
Abstract
The use of designed antimicrobial peptides as drugs has been impeded by the absence of simple sequence-structure–function relationships and design rules. The likely cause is that many of these peptides permeabilize membranes via highly disordered, heterogeneous mechanisms, forming aggregates without well-defined tertiary or secondary structure. We suggest that the combination of high-throughput library screening with atomistic computer simulations can successfully address this challenge by tuning a previously developed general pore-forming peptide into a selective pore-former for different lipid types. A library of 2916 peptides was designed based on the LDKA template. The library peptides were synthesized and screened using a high-throughput orthogonal vesicle leakage assay. Dyes of different sizes were entrapped inside vesicles with varying lipid composition to simultaneously screen for both pore size and affinity for negatively charged and neutral lipid membranes. From this screen, nine different LDKA variants that have unique activity were selected, sequenced, synthesized, and characterized. Despite the minor sequence changes, each of these peptides has unique functional properties, forming either small or large pores and being selective for either neutral or anionic lipid bilayers. Long-scale, unbiased atomistic molecular dynamics (MD) simulations directly reveal that rather than rigid, well-defined pores, these peptides can form a large repertoire of functional dynamic and heterogeneous aggregates, strongly affected by single mutations. Predicting the propensity to aggregate and assemble in a given environment from sequence alone holds the key to functional prediction of membrane permeabilization.
Graphic Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Recent years have seen a renewed interest in antimicrobial peptides (AMPs) as potential successors to small-molecule antibiotics (Lazzaro et al. 2020; Chen and Lu 2020; Lei et al. 2019; Lee et al. 2019; Biswaro et al. 2018; Magana et al. 2020; Cardoso et al. 2019; Huang 2020). The advantages of AMPs are clear: peptides are getting cheaper to synthesize on an industrial scale (Simon et al. 2014; Mijalis et al. 2017; Cao et al. 2018; Wibowo and Zhao 2019; Hartrampf et al. 2020), offer a near infinite chemical repertoire to target different species and cellular processes (Guha et al. 2019), and can be rapidly screened using high-throughput methodologies (Rathinakumar and Wimley 2008; Carney et al. 2017; Vinogradov et al. 2017; Quartararo et al. 2020). AMPs are also proven, being a ubiquitous part of the innate immune defense of most branches of life. Although some AMPs are toxic to mammalian cells, many of these amphiphilic peptides selectively target and kill bacteria at low micromolar concentrations without harming host cells (Zasloff 1987; Lehrer et al. 1989; Yeaman and Yount 2003; Chen et al. 2020a). Sequence analysis of > 3000 of known AMPs reveal a wide variation in amino acid composition, peptide length, and secondary structure; however, no clear functional motifs associated with antimicrobial activity have been identified to date, impeding rational optimization and de novo design (Mishra and Wang 2012; Chen et al. 2019; Chen and Lu 2020; Ablan et al. 2016). Despite this, there has been considerable progress in rational design and re-engineering of AMPs (Krauson et al. 2012; Wiedman et al. 2017; Chen et al. 2019; Torres et al. 2018; Porto et al. 2018; Li et al. 2018; Haney et al. 2018; Mishra et al. 2019; Hu et al. 2020; Gong et al. 2019; Huang and Charron 2017). These studies have shown that a small number of amino acid mutations in a given sequence can significantly change functional properties such as pore stability (Wiedman et al. 2014), antimicrobial activity (Krauson et al. 2012; Krauson et al. 2015; Libardo et al. 2017; Lakshmaiah Narayana et al. 2020), pore size (Wiedman et al. 2014; Li et al. 2018), membrane selectivity (Krauson et al. 2015), and pH-dependent activity (Wiedman et al. 2015, 2017; Kim et al. 2019; Mihailescu et al. 2019). Peptide length also acts as an important factor. Ulrich et al. reported several rationally designed helical peptides with repeated KIAGKIA motifs with peptide length between 14 and 28 amino acids and showed that the peptide length can affect its ability to penetrate and disrupt cell membranes (Grau-Campistany et al. 2015, 2016).
The ultimate goal is to develop AMPs that can selectively target specific membrane types in order to target pathogens with high potency, without harming host cells. A rational joint in silico/experimental process has great potential for such de novo AMP design (Chen et al. 2020a, b). In the absence of reliable predictive rules for engineering the activity of membrane permeabilizing peptides, a recent breakthrough has been the use of synthetic molecular evolution, which is accomplished with orthogonal screening of a designed, iterative, combinatorial peptide library (Wiedman et al. 2014, 2017; Li et al. 2018; Kim et al. 2019; Krauson, He, and Wimley 2012). Another strategy has been a simulation-guided design approach (Chen et al. 2019), which we have applied to develop a potent pore-forming AMP starting from a membrane-spanning polyleucine helix (Ulmschneider et al. 2014). This new synthetic 14-residue AMP (sequence = GLLDLLKLLLKAAG), called LDKA, consists of only five amino acids (glycine, aspartic acid, lysine, leucine, and alanine) and shares similar sequences to many short antimicrobial peptides (Chen et al. 2019; 2020a). LDKA exhibits low micromolar antimicrobial activity and forms pores in both anionic POPG (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol) and neutral POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) lipid vesicles at low peptide-to-lipid ratios (P:L = 1:1000) (Chen et al. 2019), which is comparable to the potent pore-forming peptide, melittin, and its gain-of-function analogue MelP5 (Wiedman et al. 2014; Krauson et al. 2015).
Here, we explore whether we can rationally develop a general pore-forming peptide into a selective pore-former via a joint molecular dynamics (MD) simulation and an experimental library-screening approach. We start by tuning the hydrophobic moment and charge distribution to introduce preferential binding and pore formation in charged and neutral lipid bilayers and that this preferential binding correlates to activity against human versus bacterial cells. We demonstrate that relatively conservative sequence changes of the LDKA template can indeed modulate the induced preferential pore-forming potency in anionic versus neutral lipid bilayers as well as the size of the pores formed. We further demonstrate that these properties correlate well with antimicrobial activity for specific bacteria and selectivity for bacterial over human red blood cells.
Our results suggest that in vitro activity, lipid selectivity, and aggregation propensities of AMPs depend highly on even the most conservative sequence changes. While the broad underlying properties correlate with simple descriptors that can be directly derived from the peptide sequence (e.g., hydrophobic moment, overall charge, and amphiphilicity), these quantities do not allow us to directly determine which sequence will be selective, or porate membranes at all. The peptides form a large repertoire of functional dynamic and heterogeneous structures in the membrane, and each sequence change can dramatically affect the oligomerization propensity, structure of the aggregates, ability to porate, and selectivity for different membrane compositions so desired for pharmaceutical application. This suggests that ultimately only structure (rather than sequence) based approaches, such as direct pore aggregation and equilibrium simulations, will enable predictive, rather than descriptive de novo AMP design.
Methods
Combinatorial Peptide Library Synthesis
The synthesis of combinatorial peptide library was modified from the method described by Krauson et al. ( 2012). Peptides library synthesis was performed using Tentagel® NH2 macrobeads (280–320 µm bead diameter) particle size (~ 65,550 beads/g) using Fmoc solid-phase peptide synthesis. Each bead only has one peptide sequence. A photolinker is attached between peptide and bead to allow the UV light-induced cleavage of homogenous peptide from bead in each well. The quality of the peptide library was verified by mass spectrometry (e.g., MALDI) and Edman sequencing. After placing one bead in each well of 96-well microplate, the photolinker between peptide and bead was cleaved with 5 h of low-power UV light on dry bead, which was spreading to a dispersed single layer in a glass dish. The peptides were each dissolved in DMSO, quantified by tryptophan absorbance using nanodrop, and stored in − 20 °C freezer.
Bulk Peptides and Chemicals
The selected LDKA-like peptides were synthesized using standard Fmoc chemistry and purified to 98% purity using reverse phase HPLC by GenScript, Inc (Piscataway, NJ, USA). The N-terminus was positively charged amine group and C-terminus is neutral amide group. Peptide purity and identity were confirmed by HPLC and ESI mass spectrometry. The solubility test was performed by GenScript, Inc (Table S1).
Large Unilamellar Vesicle (LUV) Preparation
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (POPG), and hexadecanoyl sphingomyelin (Egg SM; PSM) were purchased from Avanti Polar Lipids. Lipids were dissolved in chloroform, mixed, and dried under nitrogen gas in a glass vial. Any remaining chloroform was removed under vacuum overnight. To make LUVs, lipids were resuspended in 10 mM sodium phosphate buffer (pH 7) with 100 mM potassium chloride. LUVs were generated by extruding the lipid suspension ten times through 0.1 µm nuclepore polycarbonate filters to give LUVs of 100 nm diameter (MacDonald et al. 1991).
ANTS/DPX Leakage Assay
5 mM ANTS and 12.5 mM DPX were entrapped in 0.1 µm diameter extruded vesicles with lipids (Ladokhin et al. 1995; Wimley et al. 1994). Gel filtration chromatography of Sephadex G-100 (GE Healthcare Life Sciences Inc) was used to remove external free ANTS/DPX from LUVs with entrapped contents. LUVs were diluted to 0.5 mM and used to measure the leakage activity by addition of aliquots of LDKA. Leakage was measured after 3 h incubation. 10% Triton was used as the positive control to measure the maximum leakage of the vesicle. Fluorescence emission spectra were recorded using excitation and emission wavelength of 350 nm and 510 nm for ANTS/DPX using a BioTek Synergy H1 Hybrid Multi-Mode Reader.
Macromolecule Release Assay
Several different size dextrans were prepared and labeled with both TAMRA and biotin. Conjugated dextran was entrapped in POPC LUVs as described above (Wiedman et al. 2014). External dextran was removed by incubation with immobilized streptavidin. Streptavidin labeled with an Alexa-488 fluorophore was added during the leakage experiment with the peptide. The sample was incubated for 3 h before measuring Alexa-488 fluorescence. A control without added peptide served as the 0% leakage signal and addition of 0.05% vol. Triton X-100 was used to determine 100% leakage.
Circular Dichroism (CD) Spectroscopy
LDKA solutions (50 μM) in 10 mM phosphate buffer (pH 7.0) were co-incubated with 800 μM POPC:POPG (1:1) and POPC:CHOL (7:3) LUVs in identical buffer (see LUV preparation above). CD spectra were recorded using the synchrotron radiation circular dichroism beamline on ASTRID at Aarhus University. Spectra were recorded from 270 to 170 nm with a step size of Δλ = 0.5 nm, a bandwidth of 0.5 nm, and a dwell time of 2 s. Each spectrum was averaged over 3 repeat scans. The averaged spectra were normalized to molar ellipticity per residue. The raw data were analyzed using DichroWeb <http://dichroweb.cryst.bbk.ac.uk/> (Whitmore and Wallace 2004, 2008; Lobley et al. 2002).
