
Abstract
Major intrinsic protein (MIP) superfamily contains water-transporting AQP1 and glycerol-specific GlpF belonging to two major phylogenetic groups, namely aquaporins (AQPs) and aquaglyceroporins (AQGPs). MIP channels have six transmembrane helices (TM1 to TM6) and two half-helices (LB and LE). LE region contributes two residues to the aromatic/arginine (Ar/R) selectivity filter (SF) within the MIP channel. Bioinformatics analyses have shown that all AQGPs have an intra-helical salt-bridge (IHSB) in LE half-helix and all AQGPs and majority of AQPs have helix destabilizing Gly and/or Pro in the same region. In this paper, we mutated in silico the acidic and basic residues in GlpF to Ser and introduced salt-bridge interaction in AQP1 LE half-helix by substituting Ser residues at the equivalent positions with acidic and basic residues. We investigated the influence of IHSB in LE half-helix on the transport properties of GlpF and AQP1 mutant channels using molecular dynamics simulations. With IHSB abolished in LE half-helix, the GlpF mutant exhibited a significantly reduced water transport. In contrast, the introduction of IHSB in the two AQP1 mutants has increased water transport. Absence of salt-bridge in LE half-helix alters the SF geometry and results in a higher energy barrier for the solutes in the Ar/R selectivity filter. Presence/absence of IHSB in LE half-helix influences the channel transport properties and it is evident especially for the AQGPs. By modulating its helical flexibility, LE half-helix can perhaps play a regulatory role in transport either on its own or in conjunction with other extracellular regions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Water-transporting aquaporin (AQP) AQP1 and glycerol-specific aquaglyceroporin (AQGP) GlpF are among the most characterized channels belonging to the superfamily of major intrinsic proteins (MIPs) (Fu et al. 2000; Sui et al. 2001). Members of MIP channels transport neutral solutes across the membranes and they are present in all three domains of life. Bioinformatics studies of several genome sequences identified more than 1500 MIPs from bacteria, archaea, fungi, plants, non-mammalian metazoans, and mammalians (Abascal et al. 2014; Gupta and Sankararamakrishnan 2009; Gupta et al. 2012; Verma et al. 2014). Phylogenetic analysis of non-plant MIP sequences identified two major families, AQPs and AQGPs. Structures of more than 20 MIPs from different species have been determined at atomic resolution. All MIP channels adopt a unique hourglass helical fold which is characterized by six transmembrane helical segments (TM1 to TM6) and two half-helices (LB and LE). The half-helices meet at the middle of the membrane to form the seventh pseudo-transmembrane helix (Verma et al. 2015a) (Fig. 1A). There are two narrow constriction regions within the channel, one formed by the two conserved NPA motifs and the other by four residues from TM2, TM5 and the LE segment of the channel called aromatic/arginine (Ar/R) selectivity filter (SF).

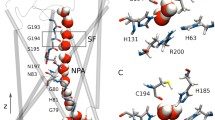
A Structure of GlpF (PDB ID: 1FX8) (Fu et al. 2000) shown as a representative structure of a MIP channel. The LB and LE half-helices are displayed in blue and orange respectively. Rest of the structure is shown in gray color. The acidic and basic residues at LE.54 and LE.58 are shown in magenta and green color respectively. LE half-helices from each of the four monomers are superposed on the same region from the starting structure (green) for B GlpF-mut, C AQP1-mut1 and D AQP1-mut2. The side-chains of acidic and basic residues mutated at positions LE.54 and LE.58 are shown in stick representation in AQP1 mutant structures. The Ser residues substituted at the same positions are also displayed in stick representation in GlpF-mut. Loop B half-helical region from all four monomers is superposed on the starting structure for E GlpF-mut, F AQP1-mut1 and G AQP1-mut2. UCSF Chimera (Pettersen et al. 2004) was used in all molecular plots. (Color figure online)
Experimental and computer simulation studies have investigated the mechanism of water and/or glycerol transport and proton blockage in AQP1 and GlpF channels. Molecular dynamics simulations of human AQP1 suggested that the Ar/R selectivity filter is likely to act as a proton filter (de Groot and Grubmuller 2001). Computer simulation studies have also explored the factors in pure water channels like AQP1 that are responsible for excluding glycerol (Wang et al. 2005). Calculation of potential of mean force profiles using steered molecular dynamics of two MIP channels from the same species, Escherichia coli, revealed a large energy barrier due to steric constraints for the water channel AqpZ at the Ar/R constriction compared to the glycerol-specific GlpF. The question of permeation of gases such as CO2, O2 and NH3 was investigated by calculating the free energy profiles along AQP1 and GlpF channels and the energy barriers were compared with those calculated across the lipid bilayers (Hub and de Groot 2006, 2008). Both hydrophobicity and the size of the molecule seem to be the major factor for the solute permeability and the Ar/R selectivity filter is the site of major energy barrier in AQP1 channels. The role of arginine and its interactions with the solute is suggested to be especially important in the solute selectivity. Apart from the well studied AQP1 and GlpF, molecular dynamics simulations have also been performed on other mammalian aquaporins including AQP0 (Jensen et al. 2008), AQP4 (Alberga et al. 2014), and AQP5 (Janosi and Ceccarelli 2013). MIP members from other species such as E. coli AqpZ (Xin et al. 2011), yeast Aqy1 (Fischer et al. 2009), plant SoPIP2;1 (Khandelia et al. 2009), PfAQP from Plasmodium falciparum (Aponte-Santamaria et al. 2010), and archaeal AqpM (Araya-Secchi et al. 2011) have also been investigated using simulation techniques. Several of these simulation studies compared the rate of water transport with AQP1 and solute selectivity with both AQP1 and GlpF. The possible role played by the respective Ar/R SF regions has been explored in these studies.
