, 9]. Interestingly, an extremely wide range of mutations have been identified (> 1000), most of which occur de novo and are found throughout the gene with the exception of exon 1 and the 3′ terminal end of exon 38. Most pathogenic variants are frameshift or nonsense and hence associated with haploinsufficiency. A smaller proportion is missense variants, which may be harder to classify and the mechanism of pathogenicity less clearly established [4, 10]. As such, it has been suggested that different variants in CHD7 should be incorporated into the major diagnostic criteria [11].
Although there are common phenotypic features of CHARGE important for diagnosis [2, 5], variability in end organ involvement is increasingly recognised, which may have implications for management and outcome. Some studies have suggested that more severe clinical presentations of CHARGE syndrome are associated with variants that cause haploinsufficiency, with mild cases more likely to be seen with missense variants, potentially due to a hypomorphic allele [3]. Radiological studies can identify and allow for detailed depiction of specific phenotypic manifestation in each individual patient with CHARGE, and are therefore a powerful tool in contributing to precision management. Analysing and comparing imaging features among a large population of CHARGE patients may also provide insight into the pathogenesis of some of these features.
With regard to the temporal bone, morphological abnormalities and variability in CHARGE patients have been previously characterised in multiple studies using both CT and MRI [12,13,14,15]. The constellation of findings of cochlear hypoplasia and total or near-total absence of the semicircular canals is sensitive and specific for CHARGE syndrome [15]. In particular, semicircular canal (SCC) aplasia is considered a hallmark of CHARGE syndrome, in accordance with the fact that CHD7 is highly expressed in the developing ear and is required for SCC development [16, 17].
Appropriate classification of cochlear malformations is important for the management of sensorineural deafness, in both providing prognostic information and planning cochlear implantation [18]. Investigation into the range of cochlear malformations in CHARGE has been limited, but is important in light of the updated classification system for inner ear malformations that has linked management recommendations related to the feasibility and efficacy of cochlear implantation [19, 20]. More precise phenotypical description of the cochlea in CHARGE may also contribute to better understanding of the role of CHD7 in cochlear development [21, 22].
The aim of this article is to characterise the range of cochlear malformations in a large population of patients with confirmed CHARGE syndrome. A secondary aim is to gain insight into the role of CHD7 in inner ear development through observation of the spectrum of cochlear malformations.
Materials and methods
From a prospectively maintained database, we retrospectively reviewed CT and (when available) MR imaging for patients referred to two institutions between 2005 and 2022 with a clinical and/or genetic diagnosis of CHARGE syndrome. This study was approved by the institutional review board/ethics committee of each institution. The need for informed consent was waived due to the retrospective nature of this work.
Inclusion criteria for the diagnosis of CHARGE syndrome were based on the criteria set forth by Verloes et al. [3,4,5]. In addition, a proportion of these patients had confirmed mutations in CHD7.
Imaging evaluation
Given the multi-site design and retrospective nature of the study, CT and MR imaging protocols were variable, and therefore, not all structures were completely evaluated in all patients (in part dependent on whether CT and/or MRI was available), which is reflected in the final data.
Imaging studies from 26 patients were reviewed in consensus by 3 neuroradiologists, two of them with subspecialty expertise in paediatric head and neck imaging.
Each ear was evaluated for CT and/or MR imaging features of CHARGE syndrome, with a focus on cochlear morphology. A general assessment of the extra-temporal structures visible on those imaging studies, including the eyes, olfactory system, face, skull base and brain, was also performed. Non-neurological manifestations were mined from patient notes in chart review.
Cochlea
We attempted to classify the cochleae within our patient population according to the current morphological classification system of cochlear hypoplasia (CH) by Sennaroglu et al. [19, 20]. However, during the analysis of the cochlear phenotypes in our cohort, it was clear that fitting these cochleae into the 4 discrete groups described in that classification system was challenging, given that the CHARGE cochleae spanned a continuous spectrum of morphologic disturbance, and varied in the manner in which they were underdeveloped and hypoplastic. As such, we opted for a descriptive morphological grouping, and we graded each cochlea based on its degree of deviation from a normally developed cochlea.