Peptide Thermostability Enables Advanced Sampling at High Temperatures
The LDKA peptide is resistant to thermal denaturation when bound to the membrane and the simulated helicity is comparable to the experiments (Figure S1, Tables S2, and S3) (Ulmschneider and Ulmschneider 2008; Ulmschneider et al. 2010, 2011, 2014; Chen et al. 2014, 2019; Wang et al. 2016a, b). This allows all simulation to be run at 120 °C, increasing pore formation kinetics. We have previously demonstrated that elevating the temperature does not change conformational equilibria or partitioning free energies of helical membrane-active peptides, provided they are stable against thermal denaturation (Figure S1 and Table S2); however, the vast increase in sampling kinetics at high temperatures allows simulation of peptide folding, bilayer partitioning, and pore assembly without the need for advanced sampling techniques that require additional information or may bias the system (Ulmschneider and Ulmschneider 2008; Ulmschneider et al. 2010, 2011, 2014; Chen et al. 2014, 2016, 2019; Wang et al. 2016a, b).
Tryptophan Fluorescent Binding Assay
The protocol was modified from the original methods (Ladokhin et al. 2000; White et al. 1998). LDKA peptides (50 µM) and POPC/POPG (600 µM) were prepared in 10 mM phosphate buffer (pH 7.0). The solutions were incubated and measured after 60 min. Excitation was fixed at 280 nm (slit 9 nm) and emission was collected from 300 to 450 nm (slit 9 nm). The spectra were recorded using Synergy H1 Hybrid Multi-Mode Reader and Cytation™ 5 Cell Imaging Multi-Mode Reader from BioTek and were averaged by 3 scans.
Isothermal Titration Calorimetry (ITC)
ITC measurements with a MicroCal VP-ITC microcalorimeter (Microcal, Inc) were performed to determine the thermodynamic parameters of the peptide–lipid interactions (Breukink et al. 2000; Abraham et al. 2005). The lipid vesicle suspension (16.23 mM) was titrated into peptide solution (100 µM) in the sample cell. All the samples were prepared in 10 mM phosphate buffer (pH 7.0) and had been degassed under vacuum for 30 min. The lipid solution was added in 6 µL aliquots into the reaction cell (volume = 1.46 mL) containing a 50 µM peptide solution with an injection duration of 12 s. The equilibration time between each titration step was 15 min. A first titration of 6 µL was disregarded to ensure that a premixing of both solutions during the equilibration time did not affect the first titration step. The stirring speed of the injection syringe was 307 rounds per minute. The thermodynamic parameters were calculated using the standard ITC software that utilizes stoichiometric model of binding. Membrane partitioning, however, is not stoichiometric (White et al. 1998); consequently, the actual errors in free energy determination might be larger due to cross correlation of "n" and "Kd" fitting parameters.
Bacterial Minimum Inhibitory Concentration (MIC)
Escherichia coli strain ATCC 25922, Staphylococcus aureus strain ATCC 25923, and Pseudomonas aeruginosa strain ATCC PAO1 were used in this study. Overnight cultures were sub-cultured and diluted to an initial bacterial cell density of ~ 3 × 105 colony forming units (CFU) per mL in Lysogeny broth. Cell counts were determined by measuring optical density at 600 nm (OD600), with an optimal sensitivity at OD600 = 0.3–0.6 in a 1 cm path-length cuvette. OD600 = 1 corresponds to 1.5 × 108 CFU/mL for S. aureus, 5 × 108 CFU/mL for E. coli, and 2.04 × 108 CFU/mL for P. aeruginosa. Bacteria were added to peptide (LDKA and indolicidin) dilutions (1.3, 2.0, 2.9, 4.4, 6.6, 9.9, 14.8, 22.2, 33.3, 50.0, and 75.0 µM) and co-incubated at 37 °C. After 12 h of incubation, the optical density of the wells was recorded on a plate reader to determine whether they were sterilized (OD600 < 0.08) or were at stationary phase growth (OD600 > 0.5). Intermediate values, which were rare, were considered positive for growth. Average minimum sterilizing concentrations were calculated from the lowest peptide concentration that sterilized the bacteria in each serial dilution. The samples were done in sextuplet.
Biofilm
The formation of biofilm and quantification was modified from the method described by O'Toole et al. (2011). Escherichia coli strain ATCC 25922, Staphylococcus aureus strain ATCC 25923, and Pseudomonas aeruginosa ATCC PAO1 were overnight cultured to log phase OD600 = 0.3–0.6. The overnight culture 1:100 was diluted into fresh medium for biofilm assays and100 µL dilution was added to each well and cultured at room temperature without shaking. After 48 h of incubation, the media were removed and each well was rinsed with 150 µL water for three times. Elevated concentration of AMPs was prepared and the biofilm was treated using total 150 µL volume in each well and incubated for 3 h at room temperature and the supernatant was removed. Each well was rinsed for three times using water. 150 µL of 1% crystal violet was added in water to each well and the plate was incubated at room temperature for 15 min. The plate was rinsed three times with water to remove the free crystal violet. The plate was turned upside down and dried for overnight. 150 µL of 30% acetic acid was added in water to each well of the plate to solubilize the crystal violet on the cells and the plate was incubated at room temperature for 15 min. 100 µL of the solubilized crystal violet was tranferred to another plate andthe absorbance was quantified at 550 nm using Cytation™ 5 Cell Imaging Multi-Mode Reader from BioTek.
Drug-Resistant Escherichia coli
Escherichia coli strain ATCC 25922 was overnight cultured to log phase OD600 = 0.3–0.6. Initial bacterial cell density was prepared with ~ 3 × 105 CFU/mL in LB broth in 96-well plate. Bacteria were added to serially diluted antibiotics (e.g., ceftazidime, ciprofloxacin, streptomycin, and gentamicin) and co-incubated at 37 °C. After 12 h of incubation, the optical density of each well was recorded on a plate reader to determine whether they were sterilized or were at stationary phase growth. The E. coli which survived at the highest antibiotic concentration (below or near the MIC) was collected and cultured for another generation. This cycle is repeated for 10 times until the E. coli gained resistance (twofold higher MIC than its wildtype) against the antibiotics.
Hemolysis Assay
Fresh human red blood cells were obtained from Interstate Blood Bank, Inc., and thoroughly washed in PBS until the supernatant was clear. hRBC concentration was determined using a standard hemocytometer. In hemolysis assays, serial dilutions of peptide were prepared, followed by the addition of 2 × 108 hRBC/mL. After incubation for 1 h at 37 °C, the cells were centrifuged, and the released hemoglobin was measured by optical absorbance of the heme group (410 nm). Negative control was buffer only (0% lysis), and the positive controls were 20 µM melittin and distilled water (100% lysis). The measurements were made in triplicate.
Molecular Dynamics Simulations and Analysis
Unbiased all-atom MD simulations were performed and analyzed using GROMACS 5.0.4 (Pronk et al. 2013) and Hippo BETA simulation packages <http://www.biowerkzeug.com>, and VMD molecular visualization program (Humphrey et al. 1996) <http://www.ks.uiuc.edu/Research/vmd/>. The pdb structure of extended peptides (GL5KL6G, LDKL, and LDKA) was generated using Hippo BETA (see Tables S1, S2, and S3). These initial structures were relaxed via 200 Monte Carlo steps, with water treated implicitly using a Generalized Born solvent.
After relaxation, the peptides were placed in all-atom peptide/lipid/water systems containing model membranes with 100 mM K and Cl ions using CHARMM-GUI (Lee et al. 2016) <http://www.charmm-gui.org/>. Four helical peptides were initially placed on both interfaces of the bilayer and equilibrated and relaxed for ~ 600 ns. After equilibration, the system was multiplied by 2 × 2 matrix in both the x and y directions and results in a bigger system with total 16 surface-bound peptides on the bilayer, 192 total lipid molecules, 8200 water molecules, and typical box dimensions of 92 × 92 × 67 Å. The simulations were performed at 120 °C to speed up the kinetics, and we confirmed their simulated helicity with the liquid-state circular dichroism spectroscopy (Figure S1 and Table S2). MD simulations were performed with GROMACS 5.0.4 using the CHARMM36 force field (Huang and MacKerell 2013), in conjunction with the TIP3P water model (Jorgensen et al. 1983). Electrostatic interactions were computed using PME, and a cut-off of 10 Å was used for van der Waals interactions. Bonds involving hydrogen atoms were constrained using LINCS. The integration time-step was 2 fs and neighbor lists were updated every five steps. All simulations were performed in the NPT ensemble, without any restraints or biasing potentials. Water and the protein were each coupled separately to a heat bath with a time constant τT = 0.5 ps using velocity rescale temperature coupling. The atmospheric pressure of 1 bar was maintained using weak semi-isotropic pressure coupling with compressibility κz = κxy = 4.6 · 10−5 bar−1 and time constant τP = 1 ps.
Oligomer Population Analysis
In order to reveal the most populated pore assemblies during the simulations, a complete list of all oligomers was constructed for each trajectory frame. An oligomer of order n was considered any set of n peptides that are in mutual contact, defined as a heavy-atom (N, C, O) minimum distance of < 3.5 Å. Frequently, this definition overcounts the oligomeric state due to numerous transient surface-bound (S-state) peptides that are only loosely attached to the transmembrane inserted peptides that make up the core of the oligomer. These S-state peptides frequently change position or drift on and off the stable part of the pore. To focus the analysis on true longer-lived TM pores, a cut-off criterion of 65° was introduced for the tilt angle τ of the peptides. Any peptide with τ ≥ 65° was considered in the S-state and removed from the oligomeric analysis. This strategy greatly reduced the noise in the oligomeric clustering algorithm by focusing on the true longer-lived pore structures. Population plots of the occupation percentage of oligomer n multiplied by its number of peptides n were then constructed. These reveal how much peptide mass was concentrated in which oligomeric state during the simulation time.
Results
Peptide Design
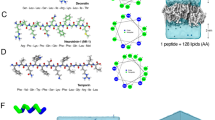
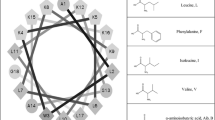
LDKA is a small pore-former in neutral POPC and anionic POPG vesicles and has low micromolar antimicrobial activity against bacteria. The goal of this library is to explore whether simple rearrangements of the LDKA sequence using four amino acids (Leu, Asp, Lys, and Ala) will allow modulation of pore-forming potential, pore size, and targeting of specific membrane compositions. To achieve this, we have designed a combinatorial peptide library containing 2916 LDKA analogues (Fig. 1a, b). The LDKA template sequence was mutated in order to: (i) adjust peptide hydrophobicity, (ii) promote more salt bridge formation between the peptides, (iii) introduce a central proline kink to the structure, and (iv) substitute more positively charged residues on the C-terminus, which is one of the common motifs in the Antimicrobial Peptide Database (APD; http://aps.unmc.edu/AP) (Wang et al. 2016a).