In addition to the selectivity and transport of solutes, gating and regulation of channel transport are important features in a channel’s function. Experimental and computational studies have examined the gating mechanisms of several aquaporin channels. MIP channel’s regulation has been investigated in AQP1, GlpF, and other members of MIP superfamily. In a recent study, our group has shown that almost 99% of MIP channels in a dataset of 1468 MIPs contain at least one helix destabilizing residues (Gly and/or Pro) in the functionally important LE half-helix (Verma et al. 2015b). This LE region contributes two out of four residues to the formation of Ar/R selectivity filter. In the same bioinformatics study, it was also shown that an intra-helical salt-bridge (IHSB) is present in almost all the AQGP members but this stabilizing interaction is conspicuously absent in non-AQGPs. The acidic residue participating in IHSB is located immediately after the conserved NPA motif. The acidic and basic residues are separated by four positions. The helical geometry in the LE half-helix will position the oppositely charged residues one above the other that will enable them to participate in a salt-bridge interaction. The same helical turn also contains at least one helix destabilizing residue in AQGPs. Subsequent molecular dynamics simulations demonstrated that the LE half-helix is more stable in GlpF compared to AQP1 (Verma et al. 2015b). Potential of mean force profiles (PMF) were calculated for AQP1 wild-type and its mutant in which the helix destabilizing Gly in the LE half-helical region was substituted by helix-promoting Ala. PMF profiles supported by number of water permeation events indicated a correlation between LE half-helix stability and water transport in AQP1 (Verma et al. 2015b). In this paper, we have carried out MD simulations of three mutant MIP channels. In GlpF mutant, the salt-bridge forming acidic and basic residues were substituted by Ser. In AQP1 mutants, Ser residues in the equivalent positions were substituted by charged residues so that they can form a salt-bridge. The two AQP1 mutants differed in the initial structures, one with the IHSB and the other without the IHSB to begin with. Our simulation results demonstrate that the presence/absence of IHSB in LE half-helix can be correlated with the rate of water transport across the channels.
Materials and Methods
Initial Structures
The structures of GlpF (PDB ID: 1FX8) (Fu et al. 2000) and AQP1 (PDB ID: 1J4N) (Sui et al. 2001) were downloaded from the Protein Data Bank (PDB) (Berman et al. 2000). In the generic structure-based numbering scheme proposed for the MIP superfamily (Verma et al. 2015a), the acidic and basic residues of LE half-helix in GlpF correspond to D207LE.54 and K211LE.58 respectively (Fig. 1A). According to this numbering scheme, the reference position for the LE half-helix is the highly conserved Asn of NPA motif and is given the number 50. The other residue numbers in loop LE are relative to this number. The numbers 207 and 211 are the actual residue numbers of GlpF protein. This numbering scheme is similar to that proposed for G protein-coupled receptors (Ballesteros and Weinstein 1995), transporters (Beuming et al. 2006), and SWEET family members (Gupta and Sankararamakrishnan 2018) and this will be followed throughout this paper. The residues S198LE.54 and S202LE.58 in AQP1 channel are at the equivalent positions of acidic and basic residues of GlpF. The residue numbers of conserved arginine that is part of the Ar/R selectivity filter are R206LE.53 and R197LE.53 in GlpF and AQP1 respectively. The following mutants were generated in silico from GlpF and AQP1 wild-type channels using UCSF Chimera program (Pettersen et al. 2004). In GlpF, the residues D207LE.54 and K211LE.58 were both replaced by Ser and henceforth this mutant will be referred as GlpF-mut. In AQP1, S198LE.54 and S202LE.58 residues were substituted by Asp and Arg respectively. Two AQP1 mutant models were considered for further studies. In AQP1-mut1, the side-chain conformations of Asp and Arg were chosen such a way that they were away from each other. In AQP1-mut2, the Asp and Arg residues formed salt-bridge interaction in the initial structure. All the three mutant models were the starting structures for our molecular dynamics (MD) simulations.
Simulation Protocol
We followed the same MD protocol which we used in our earlier simulations of GlpF and AQP1 wild-type channels (Verma et al. 2015b). The channel-bilayer complex system was constructed using a pre-equilibrated, pre-hydrated POPE bilayer containing 340 lipids (Tieleman and Berendsen 1998). Since GlpF and AQP1 are observed as tetramers under physiological conditions, the channel tetramers were generated and inserted in the lipid patch as per the protocol explained in Kandt et al. (Kandt et al. 2007). The systems were simulated using the GROMACS suite of software (Version 4.5.5) (Hess et al. 2008). We have used Berger’s united atom force-filed for lipids (Berger et al. 1997) compatible with the all atom OPLS force-field for the proteins (Jorgensen et al. 1996) (Dr. Bert de Groot, Personal Communication). TIP3P water model (Jorgensen et al. 1983) was used. The systems were neutralized by adding chloride counter ions and the ion parameters were used from OPLS-AA force-field. The size of the systems varied from 75,300 to 79,500 atoms. All of them were energy minimized before equilibration.