This is in accordance with the known fact that the current classification scheme is a simplification designed to facilitate stratification of patients for determination of cochlear implant feasibility and does not encompass nor reflect the entire spectrum of cochlear anomalies, including several inner ear malformations with known genetic causes such as the unwound cochlea in EYA1 mutation-associated branchiootorenal syndrome, the type of hypoplastic cochlea associated with SOX10 mutation, or the distinctive type of hypoplastic cochlea observed in Walker-Warburg syndrome [23,24,25].
In our patient cohort, the size of the cochlea was measured in the coronal plane (cochlear height: maximal height of the cochlea on a coronal plane perpendicular to the oval window) to determine whether there was cochlear hypoplasia [26]. If cochlear hypoplasia was present, the type was recorded according to our descriptive categories, detailed below.
Retrospective review of our cohort revealed 4 cochlear phenotypes:
-
1.
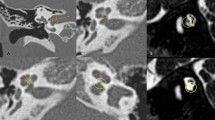
Normal size and normal contour of all the turns of the cochlea, with or without dysmorphism of the modiolus and stenotic cochlear aperture (Fig. 1A)
-
2.
Normal complete basal turn, with a complete 360° turn that is normal in contour and calibre, but a hypoplastic upper part of the cochlea; we designated this as the “mild” phenotype (Fig. 1B and C). This is similar to the description of CH type 4 by Sennaroglu and colleagues
-
3.
Normal first half of the basal turn (180°) but incomplete or dysmorphic second half of the basal turn, resulting in a “truncated” appearance of the basal turn, and a hypoplastic upper part of the cochlea; we designated this as the “moderate” phenotype (Fig. 1D and E)
-
4.
Abnormal/dysmorphic (most often dilated) appearance of the first half of the basal turn, and minimal or no second half of the basal turn, with the remainder of the upper part of the cochlea not recognizable; we designated this as the “severe” phenotype (Fig. 1F)

Axial CT images of representative cochleae along the spectrum of morphological disturbance encountered in patients with CHARGE syndrome in our cohort. A Normal size and normal external contour of the cochlea with all turns present, and only a misshapen, thickened modiolus (black arrow). The cochlear aperture is stenotic. B, C ”Mild” phenotype: the entire basal turn is normal with a complete 360° turn, and hypoplastic upper part of the cochlea (red arrows). D, E “Moderate” phenotype: normal first half of the basal turn (blue arrowhead in E), partially present second half of the basal turn showing a “truncated” appearance (curved blue arrow in D) and small upper cochlea (yellow arrow in D). F “Severe” phenotype: only a partial portion of the basal turn is present, and it is dysmorphic and dilated (short blue arrow); the upper part of the cochlea is not well developed and dysmorphic
Other radiological abnormalities
Vestibular system
For all the imaging studies in our cohort, we recorded whether the lateral semicircular canal was absent, the posterior/superior semicircular canals were absent or bud-like, and the vestibule was present/dysplastic. The size of the vestibular aqueduct was recorded as normal or enlarged with criteria based on current literature [27].
Middle ear
The ossicles were recorded as normal, malformed (abnormal morphology or size) or fused (ankylosis to the wall of the epitympanum and/or interossicular inseparability). The stapes footplate was recorded as normal or misshapen. Atresia/absence of the oval and round windows was recorded when the aperture was narrow or completely stenotic.
Facial nerve
The path of the facial nerve canal was recorded as normal, absent, or aberrant, with the course evaluated on CT and/or MRI. When MRI was available, aplasia/hypoplasia of the cisternal or canalicular portion of the facial nerve was recorded.
Venous drainage
Persistent petrosquamous sinus (PPS) is defined as an emissary vein coursing from the dorsolateral sinus to the confluence with the superior petrosal sinus [28]. When this direct vascular channel was not seen, but there were other small veins, they were recorded as emissary veins.
Extra-temporal structures
Based upon previously reported associations with CHARGE syndrome, notable extra-temporal structures recorded were choanal atresia, chorioretinal coloboma, olfactory bulb hypoplasia, vermian/pontine hypoplasia, and craniofacial abnormalities [13]. The assessment of the clivus was not possible because of the limited field of view of the CT scans.