Peptide design and screening of the LDKA library contains 2916 variants. a Amino acid sequence of LDKA and its variants in the combinatorial peptide library. b Helical wheel projection of LDKA showing charged and hydrophobic faces of the helix, which assumes it is a 100% helical configuration. Red and blue symbols present charged residues: negative charged and positive charged, respectively. Proline acts as a kink in the helix, and it is shown as green symbols. Other hydrophobic (leucine) and small (glycine and alanine) residues are indicated as gray symbols. c High-throughput screen of LDKA peptide library induces fluorescent dye (ANTS/DPX) leakage from each POPC (x-axis) and POPG (y-axis) LUVs in 10 mM phosphate buffer at pH 7.0. Fluorescent dye release above 90% from POPC and POPG LUVs are highlighted in green areas, respectively, and the selected peptides were further analyzed their pore sizes using macromolecular fluorescent dye (3-kDa dextran). The selected LDKA library variants induce 3-kDa dextran releasing from each d POPC and e POPG LUVs. 10% Triton was used as a positive control to measure the maximum leakage of the vesicle (100% leakage), and the vesicle incubated in the phosphate buffer was used as a negative control (0% leakage). Hylaseptin-P1 (HSP1), a natural antimicrobial peptide from Polka-dot tree frog and has a similar sequence to LDKA, was used as a reference for comparison (Color figure online)
Peptide hydrophobicity is modulated by interchanging leucine and alanine residues as well as substituting more positive (lysine) and negative charges (aspartic acid) in the sequence. The goal of these mutations is to fine-tune the peptide solubility and membrane partitioning. To further allow for more structural plasticity of the peptide, we introduced a proline near the center of the peptide sequence, which is common in naturally occurring AMPs (Sani et al. 2015; Fernandez et al. 2013). More charged residues (aspartic acid and lysine) were introduced to both facilitate inter-peptide salt bridge formation and strengthen the peptide–peptide interface (Walther and Ulrich 2014), as well as to allow for a more polar central pore enabling larger multimeric channel structures (Chen et al. 2019; Wang et al. 2016a, b). Additional positive charges were introduced at the C-terminus to enhance peptide binding to anionic lipids, which is a common motif in many antimicrobial peptides from natural sources, such as Hylaseptin-P1 (Chen et al. 2020a; Prates et al. 2004), Hylain 2 (Wu et al. 2011), melittin (Dempsey et al. 1991; Rex 2000), and maculatin (Fernandez et al. 2013; Sani et al. 2015).
Membrane-Specific Poration and Pore Size
The potency and membrane selectivity of the 2,916 LDKA library peptides for zwitterionic (POPC) and anionic (POPG) large unilamellar vesicles (LUVs) were evaluated using a high-throughput liposome leakage screen. This approach allows us to detect and quantify the release of small fluorescent dye ANTS (8-aminonaphthalene-1,3,6-trisulfonic acid disodium salt; MW = 427 Da) and its fluorescent quencher DPX (p-xylene-bis-pyridinium bromide; MW = 422 Da) encapsulated in LUVs after addition of the library peptides. Neutral POPC LUVs serve as a simple model for mammalian membranes, while charged POPG LUVs serve as a very simplistic model for bacterial membranes enriched in anionic lipids.
Figure 1c demonstrates the fluorescent dye leakage fraction from both neutral and charged LUVs after the addition of the library peptides. In this study, 11.2% of the LDKA analogues have POPG-favorable selectivity and induce > 50% encapsulated dye leakage from charged POPG LUVs at low peptide concentration (P:L = 1:1000), while 0.4% cause leakage from neutral POPC LUVs only, and 6.6% disrupt both POPC and POPG LUVs. LDKA analogues that induce > 90% dye leakage from POPC and POPG LUVs were screened for their ability to induce leakage of a larger 3-kDa TAMRA-biotin-dextran (TBD) dye (Wiedman et al. 2014). Several LDKA-like peptides form larger pores in POPG vesicles, while the pores induced in POPC vesicles are generally smaller (Fig. 1d, e).
Eight LDKA peptides with different lipid selectivity and pore sizes were selected from the high-throughput screen and sequenced using Edman degradation (Laursen 1971). Table 1 shows that these peptides have 1–4 mutations compared to the LDKA template sequence. The most common mutation is leucine to alanine, occurring 13 times and in a total of 7 of the 8 peptides. Alanine to leucine occurred 6 times in 5 peptides, leucine to aspartic acid occurred 3 times in 3 peptides, and leucine to proline occurred once.
The analysis of selected peptide sequences showed positive-charged lysine is not a favorable substitution in the non-polar face of the LDKA template helix and the C-terminal motif (positions 6, 8, 10, 12, and 13). Instead, hydrophobic leucine and alanine are more preferable. This is in agreement with the evolutionary derivatives of 26-residue melittin, in that the positively charged amino acids (lysine and arginine) are less likely to be favored in the non-polar face (Krauson et al. 2012, 2015).
Other than fixed lysine at positions 7 and 11, no additional lysine residues were observed in the analogues. Additional aspartic acids were observed at position 3 and 5, which is right next to the fixed aspartic acid at position 4 that can further promote salt bridge formation in peptide-peptide interactions. The net charges of these analogues are between + 1 and + 2, and they are consistent with the majority (net charge + 1) of AMPs in the APD (Mishra and Wang 2012; Wang et al. 2016a). This shows that cationic residues can promote peptide binding to anionic bacterial membranes; however, more cationic charges may result in lower hydrophobicity and higher energy barriers to cross the hydrophobic core of membranes. Therefore, a longer peptide length is needed to strengthen the hydrophobicity when the sequence contains more charges. A natural membrane-active peptide, melittin (sequence: GIGAVLKVLTTGLPALISWIKRKRQQ-Amide), is a good example. Although it has four positive charges (-KRKR-) in its C-terminus, longer peptide length (26 amino acids) and the hydrophobic N-terminus (GIGAVLKVL-) make it hydrophobic enough to span cell membranes.
Table 1 reveals that leucine to alanine mutations are generally sufficient to prevent poration in neutral POPC membranes, while the peptides still porate charged POPG membranes, which is similar to the L16G mutation of melittin (Krauson et al. 2015). More specifically, the LDKA analogues that only induce ANTS/DPX leakage from anionic POPG LUVs have 4–5 leucines, while the analogues that can porate both POPC and POPG LUVs have 6–7 leucines in their sequences. The net charge of all LDKA wildtype and analogues is between + 1 and + 2, and we did not observe any anionic peptide, neutral peptide, or peptide that has net charge greater than + 2. This suggests that the membrane selectivity is driven by hydrophobic moment to POPC but including electrostatics on POPG.
Binding to Mixed Membranes
To investigate the root cause of the different leakage preferences of LDKA analogues for POPC and POPG membranes, we studied the binding and secondary structural properties of LDKA analogues using tryptophan fluorescence and circular dichroism (CD) spectroscopy, respectively. Peptide solutions (50 µM peptide concentration) were titrated with POPC and POPG LUVs (between 0 and 5 mM) and the corresponding changes of tryptophan fluorescent spectra were collected, yielding binding free energies and helicities of the peptides, albeit without any structural information on the underlying poration process (Fig. 2).

Tryptophan fluorescence binding assay shows the binding strength of the LDKA analogues (50 µM peptide concentration) with each titrated zwitterionic POPC and anionic POPG LUVs from 0 to 5 mM lipid concentration in 200 mM phosphate buffer at pH 7.4. Different colors of the spectra from dark red to light red indicate the increased peptide-to-lipid (P:L) ratio between P:L = 1:0.5 and P:L = 1:100, and the peptide-only without any lipid (P:L = 1:0) in the buffer is shown as dark gray. The vertical dashed line at 350 nm wavelength represents as a reference, which is the emission wavelength of free tryptophan (Color figure online)
Further studies were performed to answer why some peptides (i.e., 7D12, 7G6, 28H6, 11D12, and 24F1) show selectivity for either membrane type. First, we characterized peptide secondary structural changes and binding to LUVs containing binary mixtures of POPC and POPG lipids. Figure 3 (7D12, 7G6, and 28H6) and Fig. 4 (11D12 and 24F1) show changes in the tryptophan fluorescence and CD spectra for these peptides upon addition of LUVs for whom the ratio of POPG was elevated from 0 to 100% with 20% increments (0, 20, 40, 60, 80, and 100% POPG). These analogues are sensitive to the anionic POPG lipid and have significant structural change with small PG fraction (20% POPG), except 7D12 which is less sensitive to anionic lipid. These membrane-selective peptides only bind to POPG and show little or no binding to POPC, which is consistent to our liposome leakage assay.

Peptide binding and folding of 7G6 (left), 7D12 (middle), and 28H6 (right) onto binary mixtures of charged lipid (POPG) and neutral lipid (POPC) LUVs. a Circular dichroism spectroscopy and b tryptophan fluorescent binding assay of LDKA peptides (50 µM) at P:L = 1:12 in 600 µM POPC/POPG LUVs with different lipid compositions: no lipid (black), 100% POPC (red), 80% POPC and 20% POPG (green), 60% POPC and 40% POPG (purple), 40% POPC and 60% POPG (dark blue), 20% POPC and 80% POPG (orange), and 100% POPG (light blue). The experiments were performed in 10 mM phosphate buffer at pH 7.0. The ellipticity of 222 nm wavelength versus POPG content was shown in the upper right in the circular dichroism panels (part a) (Color figure online)

Peptide binding and folding of 11D12 (left) and 24F1 (right) onto binary mixtures of charged lipid (POPG) and neutral lipid (POPC) LUVs. a Circular dichroism spectroscopy and b tryptophan fluorescent binding assay of LDKA peptides (50 µM) at P:L = 1:12 in 600 µM POPC/POPG LUVs with different lipid compositions: no lipid (black), 100% POPC (red), 80% POPC and 20% POPG (green), 60% POPC and 40% POPG (purple), 40% POPC and 60% POPG (dark blue), 20% POPC and 80% POPG (orange), and 100% POPG (light blue). The experiments were performed in 10 mM phosphate buffer at pH 7.0 (Color figure online)
Peptide solutions (50 µM peptide concentration) were titrated with POPC and POPG LUVs (between 0 and 5 mM) and the corresponding changes of tryptophan fluorescent spectra were collected (Fig. 2). Most of the peptides have tryptophan fluorescence peaks at ~ 348 nm, indicative of monomeric peptides or low multimeric soluble aggregates. Change of the maximum wavelength indicates the tryptophan is either in the aqueous phase or surrounded in the hydrophobic region. It can correlate to the partitioning between water and lipid phases and gives a rough measure of the binding for each peptide with different lipids. This technique can be limited because some peptides (e.g., 7F3 and 28H6) can fold as helix and form multimeric soluble aggregate in the aqueous phase or at higher concentration that bury the tryptophan in the hydrophobic core. 7F3 and 28H6 show maximum fluorescent emission of ~ 331 nm in phosphate buffer, suggesting aggregate formation due to the potential concentration-dependency of the helical fold, and it results in the blue-shifted spectra and smaller spectral widths.