Equilibration of each system consisted of two stages. In the first stage, while positions of all the lipid atoms were restrained, a harmonic force constant of 10,000 kJ/mol/nm2 was applied on all protein atoms to restrain them. The positional restraints on lipid atoms were gradually removed in steps of 100 ps and NVT ensemble was used at this stage. The second stage used NPT ensemble. At this stage, the system was then equilibrated for a period of 1 ns with restraints only on the protein atoms. A further 10 ns equilibration was carried out without any restraints on any atoms. Membrane normal (Z-axis) and membrane plane (X − Y) were coupled separately using semi-isotropic coupling system.
Particle Mesh Ewald (PME) method (Essmann et al. 1995) was used to calculate long-range electrostatic calculations. A cut-off of 12 Ǻ was employed to determine VDW interactions. Constant temperature (T = 310 K) and constant pressure (P = 1 bar) were maintained using Nose–Hoover coupling algorithm (Cheng and Merz 1996) and Parrinello–Rahman algorithm (Parrinello and Rahman 1981) respectively. Production runs consisted of 100 ns simulation and periodic boundary conditions were applied in all three directions.
Analysis of Water Transport Properties
Water permeation events, potential of mean force (PMF) profiles and pore radius profiles were calculated for all four monomers of the GlpF and AQP1 mutant channels as described in the previous studies (Aponte-Santamaria et al. 2010; Verma et al. 2015b). The results obtained for the mutant channels were compared with that of wild-type. Average number of water molecules at each position along the pore axis (Z-axis) was considered for evaluating the PMF profiles for the entire 100 ns MD simulation. For each monomer of GlpF/AQP1 wild-type or mutant, the PMF profile was calculated using Eq. (1).
Where kB, T and < ni(z) > are Boltzmann constant, temperature and average number of water molecules at a particular pore coordinate along the pore axis respectively. NPA motif was considered as a reference point with Z = 0 Ǻ and the PMF profiles were calculated from − 30 to + 30 Ǻ with positive and negative sides towards cytoplasmic and extracellular regions respectively. This method of calculating PMF underestimates the free energy of bulk water at the entrance and exit of the channels of each monomer and hence a trapezoidal correction (Aponte-Santamaria et al. 2010) was applied to account for this. The computed correction values were: 6.43, 6.30 and 6.46 for GlpF-mut, AQP1-mut1 and AQP1-mut2 respectively. Bert de Groot and his co-workers have demonstrated that the PMF profiles calculated using this approach shows an excellent agreement with the free energy profiles obtained using umbrella simulations (Hub and de Groot 2006, 2008).
To find out the number of water permeation events in each monomer, a cylinder of 18 Ǻ length and 5 Ǻ radius was aligned along the pore axis with the centroid of NPA motif lying at the position Z = 0 Ǻ. The cylindrical axis varied from Z = + 13 Ǻ (extracellular side) and Z = − 5 Ǻ (cytoplasmic side). A permeation event is counted when a water molecule completely passes through the cylinder from one end to the other end entering from any direction. The pore radius profile for each monomer was calculated using the program HOLE (Smart et al. 1996). The procedure to calculate the pore radius profile is described in detail in our earlier studies (Bansal and Sankararamakrishnan 2007).
Results
Transport properties such as number of water permeable events and channel characteristics including potential of mean force (PMF) profiles and pore radius profiles were determined from the simulations for the mutant channels. Results obtained for the mutant channels were compared with that found in the corresponding wild-type channels. Initial results of wild-type simulations have been reported in our earlier studies by Verma et al. (Verma et al. 2015b). We performed additional analyses on wild-type channels wherever necessary and the results have been compared with those obtained for the mutant channels.
Loop E Half-Helix Stability in GlpF and AQP1 Channels: Wild-Type Versus Mutants
We have calculated the root mean-square deviation (RMSD) of GlpF and AQP1 mutant channels and compared them with that of wild-type simulations (Verma et al. 2015b). The RMSD varied from 0.7 to 1.8 Ǻ for wild-type and mutant channels of GlpF and AQP1 (Table 1). The stability of all six transmembrane helices and the two half-helices was examined and compared between the wild-type and mutant channels. DSSP (Kabsch and Sander 1983) plots were used for this purpose and all six transmembrane helices were found to be stable in both wild-type and mutant channels (data not shown). However, we have found marked difference in the stability of loop E half-helix. In almost all four monomers of GlpF-mut, the loop E half-helix was moderately or severely disrupted indicating that the absence of IHSB and the presence of two helix destabilizing residues played a major role in LE half-helix stability and disrupted the helical character of this region (Fig. S1 in the Supporting Material). When an IHSB was introduced through acidic and basic residues at LE.54 and LE.58 positions in the loop E half-helix of AQP1, the two mutant channels displayed relatively more stable helical character compared to the wild-type especially in the middle region of the helix (Fig. S1). However, it should be noted that LE half-helix in all four monomers exhibited certain amount of flexibility in both wild-type and mutant channels of GlpF and AQP1. This is further confirmed by comparing the stable helical nature of the other half-helix from loop LB (Fig. S2 in the Supporting Material). DSSP plots of LB half-helix for all the wild-type and mutant AQP1 and GlpF channels revealed that the helical segment of LB half-helix is extremely stable. The initial and final structures of LE and LB half-helices from all monomers are superposed and are shown in Fig. 1. Hence, with both GlpF and AQP1 possessing helix-breaking residue(s), presence or absence of IHSB appears to decrease or further increase the flexibility of LE half-helix.
Comparison of Water Transport in GlpF Wild-Type and Mutant Channels
We then wanted to find out whether the loss of IHSB or its introduction in loop E half-helix has any implication on the transport properties of the GlpF and AQP1 channels. We calculated the number of water permeation events for all four monomers of mutant channels and compared with that found in the wild-type channels. In all the four monomers, the number of water permeation events in GlpF mutant is 40 compared to 180 in GlpF wild-type (Table 1). Thus the absence of IHSB seems to have consequence in the transport properties of GlpF channel.