Results
Overall, we evaluated 52 ears from 26 patients (12 males, 14 females) with a clinical and/or genetic diagnosis of CHARGE syndrome. Eight patients underwent CT and 18 underwent both CT and MR imaging. The mean age of the patients was 5.9 years (min: 9 days; max: 12 years).
CHD7 mutation reports were available for 10 patients. Nine patients had stop or frameshift mutations consistent with loss of CHD7 function; 1 had a splice site mutation (c.3990-2A > G) that it is predicted to abolish normal splicing between intron 16 and 17.
Cochlear phenotype (Table 1)
Of the 52 cochleae, 9 (4 bilateral and 1 unilateral) had normal external contours (17%), with only mild dysmorphism (evaluated subjectively since there are no reliable measurements available in literature) of the modiolus in 2 patients (bilateral) and otherwise normal size and number of turns. The other cochleas were hypoplastic (cochlear height < 4.3 mm; range 3.4–4.0).
Seven patients (14/52 cochleae, 27%) had asymmetrical cochlear appearances.
The mild cochlear hypoplasia phenotype was by far the most common morphology, found in 32/52 cochleae (61%) and characterised by a normal complete 360° basal turn and hypoplastic upper part of the cochlea.
Five/52 (9.6%) demonstrated a moderate phenotype, characterised by preserved first half of the basal turn (180°) with partially present second half of the basal turn, resulting in a “truncated” appearance of the basal turn, and hypoplastic upper part of the cochlea. Only one patient had bilateral moderate phenotype; the others had moderate phenotype on one side and mild phenotype on the contralateral side.
Six cochleae (6/52, 11.5%) showed severe hypoplasia phenotype (first half of the basal turn present but dysmorphic, minimal or no upper part of the cochlea appreciable).
MRI imaging was available for 18 patients, which allowed better definition of the internal cochlear partitioning and direct assessment of the nerves with the following results: 29 absent or hypoplastic cochlear nerves (1 with additional vestibular nerve absence), and 1 absent facial nerve.
In cases where MRI was available, there was no clear or consistent relationship between the cochlear phenotype and the presence or absence of the cochlear nerve (apart from the cochlear aperture stenosis associated with cochlear nerve hypoplasia or aplasia), stressing the important role of MRI in cochlear implant planning.
The prevalence of aberrant facial nerves among our cohort was high, either bowed or having a horizontal orientation.
All our patients demonstrated complete or near-complete aplasia of the semicircular canals and small misshapen appearance of the vestibule, pathognomonic of CHARGE, despite having different degrees of cochlear hypoplasia (Fig. 2). Other temporal bone and extra temporal findings are summarised in Table 2.

MRI appearances of the cochleae and vestibule/semicircular canals in 3 CHARGE patients with different degrees of cochlear hypoplasia (CH). A Mild phenotype: the first half of the basal turn is present and normal, the second half of the basal turn is complete with well-visualized basilar membrane/osseous spiral lamina complex (thin arrow), the upper part of the cochlea is hypoplastic (thick arrow). B Moderate phenotype: the first half of the basal turn is present and normal; the second half of the basal turn is present but incomplete (thin arrow). Note the very hypoplastic upper part of the cochlea (thick arrow). C Severe phenotype: only the first half of the basal turn is present (thin arrow) with almost completely absent remainder of the cochlea. Note the narrow internal auditory canal (thick arrow). MR images in the lower row (D, E, F from the same patient as the respective image in the upper row) show dysmorphic appearance of the vestibules and absent semicircular canals (double arrows)
Summary of temporal bone findings
In descending order of frequency, the most common findings were:
-
1.
Vestibular system involvement: aplasia or marked dysplasia of the semicircular canals (52/52 inner ears), invariably associated with a misshapen vestibule
-
2.
Malformed/abnormally orientated long and lenticular processes of incus and misshapen stapes with associated oval window atresia (51/52 inner ears; associated with epitympanic hypoplasia in 35/52 inner ears and fusion of the head of the malleus to the anterior epitympanic wall in 18/52 inner ears)
-
3.
Cochlear hypoplasia (as described above)
-
4.
Cochlear nerve hypoplasia/aplasia 29/40 ears
-
5.