We further performed CD spectroscopy to study the secondary structure of these peptides with each POPC and POPG LUVs at elevated temperature (Figure S1 and Table S2). CD spectroscopy shows that all the toxic peptides are helical structure (54–75% helicity) in the solution, and membrane-selective peptides are mostly coiled structure (22–38% helicity). The secondary structure of the peptides in solution explains why these LDKA analogues have selectivity toward different membrane types and result in different binding free energy. Coiled structure exposes its intramolecular hydrogen bonds to water that make the compound more polar; in opposite, helical structure makes it more hydrophobic. Therefore, toxic peptides have higher helical content and strong interaction with both membrane types. Interestingly, 28H6 only folds as beta-strand structure in POPG LUV, and the temperature at 95 °C can break the intermolecular hydrogen bonds and reverse it to helix. As expected, the membrane-selective peptides fold as helix in POPG LUV and have no response to POPC LUV. Most of the helical structures are highly resistant to thermal denaturation (at 95 °C) when they once fold in the membrane (Figure S1).
The linear regression analysis shows strong correlation between hydrophobic moments, helicity in POPC LUV, and ANTS/DPX leakage fraction from POPC LUV (Fig. 5a). It confirms the interaction between peptide and POPC LUV is strongly dependent on the peptide’s hydrophobic moment; however, it does not correlate to the membrane pore size. Figure 5b shows that the helicity of a peptide is linearly correlated to the hydrophobic moment, which is promoted by the hydrophobicity. We further analyzed the AMPs from APD that have peptide length between 5 and 30 amino acids, which dominate > 50% peptide population (1500 AMPs) in APD. We grouped the AMPs by their peptide length and averaged each of their net charge and hydrophobic moment. It shows that increased hydrophobic moment corresponds to higher net charge (Fig. 6a).

Linear regression analysis of hydrophobic moment with several experimental factors includes a hydrophobic moment and fluorescent dye leakage fraction from POPC vesicle (y = 37.249x – 74.292; R2 = 0.801, where x is hydrophobic moment and y is the ANTS/DPX leakage fraction from POPC vesicle in percentage), and b hydrophobic moment and helical structure in phosphate buffer (y = 14.635x – 0.877; R2 = 0.555) and with POPC vesicle (y = 20.853x − 13.72; R2 = 0.907). Blue dots represent the peptide in POPC vesicle, and orange squares represent the peptide in buffer. x means hydrophobic moment, and y means helicity (%) (Color figure online)

Sequence analysis of antimicrobial peptide database and LDKA peptide library. a Net charge versus hydrophobic moment of AMPs (peptide length between 5 and 30 amino acids) from APD. Each data point is the average in each peptide length. b Net charge versus hydrophobic moment of LDKA peptides and comparison with the AMPs that have the same peptide length (15 amino acids) from APD. Dash gray lines mean the average of net charge and hydrophobic moment of AMPs that have 15 amino acids from APD. c Ratio of ANTS/DPX leakage from each POPC and POPG LUVs (P:L = 1:1000) versus hydrophobic moment of LDKA peptides. The value close to zero suggests the peptide has more selectivity toward POPG. d Hemolytic activity of 75 µM LDKA peptides versus hydrophobic moment. e Hydrophobic moment, f net charge, g hydrophobic moment versus net charge, and h total charged residues of the LDKA library peptides. Colors of data point present different peptides: brown means the AMPs from APD, black means the LDKA analogues, blue means the toxic LDKA peptides, and red means the membrane-selective LDKA peptides (Color figure online)
We analyzed the sequence of LDKA analogues and compared them to the AMPs from APD that have same peptide length to LDKA (Fig. 6b). It showed that the toxic LDKA peptides have higher hydrophobic moment 3.41–4.78 than membrane-selective LDKA peptides with hydrophobic moment 1.92–3.32 (Table 1), which correspond to their specificity toward different membrane types (Fig. 6c) and toxicity to human red blood cell (Fig. 6d). We found hydrophobic moment 3.37 is a cut-off between membrane-selective and toxic peptides in the LDKA library peptides. However, the cut-off may shift in different peptide length and charge distribution (Fig. 6e–h and Table S5); therefore, bigger sample size is necessary to improve this sequence-based prediction of membrane selectivity.
Simulations of Two Similar Sequences
The information gained from the experimental screen is limited in that there is an absence of a nuanced correlation between simple peptide descriptors and selectivity and leakage propensity. Since a multitude of AMP structures can cause membrane permeabilization (Guha et al. 2019), it is critical to identify which overall mechanism applies for the chosen library template. Without knowledge of pore structures in the membrane, it is difficult to explain the role of individual sequence changes on both selectivity and poration ability, rendering the design process blind. Here, computer simulations offer to fill in the missing information. We have demonstrated before for numerous peptide/membrane systems that long-scale equilibrium MD simulations are now able to directly generate aggregate structures in different membrane types from peptide sequence alone (Chen et al. 2020a, b; Wang et al. 2016a, b). The computational effort is—for now—enormous, so only a small subset of the LDKA library was chosen for structural investigation, focusing on 2 peptides that are almost similar but have very different membrane selectivity: 25B2 (sequence: GLDDLAKLLLKLAGW-Amide) and 7D12 (sequence: GLLDDAKLLAKLAGW-Amide). Despite very similar sequences, 25B2 (toxic, i.e., non-selective peptide) causes fluorescent dye leakage from both POPC and POPG LUVs, and 7D12 (membrane-selective peptide) only porates POPG LUVs without disturbing POPC membranes. We sought to observe how these library peptides lyse their target membranes, and how almost identical sequences can have vastly different binding properties.
Ab-initio pore prediction simulations are costly. In addition to limiting ourselves to the two sample sequences, several lessons from our prior simulation studies were incorporated. Firstly, sufficiently hydrophobic peptides all exhibit a surface state in the simulations, so the simulations started from folded helices in that state. Much longer simulations would be needed to directly show the adsorption from an initial solvated state distant from the membrane. Secondly, peptides were placed in both interfaces. This accelerates the prediction of the equilibrium pore states, as peptides usually translocate in the long run, even monomerically for a highly charged AMP like PGLa (Ulmschneider 2017). Attacking the membrane only from one leaflet would be the correct setup but require much longer simulations as the mass has to be first balanced in both leaflets, before equilibrium poration conditions are met.
Both peptides have a net charge + 1 and have the same C-terminal motif (-KLAGW-Amide). The only differences are (i) aspartic acid shifts from position 3 in 25B2 to position 5 in 7D12, and (ii) hydrophobic position 10 where 25B2 is leucine and 7D12 is alanine. A quick analysis shows these simple modifications result in a hydrophobic dipole moment of 4.8 in 25B2 and 1.9 in 7D12.
Mirroring the biophysical experiments, we performed peptide-assembly simulations of 7D12 and 25B2 in both POPC and POPG bilayers. (Fig. 7 and Table S3). Simulations and experiments show that 25B2 results in higher helical content than 7D12 (Tables S2 and S3). Similar to our prior simulations of LDKA, the peptides spontaneously insert and form a large number of heterogeneous oligomeric pore structures. These can range from 3 to 9 peptides, with a core of mainly 4–5 tilted TM inserted peptides, supported by several surface-bound peptides that are more loosely attached. Peptides can span the membrane under negative mismatch, but often span only 2/3rds of the membrane, and they are in partial overlap with other peptides in the opposing leaflet that are similarly only 2/3rds inserted. Full double-stacking can also occur occasionally, in which case peptides are highly tilted, each centered in one leaflet. Peptides align both parallel and anti-parallel at various levels of insertion. The large number of charged sidechains, both cationic and anionic, enables small water-filled bilayer channels with many cross-peptide salt-bridges, pulling in both lipid headgroups and ions. Peptides usually leave and join these small aggregates, resulting in no overall stable structures but rather in a wide variety of different pore assemblies. There is substantial water and ion flux across these, with higher oligomers yielding larger flux. Both cations and anions can translocate across the pores, with a preference for cations in the POPG simulations, presumably due to the more anionic environment of the pore aggregates, where PG headgroups are pulled into the membrane. The heterogenous nature of the pore aggregates indicates a highly dynamic equilibrium which is strongly influenced by individual sequence changes. 7D12 is shown to be selective: It does not insert and form aggregates in POPC, but remains on the surface, indicating that pore aggregates are not stable in this membrane, and the surface state is preferred. The most likely explanation is the strong partitioning penalty for anionic sidechains, as discussed below.

Multi-microsecond molecular dynamics simulations reveal the spontaneous self-assembly of the membrane-selective peptide 7D12 and the toxic peptide 25B2 and their oligomeric structural ensembles in POPC and POPG membranes. a and b Representative pore aggregates (peptides colored blue (N-) to red (C-) terminal, lipid phosphates as orange beads), oligomeric occupation plots (blue = S-state, yellow = single TM, red-dark = higher TM oligomers, overall distribution on right), and cross-membrane water and ion flux caused by the pore assemblies. c and d Isothermal titration calorimetry of the heat release/absorption of the peptide–lipid interactions. The integrated ITC data curve of 7D12 and 25B2 with each POPC and POPG LUVs is also shown. The concentration is fixed at 100 µM with titrated lipid LUVs in 10 mM phosphate buffer at pH 7.0. The ITC data are consistent with the simulation results for the binding selectivity of 7D12 for POPG (Color figure online)
Isothermal Titration Calorimetry
To directly compare the above simulations, we applied isothermal titration calorimetry (ITC) to further characterize their thermodynamic parameters: enthalpy (∆H) and stoichiometry (N). Titration of POPC vesicle into membrane-selective peptide 7D12 (100 µM peptide concentration) results in ∆H = 0.1 kcal/mol (Fig. 7c, d), which is consistent with the tryptophan fluorescent binding assay that it is does not bind strongly to POPC vesicle (Fig. 2). Titrating POPG vesicle into 100 µM 7D12 has significant heat release (∆H = − 3.4 kcal/mol) with the stoichiometry of 11 lipids per peptide (N = 11). On the other hand, the toxic peptide 25B2 with titrated POPC and POPG vesicles shows ∆H = − (4.7–4.9) kcal/mol, and they both have the same stoichiometry of N = 5 lipids per peptide.
The results of MD simulations and ITC are consistent. 7D12 in POPG, and 25B2 in both membrane types assembled channel-like architectures in MD simulations and showed significant heat release in ITC. In contrast, 7D12 in POPC bilayers neither formed any structure, nor induced any heat release/absorption. Thus, there is a remarkable agreement between experiments and simulations. The lower hydrophobic moment of 7D12 appears to explain the less thermostable helical structures than other peptides (Figure S1), and the unfolded structures are more disordered than the helical structure of 25B2 as compared to what we observed in ITC (Fig. 7c, d). Therefore, it suggests hydrophobic moment is a determinant to promote membrane selectivity.