Among all the monomers, M1 showed the maximum number of water permeation events in GlpF wild-type and exhibited a dramatic difference with the GlpF-mut (Table 1). If we consider the water permeation events for the individual monomers, the M2 and M3 monomers of GlpF and its mutant showed almost negligible water transport. The M4 monomer resulted in relatively less number of water permeation. A small decrease was observed in the number of water molecules transported in GlpF-mut compared to GlpF wild-type. Hence, we can conclude that M2, M3, and M4 monomers were in fully or partially closed conformation in both GlpF wild-type and the mutant irrespective of the presence or absence of IHSB in LE half-helix. Compared to the wild-type, only the M1 monomer displayed a drastic decline in the number of water molecules transported in GlpF mutant. Only two permeation events were noted in the M1 monomer of GlpF-mut compared to 127 for the same monomer in the wild-type (Table 1). Hence, we focused our attention on the M1 monomer and further examined whether the IHSB in LE half-helix had any role in the steep decline in the number of water molecules transported in the M1 monomer of GlpF-mut.
To further investigate the factors behind the reduced water transport in M1 monomer of GlpF-mut, we calculated potential of mean force (PMF) profiles and pore radius profiles for both the GlpF wild-type and GlpF-mut channels as described in the Methods section. PMF profiles and pore radius profiles of M1 monomers are shown in Fig. 2, and for the other three monomers they are provided as Supplementary Material (Fig. S3 to S5 in the Supporting Material). There is little difference in the pore radius profiles of M2 and M3 monomers of GlpF wild-type and the mutant (Fig. S3B and S4B). In both M2 and M3 monomers of GlpF wild-type and mutant, the narrowest part is at the Ar/R SF region with the average pore radius less than 0.7 Ǻ, i.e., half of the radius of water molecule. In the case of M4 monomer, the pore radius profiles display a similar behavior in both the wild-type and mutant GlpF in the two constriction regions and the pore is slightly wider than that observed for M2 and M3 monomers with minimum pore radius slightly less than 1.0 Ǻ (Fig. S5B). However, significant differences are observed in the pore radius profiles of wild-type and mutant M1 monomers (Fig. 2B). Unlike the other three monomers, the pore in M1 monomer is wider in GlpF wild-type and the average minimum pore radius in the narrow constriction region is about 1.2 Ǻ. However, the pore becomes narrower in the Ar/R SF region for the GlpF mutant and at the narrowest part, the average radius is close to 0.7 Ǻ.

A Potential of mean force (PMF) profiles and B average pore radius profiles shown for the M1 monomer of GlpF wild-type (black) and GlpF-mut (red) simulations. PMF profiles of M1 monomer were calculated as described in the Methods section. Pore radius profiles of M1 monomer were calculated using the HOLE (Smart et al. 1996) program and the average pore radius profiles for the M1 monomers of GlpF wild-type and mutant simulations are plotted. The two constriction regions due to NPA motifs and Ar/R SF region are shown in yellow and green bands respectively. MD trajectories of minimum pore radius calculated in the vicinity of Ar/R SF region (4 Ǻ above and 4 Ǻ below the Ar/R SF) shown for the M1 monomer of C GlpF wild-type and D GlpF-mut simulations. (Color figure online)
There is a clear correlation between the pattern of pore radius profiles and the PMF profiles. Comparison of PMF profiles demonstrates that there is a steep energy barrier for the M2 and M3 monomers of GlpF wild-type near the Ar/R SF region (Fig. S3A and S4A). For the mutant GlpF, the energy barrier of about 25 kJ/mol still exists for both M2 and M3 monomers at the same region. With average minimum pore radius of less than 0.7 Ǻ, this observation explains why there is absolutely no water transport in these two monomers in both GlpF wild-type and GlpF-mut (Table 1). M4 monomer also displays an energy barrier in Ar/R SF region with much reduced height of 15 kJ/mol and a wider pore for both GlpF wild-type and GlpF-mut (Fig. S5A). M4 monomer transports reasonably higher number of water molecules in both GlpF simulations. M1 monomer displays the greatest difference in the number of water molecules transported between the GlpF wild-type and mutant. Comparison of PMF profiles reveals that the M1 monomer of GlpF-mut has an increased energy barrier of nearly 25 kJ/mol at the Ar/R selectivity filter region and this is about 10 kJ/mol higher than that observed for the M1 monomer of wild-type GlpF (Fig. 2A). Correspondingly, the average pore radius profile also exhibits a smaller radius (0.7 Ǻ in GlpF-mut versus ~ 1.2 Ǻ in GlpF wild-type) for the same monomer of GlpF-mut in the selectivity filter region (Fig. 2B).