Emissary veins 18/52 temporal bones
Discussion
Here, we describe and expand upon the range of cochlear and other temporal bone findings seen in a paediatric population with clinical and/or molecularly confirmed CHARGE syndrome. To our knowledge, this is also one of the largest cohorts with combined CT/MRI imaging of the temporal bone and membranous labyrinth.
Inner ear development is a complex process involving precise temporal and spatial interaction of many different genes. In mammals, the inner ear develops from ectodermal tissue that gives rise to the otocyst. The dorsolateral otocyst forms the vestibular components of the ear including the semicircular ducts, utricle, endolymphatic duct and saccule. The ventromedial otocyst gives rise to the auditory cochlea. These are innervated respectively by the vestibular and spiral ganglia.
CHD7 is a chromatin remodelling protein involved in the epigenetic regulation of gene expression. Its role in inner ear development is not well understood, but it is known to be expressed in the developing mammalian otocyst and in the cochlea. It is thought to regulate neural crest gene expression and migration, ultimately controlling cochlear development, patterning and hair cell organisation [29, 30].
As in previous series, the findings seen in our cohort of CHARGE syndrome patients were variable in frequency, reflecting the phenotypic variation also reported [14]. Previous studies comparing temporal bone findings between truncated and non-truncated CHD7 mutations [15] did not demonstrate significant genotype–phenotype correlation; however, there was a trend toward more severely dysmorphic cochlear phenotypes in patients with truncating mutations.
A variety of cochlear phenotypes were found in the 9 cases among our cohort that had loss of function mutations. These ranged from mild to severe, suggesting that there is not one unique and consistent loss of function cochlear phenotype. The lack of missense CHD7 mutations in this study cohort precludes further comment on genotype phenotype correlation in that setting. Importantly, our cohort had a very high prevalence of molecularly confirmed CHARGE syndrome, reinforcing the increasing importance of molecular testing and confirmation as one component among the major criteria for the diagnosis of CHARGE [11].
Among our cohort, cochlear hypoplasia was highly associated with an abnormal course of the facial nerve canal, concordant with existing literature [31].
As in previous series in the literature, bilateral hypoplasia of the vestibule (appearing as a small, misshapen structure); aplasia of the SCCs; malformation of the middle ear ossicles, particularly the stapes footplate and oval window atresia were extremely frequent and are defining features of CHARGE. In fact, it has been shown in animal models that chd7 is particularly important in the development of the semicircular canals, which are absent in the vast majority of these CHARGE patients. This is probably due to the prominent polarisation of CHD7 expression toward the posterior aspect of the developing otocyst [32,33,34].
Compared to previous reports, the presence of cochlear malformations in our series was high (43/52), suggesting an important role of CHD7 also in cochlear development [13,14,15]. However, variability in the severity of dysmorphism and the resultant range of cochlear phenotypes suggest a different expressivity of this gene in the anterior developing otocyst. This is an opposite scenario when compared to other conditions such as EYA1 mutation or Walker-Warburg syndrome, where the cochlea is profoundly abnormal (hypoplastic and with anterior offset) while the posterior aspect of the otic capsule (vestibule and semicircular canals) is relatively preserved.
Combining information related to gene anomalies (detectable on genetic analysis) and morphologic phenotypes (detectable on imaging) is critical to deepening our understanding of the roles of specific genes that regulate otic vesicle development at different time points during embryological development.
Cochlear hypoplasia in CHARGE patients
Previous series used variable and sometimes inconsistent terminology to describe cochlear abnormalities, including Mondini malformation, cochlear aplasia/dysplasia, fused turns, reduced number of coils, incomplete partition and malformations of modiolus [12,13,14].
We demonstrate that cochlear malformations associated with CHD7-mutated CHARGE are almost universally within the spectrum of cochlear hypoplasia abnormalities, ranging from normal to severe phenotype, and are likely to lie along a continuum rather than in discrete groups [19, 20].
Mild phenotype is the most prevalent malformation seen, which has important implications in elucidating the time of arrest of embryogenesis of the inner ear in CHARGE, thought to lie between 6 and 8 weeks, during which the basal turn is completely developed [35, 36]. The developing cochlea begins with a bud, with progressive elongation and turning of the coils; thus, an arrest of inner ear embryogenesis after 8–10 weeks may result in a relatively preserved basal turn; however, the precise timing of development of the different portions of the cochlea remains controversial in the literature [37, 38].