Hemolysis and Antibacterial Activity
To test toxicity of the LDKA analogues against human cells, we performed a hemolysis assay (Fig. 8a). LDKA wildtype is hemolytic at moderate micromolar concentrations with a hemolytic concentration lysing 50% of red blood cells (HC50) of 55.1 µM (Table 2). The peptide-induced POPC LUV leakage is correlated with the hemolytic activity (Fig. 8b, c). The peptides that induce leakage from POPC LUV at low peptide concentration (P:L = 1:1000) are hemolytic (HC50 = 1–57 µM). More specifically, 7F3 (HC50 = 1.1 µM) and 28H6 (HC50 = 1.2 µM) are as powerful as natural toxin melittin and its gain-of-function derivative MelP5 (HC50 = 1–3 µM) (Krauson et al. 2015). All POPG-favorable peptides have no effect to human red blood cell, even at 75 µM peptide concentration.

Selected LDKA analogues and their in vitro hemolytic activity with human red blood cell. a Hemolytic activity with human red blood cell varies in peptide concentration. Linear regression analysis of b hemolysis (at 75 µM peptide concentration) versus ANTS/DPX leakage fraction from POPC LUV (at P:L = 1:1000). y = 0.954x − 4.540 and R2 = 0.866, where x = ANTS/DPX leakage fraction (%) and y = hemolytic activity (%). c Linear regression analysis of hemolytic fraction (at 75 µM peptide concentration) versus peptide helicity in POPC LUV (at P:L = 1:12). y = 1.717x − 52.791; R2 = 0.777, where x = helicity (%) and y = hemolytic activity (%)
The real test is how selectively the selected peptides target and kill various bacteria. The antibacterial activity of LDKA analogues against E. coli, S. aureus, and P. aeruginosa was tested in vitro in nutritionally rich medium. LDKA wildtype inhibits growth of all three bacteria at micromolar peptide concentrations of a similar range to potent AMPs from natural sources. From our screen, most of the POPG-favorable peptides (7G6, 11D12, and 24F1) have antibacterial activity and specificity against E. coli with 19–44 µM minimum inhibitory concentration (MIC) but are not active against other bacterial species: S. aureus and P. aeruginosa (Table 2). The toxic peptides 7F3, 25B2, and 4H9 are effective inhibitors against E. coli and S. aureus, but not P. aeruginosa. The results show that these peptides have specificity toward different bacterial species.
Activity Against Antibiotic-Resistant Strains
Bacteria can mutate and develop resistance against conventional antibiotics (Willyard 2017; Rodríguez-Rojas et al. 2018; Chen and Lu 2020; Maria-Neto et al. 2015), which mostly target specific proteins, ribosomes, or DNA. Antimicrobial peptides exert their effects through a more generalized mechanism: membrane poration. We selected four conventional antibiotics that have different mechanisms to kill bacteria: ceftazidime, ciprofloxacin, streptomycin, and gentamicin. Ceftazidime interferes with bacterial cell wall formation (Richards and Brogden 1985; Yost and Ramphal 1985). Ciprofloxacin inhibits DNA gyrase, type II topoisomerase, and topoisomerase IV to separate bacterial DNA and DNA replication, thus inhibiting cell division (Drlica and Zhao 1997). Streptomycin and gentamicin inhibit protein synthesis at the ribosome (Luzzatto, Apirion, and Schlessinger 1968; Hahn and Sarre 1969). Drug-resistant E. coli strain ATCC 25,922 cultures were grown in the presence of each antibiotic at elevated concentration and the surviving strains were selected to grow for 10 generations, resulting in a 4- to 16-fold resistance to these antibiotics compared to their 1st-generation strain (Fig. 9a).

LDKA analogues against four different drug-resistant E. coli strains and bacterial biofilms. a Minimum inhibitory concentrations (MICs) of four conventional antibiotics (ceftazidime, ciprofloxacin, streptomycin, and gentamicin) were treated with serial E. coli generations. The E. coli that survives below/near the MICs was selected for the next generation. b MICs of LDKA analogues (membrane-selective peptides: 24F1, 11D12, and 7G6; toxin peptides: 25B2, 7F3,, and 4H9) against four different strains of drug-resistant E. coli. Antibacterial activity of LDKA analogues against quantitative biofilm formation on polystyrene 96-well plate for 3 h treatment. Selected analogues were tested with each c Escherichia coli biofilm, d Staphylococcus aureus biofilm, and e Pseudomonas aeruginosa biofilm
LDKA analogues were tested against these four drug-resistant E. coli cultures. Membrane-selective analogues (7G6, 11D12, and 24F1) remain effective and consistently inhibit the growth of ceftazidime-resistant, streptomycin-resistant, and gentamicin-resistant E. coli strains with 27–44 µM peptide concentrations (Fig. 9b). Toxic peptides (4H9, 7F3, and 25B2) are effective against ceftazidime-resistant and gentamicin-resistant E. coli at low peptide concentrations (6–14 µM). Surprisingly, none of the peptides are effective against the ciprofloxacin-resistant E. coli strain.
Activity Against Biofilms
In clinical settings, bacteria are mostly found in biofilms that are the key drivers of infections (Costerton et al. 1999; Hall-Stoodley et al. 2004). We therefore challenged our LDKA analogues against bacterial biofilms, which are generally much more resistant than planktonic equivalents (Stewart and Costerton 2001). The results showed that the selected LDKA analogues (4H9, 7F3, 25B2, 7G6, 11D12, and 24F1) can eliminate ~ 50% of the E. coli biofilm in the presence of 67–150 µM peptide. Only 7F3 is capable of reducing S. aureus biofilms by ~ 50% with 100 µM peptide concentration, and none of the analogues work against P. aeruginosa biofilms (Fig. 9c–e).
Discussion and Conclusions
In this study, we used the leucine-rich membrane-active peptide LDKA as a sequence template in order to test whether the combination of database-guided combinatorial peptide library screening, and direct MD simulation of membrane aggregation, can tune the template to significantly change its secondary structure, potency, and membrane specificity. Similar rational combinatorial design has been used before to develop and tune the activity of other AMPs (Krauson et al. 2012, 2015; Nešuta et al. 2016; Sani et al. 2015). The LDKA library peptides were designed using only four different amino acids (Asp, Lys, Leu, and Ala) in a template sequence of GxxDxxKxxxKxxGW-Amide, where ‘x’ represents one of the four amino acids. Our LDKA analogues reveal that a small number of conservative substitutions (Leu to Ala) in the LDKA sequence can dramatically change the selectivity toward different membrane types (anionic and neutral LUVs), resulting in specificity to different bacteria species and human red blood cells. This is consistent with Krauson et al., who showed a single-residue change of a leucine at position 16 to glycine (L16G) can redirect the general toxicity of melittin towards bacteria only, leaving red blood cells unharmed (Krauson et al. 2015). A similar study introduced charged amino acids in the C-terminal motif of HYL-20 peptide, fine-tuning the selectivity against several bacteria strains with negligible hemolytic activity (Nešuta et al. 2016). The fact that we did not observe this feature in our LDKA peptide library suggests, again, that simple generic structure–function rules are not applicable to membrane-active peptides.
The dependence of the drastic changes in selectivity and leakage propensity upon small sequence changes demonstrates the limitation of overall macroscopic peptide descriptors such as hydrophobic moment and polar angle as design criteria. For example, the hydrophobic moment is somewhat correlated to hemolytic activity (Fig. 6d), and the LDKA peptide library suggests a hydrophobic moment of 3.37 as a cut-off for toxicity toward human red blood cells; however, this does not apply to 26-residue peptide melittin (hydrophobic moment = 3.94) and its membrane-selective analogue (hydrophobic moment = 3.44–3.46). Hydrophobic moment estimates could be limited as they are based on a single, perfectly helical peptides, and do not consider peptide–peptide aggregates and assemblies, as observed in our MD simulations (Eisenberg et al. 1982; Wimley and White 1996; Wimley et al. 1996; White and Wimley 1998, 1999; Reißer et al. 2014). Experimentally, fluorescent dye leakage from POPC vesicle is also a reliable model to predict the hemolytic activity with a linear correlation (R-squared value = 0.87; Fig. 8b). It is similar to structure–function relationship that shows R-squared value 0.78 between helicity in POPC LUV and hemolysis (Fig. 8c).
The absence of strong correlations between macroscopic peptide descriptors and selectivity and leakage propensity means detailed structural models are needed to show what is going on. Advances in computer performance have enabled long-scale (multi-µs), fully atomistic MD simulations to provide that picture (Bennett et al. 2016; Leveritt et al. 2015; Pino-Angeles et al. 2016; Pino-Angeles and Lazaridis 2018; Lipkin et al. 2017; Sepehri et al. 2020; Perrin and Pastor 2016; Perrin et al. 2016; Berkowitz 2016; Tieleman 2017; Wang et al. 2016a, b; Ulmschneider 2017). We have demonstrated before that MD simulations are now able to directly predict aggregate structures in membranes (Chen et al. 2020a, b; Wang et al. 2016a, b; Ulmschneider 2017). The simulations here show the atomic details of how these short membrane-spanning peptides selectively fold, aggregate, and form water pores in specific lipid bilayers (Fig. 7). Key to these simulations is that they are not stuck in initial conditions. The pore aggregates are predicted without any prior information and fluctuate sufficiently to reveal the major structural assemblies that these peptides are expected to populate at either equilibrium, or during a membrane permeabilization event. Structures are highly heterogenous.
The selectivity found in the experiments is reproduced in the simulations: 7D12 only folds and assembles in anionic POPG bilayers. There are no TM pores for 7D12 in POPC, with the peptides staying on the membrane surface and no noticeable water leakage. This demonstrates the extreme effect even tiny sequence changes can have on the pore-forming equilibrium. The experimental match is not perfect though: The ITC data suggest not only no poration, but also no binding to the membrane at all, i.e., not even the initial surface state. Thus while correlated, there remains an unexplained difference between the simulation and experiments. Anionic sidechains are known for their steep insertion penalties, where a single position shift can change the S to TM equilibrium of the whole peptide by 2–3 kcal/mol (Ulmschneider et al. 2018), so the reason for the lack of TM insertion likely is the shift of the 2 Asp residues one position towards the center of the peptide. The anionic membrane allows insertion of both peptides as cationic counter-ions are pulled more deeply into the interface, allowing also for deeper insertion of anionic sidechains. The propensity of a peptide sequence to aggregate and assemble in a given environment depends in a highly complex and non-linear way on the sequence. Therefore, purely sequence-based design approaches are likely not suited for peptides that can form such a large repertoire of functional structures.