The average pore radius profiles sometimes may not reflect the actual picture and the values may be the result of extreme fluctuations in the values. To rule out such possibility, we also determined the minimum pore radius in the vicinity of Ar/R selectivity filter and we considered the region 4 Ǻ above and 4 Ǻ below the Ar/R constriction. We found out the minimum pore radius in this region for each MD simulated structure and we have plotted this value as the function of time for all four monomers from both GlpF wild-type and GlpF-mut. MD trajectories of minimum pore radius values for M2, M3 and M4 monomers are shown for both the wild-type (Fig. S3C, S4C, and S5C) and the mutant GlpF (Fig. S3D, S4D, and S5D). The pore radius remained below 0.7 Ǻ for M2 and M3 monomers throughout the 100 ns simulations for both the GlpF wild-type and the mutant with the average values ranging between 0.58 and 0.66 Ǻ. As far as M4 monomer is concerned, the minimum pore radius was above 1.0 Ǻ during the initial 10–15 ns and then the pore in the Ar/R region became narrower with the radius well below 1.0 Ǻ for the remaining period in both GlpF wild-type and GlpF-mut simulations (Fig. S5C and S5D). In the case of M1 monomer, MD trajectory of minimum pore radius in Ar/R SF region clearly shows that the GlpF wild-type is open during most of the simulation time with the minimum pore radius value between 1.0 and 1.4 Ǻ (Fig. 2C). However, the GlpF-mut channel displays a reduced pore radius, below 1.0 Ǻ, for the entire duration of the simulation (Fig. 2D) clearly indicating that the channel is occluded and unable to pass the water molecules beyond the Ar/R SF region.
We have plotted the Ar/R selectivity filter residues from MD simulated structures and superposed them on the initial structure for the M1 monomer (Fig. 3). We have provided the same data for the other three monomers in the Supplementary Material (Fig. S6 to S8). For M2 and M3 monomers, change in the side-chain conformation of Trp (W482.49) blocked the channel in both GlpF wild-type and GlpF-mut resulting in reduced pore radius and high energy barrier in the respective PMF profiles (Fig. S6 and S7). A similar scenario is observed for M4 monomer of GlpF wild-type and mutant (Fig. S8). Major differences are observed between the wild-type and mutant simulations of GlpF in terms of Ar/R SF geometry and their side-chain orientations (Fig. 3) for the M1 monomer. The side-chains of Ar/R SF in GlpF wild-type adopted conformations close to the crystal structure most of the time thus having a wider pore radius in this region (Fig. 3A). However in GlpF-mut simulation, the side-chains of Ar/R SF residues assumed conformations that occluded the channel (Fig. 3B). This is especially true with the two bulky aromatic residues Trp (W482.49) and Phe (F200LE.47). The orientation and conformation of longer Arg side-chain (R206LE.53) also differed from the GlpF wild-type population. This analysis has demonstrated that the absence of IHSB and the presence of two helix destabilizing residues in LE half-helix has changed the orientation of side-chains at the Ar/R SF region and altered the SF geometry. This has marked effect on the transport properties of M1 monomer of GlpF-mut.

Superposition of the four residues (W482.49, G1915.57, F200LE.47, and R206LE.53) that form the Ar/R SF from 10 MD simulated structures plotted for the M1 monomer of A GlpF wild-type and B GlpF-mut. The MD simulated structures were saved at the interval of 10 ns. It is clear that the side-chains of residues forming Ar/R SF adopt different conformations in GlpF-mut and block the channel. The initial structure is shown in green color. (Color figure online)
Thus the small pore radius in the narrow Ar/R constriction region and the high energy barrier as observed in the PMF profiles explain why there is almost no water transport in M2 and M3 monomers and only moderate water transport in M4 monomers of both GlpF wild-type and GlpF-mut. However, the differences in the transport properties seen in the M1 of GlpF wild-type and GlpF-mut raised the question whether the absence of IHSB in LE half-helix of Glp-mut have any implications in the transport properties. Our analysis clearly demonstrated that the absence of IHSB in GlpF-mut and the presence of Gly and Pro in the LE half-helical region resulted in more flexibility of this helical region which in turn enabled different orientations of Ar/R SF residues. This has affected the positioning of the SF residues which in turn has altered the Ar/R SF geometry. With a clear indication of the role of IHSB in LE half-helix in the transport properties of GlpF, we have carried out a similar analysis for the two AQP1 mutants to find out if the introduction of IHSB in LE half-helix of AQP1 mutants has any implication on the transport properties of AQP1 and its mutants.
Comparison of Water Transport in AQP1 Wild-Type and Mutant Channels
Water permeation events for the two AQP1 mutant channels were compared with the AQP1 wild-type channel to find out if the presence of IHSB in LE half-helix has any influence on the water transport. While only 41 waters permeated AQP1 wild-type channel (Verma et al. 2015b), more than 100 water permeation events were observed in each of the AQP1 mutant channels (Table 1). This appears to reiterate the relationship between efficient water transport in GlpF and AQP1 with the presence/absence of IHSB in LE half-helix. When individual monomers were analyzed, M1 and M3 transported only handful of water molecules in both AQP1 wild-type and mutant simulations. M4 showed an increase in the number of water permeation events in AQP1-mut1 simulation in comparison with the AQP1 wild-type channel. However for the same monomer, AQP1-mut2 displayed a decrease in the number of water molecules transported relative to the wild-type. In the case of M2 monomer, the wild-type AQP1 transported only 14 waters and there is more than 3–5 fold increase in AQP1 mutants. The two AQP1 mutants, AQP1-mut1 and AQP1-mut2, respectively transported 50 and 78 water molecules through M2 monomer (Table 1).