In addition, it seems that the first half of the basal turn (180°) is the most preserved cochlear structure in genetic causes of cochlear malformations (not only in CHARGE but also in branchiootorenal syndrome and Walker-Warburg syndrome [23, 25]).
In our cohort, we demonstrated a substantial number of bilateral but asymmetrical cochlear hypoplastic findings (26%). This is important from a diagnostic perspective, because, while symmetrical anomalies are almost always genetic in nature, asymmetrical inner ear malformations can be seen in both genetic causes and prenatal insults [24, 39]. This may support the hypothesis that genetic mutations may modify environmental factors (such as blood supply to the otic vesicle) necessary for ear development [40].
Precise description of the morphology of the cochlea is of critical importance as the size, shape, and orientation of the cochlea may influence the choice of cochlear implant type [41].
Experimental murine models
Our findings should be seen in light of recent molecular/cellular insights into the function of chd7 in the development of the inner ear in mice models [30, 42]. Briefly, chd7 has been demonstrated to play an important role in neurogenesis in both the brain and inner ear, particularly spiral ganglion neurons and apparatus pertinent to inner ear hair cell function. [43, 44]. chd7 homogeneous mutants undergo rapid degeneration of inner hair cells when subjected to an external insult such as loud noise or ototoxicity. In this sense, chd7 may have a role in maintaining an epigenetic state that allows a balanced response when exposed to oxidative stress, and a mutation may result in an inability of these cells to survive postnatally [45, 46]. chd7 heterogeneous mutants demonstrate equally severe malformation phenotypes, but at a reduced frequency. Transcriptive data reveal that spiral ganglion neurons can be divided into molecularly distinct subtypes, which may have altered susceptibility to neurodegeneration caused by chd7 deletion [47]. chd7 function is critical during early embryonic development long before hair cell/neuronal degeneration occurs, having a significant effect on multiple developmental pathways, particularly neural crest cells, in its action in remodelling chromatin [8, 9].
All this information suggests that alteration in CHD7 likely results in sensorineural hearing loss by two pathogenetic pathways in CHARGE patients. CHD7 determines anatomical forms initially and epigenetic regulation of hair cell function postnatally. Both have implications for cochlear implantation planning, from anatomical and tonotopic standpoints. This also plausibly provides a mechanism for the cochlea variability seen in this study.
Teaching points
-
Cochlear malformations in CHARGE are a spectrum ranging from normal to varying degrees of cochlear hypoplasia.
-
The first half of the basal turn is the most preserved region of the cochlea in CHARGE syndrome and in other genetic causes of cochlear hypoplasia. This sheds light on embryological development of the cochlea and has important surgical implications.
-
CHD7 mutations almost invariably impact the development of SCCs and vestibule, resulting in characteristic morphological abnormalities in most of the cases (Fig. 2).
-
Cochlear nerve can be present, hypoplastic or aplastic in CHARGE syndrome independent of cochlear morphology, other than modiolar thickening and cochlear aperture stenosis/atresia being associated with cochlear nerve hypoplasia/aplasia. Thus, MRI is critical for pre-operative planning in these patients.
Conclusion
Cochlear hypoplasia is a frequent finding in CHARGE syndrome with a range of abnormalities, but favouring a milder phenotype with a preserved basal turn. This suggests a later time of arrest of embryogenesis (> 7–8 weeks) than previously thought. The abnormalities lie along a continuous spectrum rather than as discrete entities, suggesting that a detailed description of the cochlea rather than quantum categorization would be more useful for surgical planning. Recent genetic and molecular insights provide a possible pathophysiological theory for these findings, with likely a combination of genetic and environmental interactions acting on the cochlea.