How do the designed library peptides perform in killing pathogens? Several minor mutations of LDKA can fine-tune the potency and specificity to kill E. coli, but not harming human red blood cells, S. aureus, and P. aeruginosa. Most of the LDKA peptides are able to inhibit the growth of E. coli with 19–57 µM peptide concentrations, except 28H6 and 7D12 that fail to treat E. coli. 7D12 has the lowest hydrophobic moment 1.92 that may not be strong enough to fold and assemble in the bacterial membrane. Furthermore, the surface protease OmpT on the outer membrane of the E. coli may confer resistance to these leucine-rich peptides by cleaving their peptide bonds and degrading the peptides (McCarter et al. 2004). Our study suggests that small fluorescent dye leakage assays with POPC LUVs are ideal models to quickly screen the AMPs for their hemolytic activity. However, POPG LUVs did not have a good agreement with the antibacterial activity and formation of different pore size does not correlate to the in vitro activity in our study. It suggests that POPG LUVs are not an ideal model to predict antibacterial activities because the lack of bacterial capsule, cell wall, and membrane proteins, and their physical properties are completely different (Fleeman et al. 2020; Maria-Neto et al. 2015). A more realistic vesicle model is needed to better understand how antimicrobial peptides interact with the bacteria and kill the target.
Antibiotic resistance is another serious threat. Half of the LDKA analogues are able to inhibit the S. aureus, but many of them fail to eliminate the super bug, methicillin-resistant Staphylococcus aureus (MRSA) (Table 2). Although our LDKA analogues are less or not effective against S. aureus and P. aeruginosa, many of them are useful to eradicate E. coli with negligible effect to human red blood cell. Remarkably, these LDKA peptides are active against drug-resistant E. coli (Fig. 9a, b) and biofilm (Fig. 9c–e) with micromolar peptide concentrations. It suggests membrane-perforating peptides can be a route to suppress the drug resistance, but more studies of the bacterial surface and a more realistic vesicle models are needed.
Our study is just a first attempt to design membrane-selective peptides via a joint match between experimental library screening and MD simulation poration prediction. The ultimate goal is computer-guided antibiotics design (Torres and de la Fuente-Nunez 2019; Chen et al. 2019; Wu et al. 2019; Yang et al. 2019; Rodnin et al. 2020), developing potent antimicrobial peptides that have effective membrane selectivity to distinguish between human red blood cells and bacterial membranes, and even between different bacterial species. The key advantage of in silico techniques is the vastly larger combinatorial space that can be explored in comparison to experimental library screening. In this study, the large-scale all-atomistic simulation effort was limited to only a few sequences and target membranes due to the heavy resources required. However, the strong correlation to the experimental results demonstrates the maturity of these techniques. With rising computing power in the near future, the library-screening effort will be shifted towards the computational side. This combined experimental/computational approach opens the path to apply these LDKA analogues, and numerous other designed peptides to various different biomedical applications, e.g., antibiotics, biosensors, and drug delivery.
Data Availability
The data that support the findings of this study are available from the corresponding author on reasonable request.
References
Ablan FD, Spaller BL, Abdo KI, Almeida PF (2016) Charge distribution fine-tunes the translocation of alpha-helical amphipathic peptides across membranes. Biophys J 111(8):1738–1749. https://doi.org/10.1016/j.bpj.2016.08.047
Abraham T, Lewis RNAH, Hodges RS, McElhaney RN (2005) Isothermal titration calorimetry studies of the binding of a rationally designed analogue of the antimicrobial peptide gramicidin S to phospholipid bilayer membranes. Biochemistry 44(6):2103–2112. https://doi.org/10.1021/bi048077d
Bennett WF, Hong CK, Wang Y, Tieleman DP (2016) Antimicrobial peptide simulations and the influence of force field on the free energy for pore formation in lipid bilayers. J Chem Theory Comput 12(9):4524–4533. https://doi.org/10.1021/acs.jctc.6b00265
Berkowitz M (2016) Chapter one—a molecular look at membranes. In: Bennett V (ed) Current topics in membranes. Academic Press, New York, pp 1–25
Biswaro LS, da Costa Sousa MG, Rezende TMB, Dias SC, Franco OL (2018) Antimicrobial peptides and nanotechnology, recent advances and challenges. Front Microbiol 9:855. https://doi.org/10.3389/fmicb.2018.00855
Breukink E, Ganz P, de Kruijff B, Seelig J (2000) Binding of Nisin Z to bilayer vesicles as determined with isothermal titration calorimetry. Biochemistry 39(33):10247–10254. https://doi.org/10.1021/bi000915q
Cao J, de la Fuente-Nunez C, Ou RW, Torres MT, Pande SG, Sinskey AJ, Lu TK (2018) Yeast-based synthetic biology platform for antimicrobial peptide production. ACS Synth Biol 7(3):896–902. https://doi.org/10.1021/acssynbio.7b00396
Cardoso MH, Orozco RQ, Rezende SB, Rodrigues G, Oshiro KGN, Candido ES, Franco OL (2019) Computer-aided design of antimicrobial peptides: are we generating effective drug candidates? Front Microbiol 10:3097. https://doi.org/10.3389/fmicb.2019.03097
Carney RP, Thillier Y, Kiss Z, Sahabi A, Heleno Campos JC, Knudson A, Liu R, Olivos D, Saunders M, Tian L, Lam KS (2017) Combinatorial library screening with liposomes for discovery of membrane active peptides. ACS Comb Sci 19(5):299–307. https://doi.org/10.1021/acscombsci.6b00182
Chen CH, Lu TK (2020) Development and challenges of antimicrobial peptides for therapeutic applications. Antibiotics (Basel). https://doi.org/10.3390/antibiotics9010024
Chen CH, Wiedman G, Khan A, Ulmschneider MB (2014) Absorption and folding of melittin onto lipid bilayer membranes via unbiased atomic detail microsecond molecular dynamics simulation. Biochim Biophys Acta 1838(9):2243–2249. https://doi.org/10.1016/j.bbamem.2014.04.012
Chen CH, Khan A, Huang JJ, Ulmschneider MB (2016) Mechanisms of membrane pore formation by amyloidogenic peptides in amyotrophic lateral sclerosis. Chemistry 22(29):9958–9961. https://doi.org/10.1002/chem.201601765
Chen C, Starr CG, Troendle EP, Wiedman G, Wimley WC, Ulmschneider JP, Ulmschneider MB (2019) Simulation-guided rational de novo design of a small pore-forming antimicrobial peptide. J Am Chem Soc. https://doi.org/10.1021/jacs.8b11939
Chen CH, Ulmschneider JP, Ulmschneider MB (2020a) Mechanisms of a small membrane-active antimicrobial peptide from Hyla punctata. Aust J Chem 73(3):236–245
Chen CH, Melo MC, Berglund N, Khan A, de la Fuente C, Ulmschneider JP, Ulmschneider MB (2020b) Understanding and modelling the interactions of peptides with membranes: from partitioning to self-assembly. Curr Opin Struct Biol 61:160–166. https://doi.org/10.1016/j.sbi.2019.12.021
Costerton JW, Stewart PS, Greenberg EP (1999) Bacterial biofilms: a common cause of persistent infections. Science 284(5418):1318–1322
Dempsey CE, Bazzo R, Harvey TS, Syperek I, Boheim G, Campbell ID (1991) Contribution of proline-14 to the structure and actions of melittin. FEBS Lett 281(1–2):240–244
Drlica K, Zhao X (1997) DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol Mol Biol Rev 61(3):377–392
Eisenberg D, Weiss RM, Terwilliger TC (1982) The helical hydrophobic moment: a measure of the amphiphilicity of a helix. Nature 299(5881):371–374
Fernandez DI, Lee TH, Sani MA, Aguilar MI, Separovic F (2013) Proline facilitates membrane insertion of the antimicrobial peptide maculatin 1.1 via surface indentation and subsequent lipid disordering. Biophys J 104(7):1495–1507. https://doi.org/10.1016/j.bpj.2013.01.059
Fleeman RM, Macias LA, Brodbelt JS, Davies BW (2020) Defining principles that influence antimicrobial peptide activity against capsulated Klebsiella pneumoniae. Proc Natl Acad Sci USA 117(44):27620–27626. https://doi.org/10.1073/pnas.2007036117
Gong H, Zhang J, Hu X, Li Z, Fa K, Liu H, Waigh TA, McBain A, Lu JR (2019) Hydrophobic control of the bioactivity and cytotoxicity of de novo-designed antimicrobial peptides. ACS Appl Mater Interfaces 11(38):34609–34620. https://doi.org/10.1021/acsami.9b10028
Grau-Campistany A, Strandberg E, Wadhwani P, Reichert J, Bürck J, Rabanal F, Ulrich AS (2015) Hydrophobic mismatch demonstrated for membranolytic peptides, and their use as molecular rulers to measure bilayer thickness in native cells. Sci Rep 5:9388. https://doi.org/10.1038/srep09388
Grau-Campistany A, Strandberg E, Wadhwani P, Rabanal F, Ulrich AS (2016) Extending the hydrophobic mismatch concept to amphiphilic membranolytic peptides. J Phys Chem Lett 7(7):1116–1120. https://doi.org/10.1021/acs.jpclett.6b00136
Guha S, Ghimire J, Wu E, Wimley WC (2019) Mechanistic landscape of membrane-permeabilizing peptides. Chem Rev. https://doi.org/10.1021/acs.chemrev.8b00520
Hahn FE, Sarre SG (1969) Mechanism of action of gentamicin. J Infect Dis 119(4):364–369
Hall-Stoodley L, Costerton JW, Stoodley P (2004) Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol 2(2):95–108. https://doi.org/10.1038/nrmicro821
Haney EF, Brito-Sanchez Y, Trimble MJ, Mansour SC, Cherkasov A, Hancock REW (2018) Computer-aided discovery of peptides that specifically attack bacterial biofilms. Sci Rep 8(1):1871. https://doi.org/10.1038/s41598-018-19669-4
Hartrampf N, Saebi A, Poskus M, Gates ZP, Callahan AJ, Cowfer AE, Hanna S, Antilla S, Schissel CK, Quartararo AJ, Ye X, Mijalis AJ, Simon MD, Loas A, Liu S, Jessen C, Nielsen TE, Pentelute BL (2020) Synthesis of proteins by automated flow chemistry. Science 368(6494):980–987. https://doi.org/10.1126/science.abb2491
Hu X, Liao M, Gong H, Zhang L, Cox H, Waigh TA, Jian RLu (2020) Recent advances in short peptide self-assembly: from rational design to novel applications. Curr Opin Colloid Interface Sci 45:1–13. https://doi.org/10.1016/j.cocis.2019.08.003