We then calculated PMF profiles and average pore radius profiles of all four monomers and compared between wild-type and mutant AQP1 channels. PMF and average pore radius profiles of M2 monomers are presented in Fig. 4 and for the other three monomers these profiles are given in the Supplementary Material (Fig. S9 to S11). As we discussed in the case of GlpF, the reason for poor water transport in M1, M3 and M4 monomers can be generally explained by the narrow pore region and higher energy barrier in the Ar/R SF region in both AQP1 wild-type and the mutants. In the case of M2 monomer, pore radius profiles clearly demonstrated that the average radius of the wild-type AQP1 is smaller in the Ar/R constriction region (Fig. 4B). The mutant channels were relatively wider in comparison to the wild-type channel in the same region. PMF profiles between the wild-type and two mutant AQP1 channels revealed that introduction of IHSB helped to reduce the energy barrier near the Ar/R SF region in the two mutant channels (Fig. 4A). Both the pore radius profiles and PMF profiles gave indication why the M2 monomer of AQP1 wild-type showed poor water conductivity compared to the same monomer in AQP1-mut1 and AQP1-mut2.

A PMF profiles and B average pore radius profiles shown for the M2 monomer of AQP1 wild-type (black) and AQP1-mut1 (red) and AQP1-mut2 (blue) simulations. PMF profiles of M2 monomer were calculated as described in the Methods section. Pore radius profiles of M2 monomer were calculated using the HOLE program (34) and the average pore radius profiles for the M2 monomers of AQP1 wild-type and mutant simulations are plotted. The two constriction regions due to NPA motifs and Ar/R SF region are shown in yellow and green bands respectively. MD trajectories of minimum pore radius calculated in the vicinity of Ar/R SF region (4 Ǻ above and 4 Ǻ below the Ar/R SF) shown for the M2 monomer of C AQP1 wild-type, D AQP1-mut1 and E AQP1-mut2 simulations. (Color figure online)
MD trajectories of minimum pore radius values in the vicinity of Ar/R SF were plotted for all the monomers. M1, M3, and M4 monomers clearly reveal that AQP1 wild-type was closed and had an average pore radius between 0.67 and 0.78 Ǻ for all the three monomers (Table 1 and Fig. S9C, S10C, and S11C) in the Ar/R SF region. AQP1-mut1 channel was also very narrow in the same region as far as M1 and M3 monomers are concerned with an average pore radius 0.52 and 0.71 Ǻ respectively (Fig. S9D and S10D). The pore in the M4 monomer was wider for the first 40 ns of the simulation (average pore radius: 1.1 Ǻ; Table 1) in AQP1-mut1 (Fig. S11D) and this monomer also showed significant water transport. In the case of AQP1-mut2, the M4 monomer is mostly closed during the entire course of simulation with an average pore radius of 0.66 Ǻ and it also showed much reduced water transport compared to AQP1-mut1 (Table 1 and Fig. S11E). The M1 and M3 monomers of AQP-mut2 had somewhat wider pore in the SF region (Fig. S9E and S10E), but still the average pore radius was less than that of a water molecule (1.4 Ǻ). As a result, they also didn’t seem to transport large number of water molecules (Table 1). Residues from other regions also might have played a role in blocking the water molecules from entering inside the channel. We also plotted the minimum pore radius within ± 4 Ǻ of Ar/R SF region as the function of time for the M2 monomer of all three AQP1 channels (Fig. 4C–E). For AQP1 wild-type, the average pore radius value in this region is 0.78 Ǻ, well below 1.4 Ǻ, for almost all the time indicating that water molecules would have found it difficult to cross this region (Fig. 4C; Table 1). The M2 monomer of two mutant channels, AQP1-mut1 and AQP1-mut2 with IHSB interaction shows that the same constriction is wider in this region (Fig. 4D, E) with an average value of 1.2 and 1.61 Ǻ respectively (Table 1). Thus the Ar/R constriction in the mutants could allow water molecules to pass through and this also explains why the M2 monomer of AQP-mut2 with much wider pore among all the AQP1 channels showed higher water transport compared to AQP-mut1.
Molecular plots of Ar/R SF residues from MD simulated structures were superposed on the initial structure in all monomers. This representation for M2 monomer is shown in Fig. 5 for all three AQP1 channels. For all other monomers, see Fig. S12 to S14 in the Supporting Material. The side-chains of Ar/R SF residues adopted conformations different from the initial structure and they pointed towards the center of the channel in M1 and M3 monomers of both AQP1 wild-type and the mutants (Fig. S12 and S13). Such an arrangement occluded the channel and as a result negligible amount of water molecules were transported across the channel in these two monomers in AQP1 wild-type and both the AQP1 mutants (Table 1). The same is true for the M4 monomers of AQP1 wild-type (Fig. S14A) and AQP1-mut2 (Fig. S14C). However, the side-chains of residues in Ar/R SF in M4 monomer of AQP1-mut1 had conformations that were either close to the starting structure (F582.49) or they pointed away from the channel (R197LE.53). As a result, the Ar/R SF region was wider (Fig. S14B) and the channel could transport significant number of water molecules in the M4 monomer of AQP1-mut1 (Table 1). The most significant difference between the wild-type and the two mutants is observed in M2 monomers. While both AQP1 mutants permeated many water molecules across the channel, the AQP1 wild-type could transport only a fraction of water molecules (Table 1). Molecular plots of Ar/R SF residues in M2 monomer indicate that the side-chains, especially F582.49 and R197LE.53, deviated from the initial structure and blocked the channel in AQP1 wild-type (Fig. 5A). In both the mutants, the Ar/R SF side-chains stayed close to the starting structure or pointed away from the channel as in the case of R197LE.53 (Fig. 5B, C). This caused the pore in this region to become wider and enabled many water molecules to be transported.