References
Hall BD (1979) Choanal atresia and associated multiple anomalies. J Pediatr 95:395–398. https://doi.org/10.1016/s0022-3476(79)80513-2
Blake KD, Prasad C (2006) CHARGE syndrome. Orphanet J Rare Dis 1:34. https://doi.org/10.1186/1750-1172-1-34
Jongmans MCJ, Admiraal RJ, van der Donk KP, Vissers LELM, Baas AF, Kapusta L et al (2006) CHARGE syndrome: the phenotypic spectrum of mutations in the CHD7 gene. J Med Genet 43:306–314. https://doi.org/10.1136/jmg.2005.036061
Bergman JEH, Janssen N, Hoefsloot LH, Jongmans MCJ, Hofstra RMW, van Ravenswaaij-Arts CMA (2011) CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J Med Genet 48:334–342. https://doi.org/10.1136/jmg.2010.087106
Verloes A (2005) Updated diagnostic criteria for CHARGE syndrome: a proposal. Am J Med Genet A 133A:306–308. https://doi.org/10.1002/ajmg.a.30559
Basson MA, van Ravenswaaij-Arts C (2015) Functional insights into chromatin remodelling from studies on CHARGE syndrome. Trends Genet 31:600–611. https://doi.org/10.1016/j.tig.2015.05.009
Ho L, Crabtree GR (2010) Chromatin remodelling during development. Nature 463:474–484. https://doi.org/10.1038/nature08911
Bérubé-Simard F-A, Pilon N (2019) Molecular dissection of CHARGE syndrome highlights the vulnerability of neural crest cells to problems with alternative splicing and other transcription-related processes. Transcription 10:21–28. https://doi.org/10.1080/21541264.2018.1521213
Pauli S, Bajpai R, Borchers A (2017) CHARGEd with neural crest defects. Am J Med Genet C Semin Med Genet 175:478–486. https://doi.org/10.1002/ajmg.c.31584
Qin Z, Su J, Li M, Yang Q, Yi S, Zheng H et al (2020) Clinical and genetic analysis of CHD7 expands the genotype and phenotype of CHARGE syndrome. Front Genet 11:592. https://doi.org/10.3389/fgene.2020.00592
Hale CL, Niederriter AN, Green GE, Martin DM (2016) Response to correspondence to Hale et al. atypical phenotypes associated with pathogenic CHD7 variants and a proposal for broadening CHARGE syndrome clinical diagnostic criteria. Am J Med Genet A 170:3367–3368. https://doi.org/10.1002/ajmg.a.37629
Morimoto AK, Wiggins RH, Hudgins PA, Hedlund GL, Hamilton B, Mukherji SK et al (2006) Absent semicircular canals in CHARGE syndrome: radiologic spectrum of findings. AJNR Am J Neuroradiol 27:1663–1671
Hoch MJ, Patel SH, Jethanamest D, Win W, Fatterpekar GM, Roland JT et al (2017) Head and neck MRI findings in CHARGE syndrome. AJNR Am J Neuroradiol 38:2357–2363. https://doi.org/10.3174/ajnr.A5297
Ha J, Ong F, Wood B, Vijayasekaran S (2016) Radiologic and audiologic findings in the temporal bone of patients with CHARGE syndrome. Ochsner J 16:125–129
Vesseur AC, Verbist BM, Westerlaan HE, Kloostra FJJ, Admiraal RJC, van Ravenswaaij-Arts CMA et al (2016) CT findings of the temporal bone in CHARGE syndrome: aspects of importance in cochlear implant surgery. Eur Arch Otorhinolaryngol 273:4225–4240. https://doi.org/10.1007/s00405-016-4141-z
Green GE, Huq FS, Emery SB, Mukherji SK, Martin DM (2014) CHD7 mutations and CHARGE syndrome in semicircular canal dysplasia. Otol Neurotol 35:1466–1470. https://doi.org/10.1097/MAO.0000000000000260
Hurd EA, Micucci JA, Reamer EN, Martin DM (2012) Delayed fusion and altered gene expression contribute to semicircular canal defects in Chd7 deficient mice. Mech Dev 129:308–323. https://doi.org/10.1016/j.mod.2012.06.002
Paludetti G, Conti G, DI Nardo W, DE Corso E, Rolesi R, Picciotti PM et al (2012) Infant hearing loss: from diagnosis to therapy Official Report of XXI Conference of Italian Society of Pediatric Otorhinolaryngology. Acta Otorhinolaryngol Ital. 32:347–370