Huang HW (2020) DAPTOMYCIN, its membrane-active mechanism vs. that of other antimicrobial peptides. Biochim Biophys Acta 1862(10):183395. https://doi.org/10.1016/j.bbamem.2020.183395
Huang J, MacKerell AD (2013) CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem 34(25):2135–2145. https://doi.org/10.1002/jcc.23354
Huang HW, Charron NE (2017) Understanding membrane-active antimicrobial peptides. Q Rev Biophys 50:e10. https://doi.org/10.1017/S0033583517000087
Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14(1):33–38. https://doi.org/10.1016/0263-7855(96)00018-5
Jorgensen WL, Chandrasekhar J, Madura JD (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935
Kim SY, Pittman AE, Zapata-Mercado E, King GM, Wimley WC, Hristova K (2019) Mechanism of action of peptides that cause the pH-triggered macromolecular poration of lipid bilayers. J Am Chem Soc 141(16):6706–6718. https://doi.org/10.1021/jacs.9b01970
Krauson AJ, He J, Wimley WC (2012) Gain-of-function analogues of the pore-forming peptide melittin selected by orthogonal high-throughput screening. J Am Chem Soc 134(30):12732–12741. https://doi.org/10.1021/ja3042004
Krauson AJ, Hall OM, Fuselier T, Starr CG, Kauffman WB, Wimley WC (2015) Conformational fine-tuning of pore-forming peptide potency and selectivity. J Am Chem Soc 137(51):16144–16152. https://doi.org/10.1021/jacs.5b10595
Ladokhin AS, Wimley WC, White SH (1995) Leakage of membrane vesicle contents: determination of mechanism using fluorescence requenching. Biophys J 69(5):1964–1971. https://doi.org/10.1016/S0006-3495(95)80066-4
Ladokhin AS, Jayasinghe S, White SH (2000) How to measure and analyze tryptophan fluorescence in membranes properly, and why bother? Anal Biochem 285(2):235–245. https://doi.org/10.1006/abio.2000.4773
Lakshmaiah Narayana J, Mishra B, Lushnikova T, Wu Q, Chhonker YS, Zhang Y, Zarena D, Salnikov ES, Dang X, Wang F, Murphy C, Foster KW, Gorantla S, Bechinger B, Murry DJ, Wang G (2020) Two distinct amphipathic peptide antibiotics with systemic efficacy. Proc Natl Acad Sci USA 117(32):19446–19454. https://doi.org/10.1073/pnas.2005540117
Laursen RA (1971) Solid-phase Edman degradation. An automatic peptide sequencer. Eur J Biochem 20(1):89–102
Lazzaro BP, Zasloff M, Rolff J (2020) Antimicrobial peptides: application informed by evolution. Science. https://doi.org/10.1126/science.aau5480
Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, Wei S, Buckner J, Jeong JC, Qi Y, Jo S, Pande VS, Case DA, Brooks CL, MacKerell AD, Klauda JB, Im W (2016) CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J Chem Theory Comput 12(1):405–413. https://doi.org/10.1021/acs.jctc.5b00935
Lee AC, Harris JL, Khanna KK, Hong JH (2019) A comprehensive review on current advances in peptide drug development and design. Int J Mol Sci. https://doi.org/10.3390/ijms20102383
Lehrer RI, Barton A, Daher KA, Harwig SS, Ganz T, Selsted ME (1989) Interaction of human defensins with Escherichia coli. Mechanism of bactericidal activity. J Clin Invest 84(2):553–561. https://doi.org/10.1172/JCI114198
Lei J, Sun L, Huang S, Zhu C, Li P, He J, Mackey V, Coy DH, He Q (2019) The antimicrobial peptides and their potential clinical applications. Am J Transl Res 11(7):3919–3931
Leveritt JM 3rd, Pino-Angeles A, Lazaridis T (2015) The structure of a melittin-stabilized pore. Biophys J 108(10):2424–2426. https://doi.org/10.1016/j.bpj.2015.04.006
Li S, Kim SY, Pittman AE, King GM, Wimley WC, Hristova K (2018) Potent macromolecule-sized poration of lipid bilayers by the macrolittins, a synthetically evolved family of pore-forming peptides. J Am Chem Soc 140(20):6441–6447. https://doi.org/10.1021/jacs.8b03026
Libardo MDJ, Bahar AA, Ma B, Fu R, McCormick LE, Zhao J, McCallum SA, Nussinov R, Ren D, Angeles-Boza AM, Cotten ML (2017) Nuclease activity gives an edge to host-defense peptide piscidin 3 over piscidin 1, rendering it more effective against persisters and biofilms. FEBS J 284(21):3662–3683. https://doi.org/10.1111/febs.14263
Lipkin R, Pino-Angeles A, Lazaridis T (2017) Transmembrane pore structures of β-hairpin antimicrobial peptides by all-atom simulations. J Phys Chem B 121(39):9126–9140. https://doi.org/10.1021/acs.jpcb.7b06591
Lobley A, Whitmore L, Wallace BA (2002) DICHROWEB: an interactive website for the analysis of protein secondary structure from circular dichroism spectra. Bioinformatics 18(1):211–212. https://doi.org/10.1093/bioinformatics/18.1.211
Luzzatto L, Apirion D, Schlessinger D (1968) Mechanism of action of streptomycin in E. coli: interruption of the ribosome cycle at the initiation of protein synthesis. Proc Natl Acad Sci USA 60(3):873–880
MacDonald RC, MacDonald RI, Menco BP, Takeshita K, Subbarao NK, Hu LR (1991) Small-volume extrusion apparatus for preparation of large, unilamellar vesicles. Biochim Biophys Acta 1061(2):297–303. https://doi.org/10.1016/0005-2736(91)90295-j
Magana M, Pushpanathan M, Santos AL, Leanse L, Fernandez M, Ioannidis A, Giulianotti MA, Apidianakis Y, Bradfute S, Ferguson AL, Cherkasov A, Seleem MN, Pinilla C, de la Fuente-Nunez C, Lazaridis T, Dai T, Houghten RA, Hancock REW, Tegos GP (2020) The value of antimicrobial peptides in the age of resistance. Lancet Infect Dis 20(9):e216–e230. https://doi.org/10.1016/S1473-3099(20)30327-3
Maria-Neto S, de Almeida KC, Macedo MLR, Franco OL (2015) Understanding bacterial resistance to antimicrobial peptides: from the surface to deep inside. Biochim Biophys Acta 1848(11 Pt B):3078–88. https://doi.org/10.1016/j.bbamem.2015.02.017
McCarter JD, Stephens D, Shoemaker K, Rosenberg S, Kirsch JF, Georgiou G (2004) Substrate specificity of the Escherichia coli outer membrane protease OmpT. J Bacteriol 186(17):5919–5925. https://doi.org/10.1128/JB.186.17.5919-5925.2004
Mihailescu M, Sorci M, Seckute J, Silin VI, Hammer J, Perrin BS Jr, Hernandez JI, Smajic N, Shrestha A, Bogardus KA, Greenwood AI, Fu R, Blazyk J, Pastor RW, Nicholson LK, Belfort G, Cotten ML (2019) Structure and function in antimicrobial piscidins: histidine position, directionality of membrane insertion, and pH-dependent permeabilization. J Am Chem Soc 141(25):9837–9853. https://doi.org/10.1021/jacs.9b00440
Mijalis AJ, Thomas DA, Simon MD, Adamo A, Beaumont R, Jensen KF, Pentelute BL (2017) A fully automated flow-based approach for accelerated peptide synthesis. Nat Chem Biol 13(5):464–466. https://doi.org/10.1038/nchembio.2318
Mishra B, Wang G (2012) Ab initio design of potent anti-MRSA peptides based on database filtering technology. J Am Chem Soc 134(30):12426–12429. https://doi.org/10.1021/ja305644e
Mishra B, Lakshmaiah Narayana J, Lushnikova T, Wang X, Wang G (2019) Low cationicity is important for systemic in vivo efficacy of database-derived peptides against drug-resistant Gram-positive pathogens. Proc Natl Acad Sci USA 116(27):13517–13522. https://doi.org/10.1073/pnas.1821410116
Nešuta O, Hexnerová R, Buděšínský M, Slaninová J, Bednárová L, Hadravová R, Straka J, Veverka V, Čeřovský V (2016) Antimicrobial peptide from the wild bee hylaeus signatus venom and its analogues: structure-activity study and synergistic effect with antibiotics. J Nat Prod 79(4):1073–1083. https://doi.org/10.1021/acs.jnatprod.5b01129
O’Toole GA (2011) Microtiter dish biofilm formation assay. J Vis Exp. https://doi.org/10.3791/2437
Perrin BS, Pastor RW (2016) Simulations of membrane-disrupting peptides I: alamethicin pore stability and spontaneous insertion. Biophys J 111(6):1248–1257. https://doi.org/10.1016/j.bpj.2016.08.014
Perrin BS Jr, Fu R, Cotten ML, Pastor RW (2016) Simulations of membrane-disrupting peptides II: AMP piscidin 1 favors surface defects over pores. Biophys J 111(6):1258–1266. https://doi.org/10.1016/j.bpj.2016.08.015
Pino-Angeles A, Leveritt JM III, Themis L (2016) Pore structure and synergy in antimicrobial peptides of the magainin family. PLoS Comput Biol 12(1):e1004570. https://doi.org/10.1371/journal.pcbi.1004570
Pino-Angeles A, Lazaridis T (2018) Effects of peptide charge, orientation, and concentration on melittin transmembrane pores. Biophys J 114(12):2865–2874. https://doi.org/10.1016/j.bpj.2018.05.006
Porto WF, Irazazabal L, Alves ESF, Ribeiro SM, Matos CO, Pires ÁS, Fensterseifer ICM, Miranda VJ, Haney EF, Humblot V, Torres MDT, Hancock REW, Liao LM, Ladram A, Lu TK, de la Fuente-Nunez C, Franco OL (2018) In silico optimization of a guava antimicrobial peptide enables combinatorial exploration for peptide design. Nat Commun 9(1):1490. https://doi.org/10.1038/s41467-018-03746-3
Prates MV, Sforca ML, Regis WC, Leite JR, Silva LP, Pertinhez TA, Araujo AL, Azevedo RB, Spisni A, Bloch C Jr (2004) The NMR-derived solution structure of a new cationic antimicrobial peptide from the skin secretion of the anuran Hyla punctata. J Biol Chem 279(13):13018–13026. https://doi.org/10.1074/jbc.M310838200
Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29(7):845–854. https://doi.org/10.1093/bioinformatics/btt055
Quartararo AJ, Gates ZP, Somsen BA, Hartrampf N, Ye X, Shimada A, Kajihara Y, Ottmann C, Pentelute BL (2020) Ultra-large chemical libraries for the discovery of high-affinity peptide binders. Nat Commun 11(1):3183. https://doi.org/10.1038/s41467-020-16920-3
Rathinakumar R, Wimley WC (2008) Biomolecular engineering by combinatorial design and high-throughput screening: small, soluble peptides that permeabilize membranes. J Am Chem Soc 130(30):9849–9858. https://doi.org/10.1021/ja8017863
Reißer S, Strandberg E, Steinbrecher T, Ulrich AS (2014) 3D hydrophobic moment vectors as a tool to characterize the surface polarity of amphiphilic peptides. Biophys J 106(11):2385–2394. https://doi.org/10.1016/j.bpj.2014.04.020
Rex S (2000) A Pro –> Ala substitution in melittin affects self-association, membrane binding and pore-formation kinetics due to changes in structural and electrostatic properties. Biophys Chem 85(2–3):209–228