Superposition of the four residues (F582.49, H1825.57, C191LE.47 and R197LE.53) that form the Ar/R SF from 10 MD simulated structures plotted for A AQP1 wild-type, B AQP1-mut1 and C AQP1-mut2 simulations. The MD simulated structures were saved at the interval of 10 ns. It is clear that the side-chain of F582.49 forming Ar/R SF adopt conformations in AQP1 wild-type that block the channel. The initial structure is shown in green color. (Color figure online)
AQP1 has only Gly in its LE half-helical region while GlpF has Gly and Pro in the same region. Hence, when the salt-bridge interaction was removed by substituting the acidic and basic residues, the LE half-helix in GlpF-mut exhibited greater flexibility and this changed the orientation of Ar/R SF residues. Similarly when we simulated a MIP channel from the pathogenic fungi Coccidioides posadasii, the LE half-helix of this channel, with no IHSB and with both Gly and Pro, was destabilized to a great extent (Verma et al. 2015b). When only Gly is present, the destabilization of LE half-helix of AQP1 wild-type is moderate. However, when IHSB is present, the LE half-helix exhibits some flexibility in GlpF wild-type and the two AQP1 mutants (Fig. S1). However, the differences observed in water transport between GlpF wild-type and GlpF-mut were dramatic and the loosened LE half-helix in GlpF-mut altered the SF geometry and changed the characteristics of the Ar/R SF region (Fig. 3). A change in the positioning and orientation of SF residues will change the physical and chemical nature of this constriction region. Hence, the drop in the number of water permeation events in GlpF-mut and AQP1 wild-type can be clearly attributed to the absence of IHSB in the LE half-helix. These results indicate that in GlpF and in AQP1, the presence/absence of IHSB in the LE half-helix can directly influence the transport properties.
Discussion
The significance of Ar/R SF constriction region in the selectivity and transport of solutes in MIP channels has been reported in several experimental and simulation studies (Beitz et al. 2006; Hub and de Groot 2006; Mitani-Ueno et al. 2011; Wang et al. 2005). Importance of the SF Arg in selectivity or regulation of the transport has been demonstrated. Azad et al. (Azad et al. 2012) showed the importance of the position occupied by Arg in the Ar/R SF by mutation experiments in two MIP subgroups of Arabidopsis thaliana. In another simulation study, permeabilities of different solutes were compared in Plasmodium falciparum aquaglyceroporin, human AQP1 and E. coli GlpF. Free energy profiles showed that the Arg in the Ar/R SF has a crucial role in regulating the transport of solutes (Aponte-Santamaria et al. 2010). Beitz et al. (Beitz et al. 2006) have shown that replacement of the highly conserved Arg residue at the SF enabled protons and other charged species to permeate.
Experimental and computational studies indicate that both the Ar/R SF geometry and the chemical nature of this constriction are important determinants of selectivity and transport in MIP channels. Bioinformatics analysis of MIP sequences from our laboratory and the current MD simulation studies have highlighted the potential of another important factor in LE half-helix, i.e., the presence or absence of intra-helical salt-bridge in this helical region (Verma et al. 2015b). To the best of our knowledge, previous simulation studies of AQP1, GlpF, and other MIP channels from different laboratories (Aponte-Santamaria et al. 2010; Araya-Secchi et al. 2011; de Groot and Grubmuller 2001; Ho et al. 2009; Hub and de Groot 2008; Janosi and Ceccarelli 2013; Jensen et al. 2008; Tajkhorshid et al. 2002; Tornroth-Horsefield et al. 2006) did not discuss the possibility of influence of this stabilizing intra-helical interaction of LE half-helix and its functional implication. This could be perhaps due to the fact that side-chains of both the acidic and basic residues forming the IHSB interaction are not directly facing the channel. Since the important SF Arg residue is present within the LE half-helical region, any disturbance of helical character is likely to alter the physical and chemical nature of this constriction region. This is abundantly clear in the MD simulation of GlpF-mut. By substituting the acidic and basic residues with Ser, we made sure that the stabilizing IHSB interaction is absent in the mutant GlpF channel. When this mutant was simulated for a period of 100 ns, the LE half-helix of all the four monomers showed further loosening of the helical region in LE half-helix. We attributed the decrease in water permeation in Glp-mut to the increasingly flexible helical character in loop E region. Thus the absence of IHSB in GlpF-mut and the presence of two helix destabilizing residues, Gly and Pro, contributed to a more flexible LE half-helix which in turn changed the Ar/R SF geometry causing disruption in water transport. With smaller pore radius, we speculate that glycerol transport also will be seriously affected in GlpF mutant.
In the current study, IHSB was introduced in silico in the same position as observed in GlpF by substituting the Ser residues in AQP1 water channel. MD simulations of two such AQP1 mutants for a period of 100 ns were performed for each mutant. In general, the average minimum pore radius in the Ar/R constriction is higher in APQ1 mutants than that observed for the wild-type. With one helix destabilizing residue (G200LE.56), unwinding of LE half-helix is still observed to some extent even with IHSB in AQP1 mutants. GlpF-mut or the fungal MIP from C. posadasii (Verma et al. 2015b), both of which have Gly and Pro in the LE half-helical region with no stabilizing IHSB, exhibit more pronounced unwinding of LE half-helical region. When G200LE.56 was substituted by Ala in AQP1, LE half-helix was extremely stable (Verma et al. 2015b) and resulted in higher water conductivity. In our previous bioinformatics studies, among the 391 AQGP sequences analyzed, 99.4% of them contain intra-helical salt-bridge and 99.2% have Gly and/or Pro in the LE half-helical region. In the case of AQPs, no intra-helical salt-bridge is observed in any of the sequences. However, the helical stability of AQPs is modulated by having zero (8%), one (32%), or two (60%) helix destabilizing residues. An example in this case is water-transporting AqpZ and AQP1. AqpZ does not have a helix destabilizing residue in LE half-helical region in the equivalent positions of LE.56 and LE.57, while AQP1 has only Gly (G200LE.56). MD simulation results point out that AqpZ is a much more efficient water channel than AQP1 (Hashido et al. 2005; Hashido et al. 2007). This corroborated with our earlier (Verma et al. 2015b) and current results using AQP1 mutants. In our earlier simulation studies of AQP1 channel, when we mutated the Gly (G200LE.56) in the LE half-helical region to helix-promoting Ala, we found out that LE half-helix became much more stable and overall resulted in higher water permeability (Verma et al. 2015b).