Sennaroglu L, Saatci I (2002) A new classification for cochleovestibular malformations. Laryngoscope 112:2230–2241. https://doi.org/10.1097/00005537-200212000-00019
Sennaroğlu L, Bajin MD (2017) Classification and current management of inner ear malformations. Balkan Med J 34:397–411. https://doi.org/10.4274/balkanmedj.2017.0367
Li H, Helpard L, Ekeroot J, Rohani SA, Zhu N, Rask-Andersen H et al (2021) Three-dimensional tonotopic mapping of the human cochlea based on synchrotron radiation phase-contrast imaging. Sci Rep 11:4437. https://doi.org/10.1038/s41598-021-83225-w
Fouad YA (2020) Advances in surgical and anesthetic techniques for cochlear implantation. In: Zanetti D, Di Berardino F (eds). Advances in rehabilitation of hearing loss. IntechOpen. https://doi.org/10.5772/intechopen.88380
Talenti G, Robson C, Severino MS, Alves CA, Chitayat D, Dahmoush H et al (2021) Characteristic cochlear hypoplasia in patients with Walker-Warburg syndrome: a radiologic study of the inner ear in α-dystroglycan-related muscular disorders. AJNR Am J Neuroradiol 42:167–172. https://doi.org/10.3174/ajnr.A6858
D’Arco F, Sanverdi E, O’Brien WT, Taranath A, Talenti G, Blaser SI (2020) The link between inner ear malformations and the rest of the body: what we know so far about genetic, imaging and histology. Neuroradiology 62:539–544. https://doi.org/10.1007/s00234-020-02382-3
Pao J, D’Arco F, Clement E, Picariello S, Moonis G, Robson CD et al (2022) Re-examining the cochlea in branchio-oto-renal syndrome: genotype-phenotype correlation. AJNR Am J Neuroradiol 43:309–314. https://doi.org/10.3174/ajnr.A7396
D’Arco F, Talenti G, Lakshmanan R, Stephenson K, Siddiqui A, Carney O (2017) Do measurements of inner ear structures help in the diagnosis of inner ear malformations? A review of literature. Otol Neurotol 38:e384–e392. https://doi.org/10.1097/MAO.0000000000001604
Juliano AF, Ting EY, Mingkwansook V, Hamberg LM, Curtin HD (2016) Vestibular aqueduct measurements in the 45° oblique (pöschl) plane. AJNR Am J Neuroradiol 37:1331–1337. https://doi.org/10.3174/ajnr.A4735
Marsot-Dupuch K, Gayet-Delacroix M, Elmaleh-Bergès M, Bonneville F, Lasjaunias P (2001) The petrosquamosal sinus: CT and MR findings of a rare emissary vein. AJNR Am J Neuroradiol 22:1186–1193
Bajpai R, Chen DA, Rada-Iglesias A, Zhang J, Xiong Y, Helms J et al (2010) CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 463:958–962. https://doi.org/10.1038/nature08733
Balendran V, Skidmore JM, Ritter KE, Gao J, Cimerman J, Beyer LA et al (2021) Chromatin remodeler CHD7 is critical for cochlear morphogenesis and neurosensory patterning. Dev Biol 477:11–21. https://doi.org/10.1016/j.ydbio.2021.05.009
Sennaroğlu L, Tahir E (2020) A novel classification: anomalous routes of the facial nerve in relation to inner ear malformations. Laryngoscope 130:E696–E703. https://doi.org/10.1002/lary.28596
Som PM, Curtin HD, Liu K, Mafee MF (2016) Current embryology of the temporal bone, part I: the inner ear. Neurographics 6(4):250–265
Chatterjee S, Kraus P, Lufkin T (2010) A symphony of inner ear developmental control genes. BMC Genet 11:68. https://doi.org/10.1186/1471-2156-11-68
Chatterjee S, Lufkin T (2011) The sound of silence: mouse models for hearing loss. Genet Res Int 2011:416450. https://doi.org/10.4061/2011/416450
Solomon KS, Kwak S-J, Fritz A (2004) Genetic interactions underlying otic placode induction and formation. Dev Dyn 230:419–433. https://doi.org/10.1002/dvdy.20067
Driver EC, Kelley MW (2020) Development of the cochlea. Development 147. https://doi.org/10.1242/dev.162263
Jackler RK, Luxford WM, House WF (1987) Congenital malformations of the inner ear: a classification based on embryogenesis. Laryngoscope 97:2–14. https://doi.org/10.1002/lary.5540971301