Richards DM, Brogden RN (1985) Ceftazidime. A review of its antibacterial activity, pharmacokinetic properties and therapeutic use. Drugs 29(2):105–161
Rodnin MV, Vasquez-Montes V, Nepal B, Ladokhin AS, Lazaridis T (2020) Experimental and computational characterization of oxidized and reduced protegrin pores in lipid bilayers. J Membr Biol 253(3):287–298. https://doi.org/10.1007/s00232-020-00124-3
Rodríguez-Rojas A, Moreno-Morales J, Mason AJ, Rolff J (2018) Cationic antimicrobial peptides do not change recombination frequency in Escherichia coli. Biol Lett. https://doi.org/10.1098/rsbl.2018.0006
Sani MA, Lee TH, Aguilar MI, Separovic F (2015) Proline-15 creates an amphipathic wedge in maculatin 1.1 peptides that drives lipid membrane disruption. Biochim Biophys Acta 1848(10 Pt A):2277–89. https://doi.org/10.1016/j.bbamem.2015.06.013
Sepehri A, PeBenito L, Pino-Angeles A, Lazaridis T (2020) What makes a good pore former: a study of synthetic melittin derivatives. Biophys J 118(8):1901–1913. https://doi.org/10.1016/j.bpj.2020.02.024
Simon MD, Heider PL, Adamo A, Vinogradov AA, Mong SK, Li X, Berger T, Policarpo RL, Zhang C, Zou Y, Liao X, Spokoyny AM, Jensen KF, Pentelute BL (2014) Rapid flow-based peptide synthesis. ChemBioChem 15(5):713–720. https://doi.org/10.1002/cbic.201300796
Stewart PS, Costerton JW (2001) Antibiotic resistance of bacteria in biofilms. Lancet 358(9276):135–138
Tieleman DP (2017) Antimicrobial peptides in the cross hairs of computer simulations. Biophys J 113(1):1–3. https://doi.org/10.1016/j.bpj.2017.06.004
Torres MT, de la Fuente-Nunez C (2019) Toward computer-made artificial antibiotics. Curr Opin Microbiol 51:30–38. https://doi.org/10.1016/j.mib.2019.03.004
Torres MDT, Pedron CN, Higashikuni Y, Kramer RM, Cardoso MH, Oshiro KGN, Franco OL, Silva Junior PI, Silva FD, Oliveira Junior VX, Lu TK, de la Fuente-Nunez C (2018) Structure-function-guided exploration of the antimicrobial peptide polybia-CP identifies activity determinants and generates synthetic therapeutic candidates. Commun Biol 1:221. https://doi.org/10.1038/s42003-018-0224-2
Ulmschneider JP (2017) Charged antimicrobial peptides can translocate across membranes without forming channel-like pores. Biophys J 113(1):73–81. https://doi.org/10.1016/j.bpj.2017.04.056
Ulmschneider MB, Ulmschneider JP (2008) Folding peptides into lipid bilayer membranes. J Chem Theory Comput 4(11):1807–1809. https://doi.org/10.1021/ct800100m
Ulmschneider MB, Doux JP, Killian JA, Smith JC, Ulmschneider JP (2010) Mechanism and kinetics of peptide partitioning into membranes from all-atom simulations of thermostable peptides. J Am Chem Soc 132(10):3452–3460. https://doi.org/10.1021/ja909347x
Ulmschneider JP, Smith JC, White SH, Ulmschneider MB (2011) In silico partitioning and transmembrane insertion of hydrophobic peptides under equilibrium conditions. J Am Chem Soc 133(39):15487–15495. https://doi.org/10.1021/ja204042f
Ulmschneider MB, Ulmschneider JP, Schiller N, Wallace BA, von Heijne G, White SH (2014) Spontaneous transmembrane helix insertion thermodynamically mimics translocon-guided insertion. Nat Commun 5:4863. https://doi.org/10.1038/ncomms5863
Ulmschneider JP, Smith JC, White SH, Ulmschneider MB (2018) The importance of the membrane interface as the reference state for membrane protein stability. Biochim Biophys Acta (BBA) 1860(12):2539–2548. https://doi.org/10.1016/j.bbamem.2018.09.012
Vinogradov AA, Gates ZP, Zhang C, Quartararo AJ, Halloran KH, Pentelute BL (2017) Library design-facilitated high-throughput sequencing of synthetic peptide libraries. ACS Comb Sci 19(11):694–701. https://doi.org/10.1021/acscombsci.7b00109
Walther TH, Ulrich AS (2014) Transmembrane helix assembly and the role of salt bridges. Curr Opin Struct Biol 27:63–68. https://doi.org/10.1016/j.sbi.2014.05.003
Wang G, Li X, Wang Z (2016) APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res 44(D1):D1087–D1093. https://doi.org/10.1093/nar/gkv1278
Wang Y, Chen CH, Hu D, Ulmschneider MB, Ulmschneider JP (2016) Spontaneous formation of structurally diverse membrane channel architectures from a single antimicrobial peptide. Nat Commun 7:13535. https://doi.org/10.1038/ncomms13535
White SH, Wimley WC (1998) Hydrophobic interactions of peptides with membrane interfaces. Biochim Biophys Acta 1376(3):339–352
White SH, Wimley WC (1999) Membrane protein folding and stability: physical principles. Annu Rev Biophys Biomol Struct 28:319–365. https://doi.org/10.1146/annurev.biophys.28.1.319
White SH, Wimley WC, Ladokhin AS, Hristova K (1998) Protein folding in membranes: determining energetics of peptide-bilayer interactions. Methods Enzymol 295:62–87. https://doi.org/10.1016/s0076-6879(98)95035-2
Whitmore L, Wallace BA (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res 32(Web Server issue):W668–W673. https://doi.org/10.1093/nar/gkh371
Whitmore L, Wallace BA (2008) Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89(5):392–400. https://doi.org/10.1002/bip.20853
Wibowo D, Zhao CX (2019) Recent achievements and perspectives for large-scale recombinant production of antimicrobial peptides. Appl Microbiol Biotechnol 103(2):659–671. https://doi.org/10.1007/s00253-018-9524-1
Wiedman G, Wimley WC, Hristova K (2015) Testing the limits of rational design by engineering pH sensitivity into membrane-active peptides. Biochim Biophys Acta 1848(4):951–957. https://doi.org/10.1016/j.bbamem.2014.12.023
Wiedman G, Fuselier T, He J, Searson PC, Hristova K, Wimley WC (2014) Highly efficient macromolecule-sized poration of lipid bilayers by a synthetically evolved peptide. J Am Chem Soc 136(12):4724–4731. https://doi.org/10.1021/ja500462s
Wiedman G, Kim SY, Zapata-Mercado E, Wimley WC, Hristova K (2017) pH-triggered, macromolecule-sized poration of lipid bilayers by synthetically evolved peptides. J Am Chem Soc 139(2):937–945. https://doi.org/10.1021/jacs.6b11447
Willyard C (2017) The drug-resistant bacteria that pose the greatest health threats. Nature 543(7643):15. https://doi.org/10.1038/nature.2017.21550
Wimley WC, White SH (1996) Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol 3(10):842–848
Wimley WC, Selsted ME, White SH (1994) Interactions between human defensins and lipid bilayers: evidence for formation of multimeric pores. Protein Sci 3(9):1362–1373. https://doi.org/10.1002/pro.5560030902
Wimley WC, Creamer TP, White SH (1996) Solvation energies of amino acid side chains and backbone in a family of host-guest pentapeptides. Biochemistry 35(16):5109–5124. https://doi.org/10.1021/bi9600153
Wu J, Liu H, Yang H, Yu H, You D, Ma Y, Ye H, Lai R (2011) Proteomic analysis of skin defensive factors of tree frog Hyla simplex. J Proteome Res 10(9):4230–4240. https://doi.org/10.1021/pr200393t
Wu Z, Kan SBJ, Lewis RD, Wittmann BJ, Arnold FH (2019) Machine learning-assisted directed protein evolution with combinatorial libraries. Proc Natl Acad Sci USA 116(18):8852–8858. https://doi.org/10.1073/pnas.1901979116
Yang JH, Wright SN, Hamblin M, McCloskey D, Alcantar MA, Schrübbers L, Lopatkin AJ, Satish S, Nili A, Palsson BO, Walker GC, Collins JJ (2019) A white-box machine learning approach for revealing antibiotic mechanisms of action. Cell 177(6):1649-1661.e9. https://doi.org/10.1016/j.cell.2019.04.016
Yeaman MR, Yount NY (2003) Mechanisms of antimicrobial peptide action and resistance. Pharmacol Rev 55(1):27–55. https://doi.org/10.1124/pr.55.1.2
Yost RL, Ramphal R (1985) Ceftazidime review. Drug Intell Clin Pharm 19(7–8):509–513
Zasloff M (1987) Magainins, a class of antimicrobial peptides from Xenopus skin: isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc Natl Acad Sci USA 84(15):5449–5453
Acknowledgements
We thank the Karlsruhe Institute of Technology (KIT) ANKA synchrotron CD beamline staff for support and beamtime. We thank Jochen Bürck at KIT for valuable discussion and technical support for ANKA synchrotron CD beamline. We thank Guangshun Wang at the University of Nebraska Medical Center for providing the raw data of the antimicrobial peptide database. We thank Katherine Tripp at the Center for Molecular Biophysics, Johns Hopkins University for helping the experimental setup for isothermal titration calorimeter. We thank Jodie Franklin at the Synthesis and Sequencing Facility at the Johns Hopkins University School of Medicine for sequencing the LDKA peptides. We thank Kalina Hristova and Honggang Cui at Johns Hopkins University and Gregory Wiedman at Seton Hall University and Bonnie Wallace at Birkbeck, University of London and Matthew Upton at University of Plymouth and Alexey Ladokhin at University of Kansas for valuable discussions. Simulation resources were supported by the MARCC supercomputer facility at Johns Hopkins University.
Author information
Authors and Affiliations
Contributions
CHC, JPU, MBU, and WCW designed the research. CHC performed most of the experiments and MD simulations. CGS performed hemolysis assay. CGS and CHC performed in vitro bacterial minimum inhibitory concentration assay and bacterial biofilm assay. SG and CHC performed the minimum inhibitory concentration assay with drug-resistant E. coli. JPU, CHC, and MBU analyzed the simulations. CHC, JPU, MBU, and WCW wrote the paper, with input from the other authors.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file1 (DOCX 1131 KB)
Circular dichroism spectra and analysis, peptide solubility, molecular dynamic simulations setup and analysis, thermodynamic parameters from isothermal titration calorimetry, and analysis of the peptide library are presented in the Supporting information
Rights and permissions
About this article

Cite this article
Chen, C.H., Starr, C.G., Guha, S. et al. Tuning of a Membrane-Perforating Antimicrobial Peptide to Selectively Target Membranes of Different Lipid Composition. J Membrane Biol 254, 75–96 (2021). https://doi.org/10.1007/s00232-021-00174-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00232-021-00174-1