Pujol R, Lavigne-Rebillard M, Uziel A (1991) Development of the human cochlea. Acta Otolaryngol Suppl 482:7–12. https://doi.org/10.3109/00016489109128023. (discussion 13)
Davide B, Renzo M, Sara G, Elisa L, Rodica M, Irene T et al (2017) Oculo-auriculo-vertebral spectrum: going beyond the first and second pharyngeal arch involvement. Neuroradiology 59:305–316. https://doi.org/10.1007/s00234-017-1795-1
Sennaroglu L (2016) Histopathology of inner ear malformations: do we have enough evidence to explain pathophysiology? Cochlear Implants Int 17:3–20. https://doi.org/10.1179/1754762815Y.0000000016
Breitsprecher T, Dhanasingh A, Schulze M, Kipp M, Dakah RA, Oberhoffner T et al (2022) CT imaging-based approaches to cochlear duct length estimation-a human temporal bone study. Eur Radiol 32:1014–1023. https://doi.org/10.1007/s00330-021-08189-x
Ahmed M, Moon R, Prajapati RS, James E, Basson MA, Streit A (2021) The chromatin remodelling factor Chd7 protects auditory neurons and sensory hair cells from stress-induced degeneration. Commun Biol 4:1260. https://doi.org/10.1038/s42003-021-02788-6
Hurd EA, Poucher HK, Cheng K, Raphael Y, Martin DM (2010) The ATP-dependent chromatin remodeling enzyme CHD7 regulates pro-neural gene expression and neurogenesis in the inner ear. Development 137:3139–3150. https://doi.org/10.1242/dev.047894
Hurd EA, Adams ME, Layman WS, Swiderski DL, Beyer LA, Halsey KE et al (2011) Mature middle and inner ears express Chd7 and exhibit distinctive pathologies in a mouse model of CHARGE syndrome. Hear Res 282:184–195. https://doi.org/10.1016/j.heares.2011.08.005
Dawson WJ, Henderson D, Bielefeld EC, Harris KC, Hu BH (2006) The role of oxidative stress in noise-induced hearing loss. Ear Hear. 27(1):1–19. https://doi.org/10.1097/01.aud.0000191942.36672.f3
Jiang H, Talaska AE, Schacht J, Sha S-H (2007) Oxidative imbalance in the aging inner ear. Neurobiol Aging 28:1605–1612. https://doi.org/10.1016/j.neurobiolaging.2006.06.025
Grandi FC, De Tomasi L, Mustapha M (2020) Single-cell RNA analysis of type I spiral ganglion neurons reveals a Lmx1a population in the cochlea. Front Mol Neurosci 13:83. https://doi.org/10.3389/fnmol.2020.00083
Funding
Research reported in this publication was supported by the National Institute of Health Biomedical Research Center at Great Ormond Street Hospital, London UK (unfunded).
Author information
Authors and Affiliations
Contributions
Martin A Lewis1, Amy Juliano, and Felice D’Arco: conceived the study, did the data analysis, wrote the manuscript. Caroline Robson: revised the manuscript. Emma Clement, Robert Nash, Kaukab Rajput: helped in the data collection and in writing the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in the studies involving human participants were in accordance with the ethical standards of the institutional research committee (Clinical Research Adoption Committee) and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed consent
Standard clinical informed consent was obtained at the time of the scan. Informed consent for this specific study was not applicable given the retrospective nature of this research.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Martin A. Lewis and Amy Juliano shared first authorship.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lewis, M.A., Juliano, A., Robson, C. et al. The spectrum of cochlear malformations in CHARGE syndrome and insights into the role of the CHD7 gene during embryogenesis of the inner ear. Neuroradiology 65, 819–834 (2023). https://doi.org/10.1007/s00234-023-03118-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00234-023-03118-9