Results of all the above studies and the current simulations point to a possible role of IHSB in LE half-helix in influencing the transport properties of MIP channels. Loop E half-helix, especially those with the helix destabilizing residues Gly and Pro and without the stabilizing IHSB, can exert great influence on MIP channel transport. Experimental and computational studies have mostly focused on Ar/R SF residues, other residues in the vicinity of Ar/R SF and loop A connecting TM1 and TM2 in the gating of MIP channels (Alberga et al. 2014; Gonen et al. 2004; Gonen and Walz 2006; Hu et al. 2012; Jiang et al. 2006; Li et al. 2011; Xin et al. 2011). Depending upon the environment and signals which can be species-specific, these loop regions, LE half-helix and other specific positions can act either independently or in conjunction with each other. Introduction of salt-bridge in the LE half-helix of AQP1 by site-directed mutagenesis and subsequent functional studies will help to elucidate the functional role of IHSB in MIP channels. Mutation of helix destabilizing Gly and/or Pro to Ala coupled with the introduction IHSB in LE half-helix can unequivocally establish the biological significance of helix stability in LE half-helix of MIP family channels.
Conclusions
In this paper, we have investigated the functional significance of highly conserved intra-helical salt-bridge in the loop E half-helix of AQGPs using molecular dynamics studies of in silico mutants. The side-chains of IHSB-forming residues do not directly point towards the channel interior. Since LE half-helix possesses the important Arg residue of Ar/R SF, we hypothesized that the presence or absence of IHSB in the LE half-helix is likely to influence the transport properties of MIP channels. The current MD simulations of AQP1 and GlpF mutant channels with and without IHSB interactions clearly demonstrated that the stabilizing salt-bridge interaction along with the helix destabilizing residues in the same helical region can modulate the helical character of LE half-helix. This in turn can alter the selectivity filter geometry affecting the physical and chemical nature of the narrow Ar/R SF region. The current study has demonstrated that the presence or absence of IHSB and/or Gly/Pro residues both have direct consequences for the transport properties of the channel. With several positions in the extracellular region of MIP channels implicated in the regulation of the channel transport, it is possible that the two factors in LE half-helix (presence or absence of IHSB and presence of Gly and/or Pro) can act as additional means that can be involved independently or in concurrence with other extracellular residues in regulating the MIP channel transport. Mutational studies in LE half-helical region that promote or destabilize the helical character can be performed to the test the hypothesis proposed in this study.
References
Abascal F, Irisarri I, Zardoya R (2014) Diversity and evolution of membrane intrinsic proteins. Biochim Biophys Acta 1840:1468–1481
Alberga D, Nicolotti O, Lattanzi G, Nicchia GP, Frigeri A, Pisani F, Benfenati V, Mangiatordi GF (2014) A new gating site in human aquaporin-4: insights from molecular dynamics simulations. Biochim Biophys Acta 1838:3052–3060
Aponte-Santamaria C, Hub JS, de Groot BL (2010) Dynamics and energetics of solute permeation through the Plasmodium falciparum aquaglyceroporin. Phys Chem Chem Phys 12:10246–10254
Araya-Secchi R, Garate JA, Holmes DS, Perez-Acle T (2011) Molecular dynamics study of the archaeal aquaporin AqpM. BMC Genom 12(4):S8
Azad AK, Yoshikawa N, Ishikawa T, Sawa Y, Shibata H (2012) Substitution of a single amino acid residue in the aromatic/arginine selectivity filter alters the transport profiles of tonoplast aquaporin homologs. Biochim Biophys Acta 1818:1–11
Ballesteros JA, Weinstein H (1995) Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci 25:366–428
Bansal A, Sankararamakrishnan R (2007) Homology modeling of major intrinsic proteins in rice, maize and Arabidopsis: comparative analysis of transmembrane helix association and aromatic/arginine selectivity filters. BMC Struct Biol 7:Art. No. 27
Beitz E, Wu B, Holm LM, Schultz JE, Zeuthen T (2006) Point mutations in the aromatic/arginine region in aquaporin 1 allow passage of urea, glycerol, ammonia, and protons. Proc Natl Acad Sci USA 103:269–274
Berger O, Edholm O, Jahnig F (1997) Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys J 72:2002–2013
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The Protein Data Bank. Nucleic Acids Res 28:235–242
Beuming T, Shi L, Javitch JA, Weinstein H (2006) A comprehensive structure-based alignment of prokaryotic and eukaryotic neurotransmitter/Na + symporters (NSS) aids in the use of the LeuT structure to probe NSS structure and function. Mol Pharmacol 70:1630–1642
Cheng AL, Merz KM (1996) Application of the Nose-Hoover chain algorithm to the study of protein dynamics. J Phys Chem 100:1927–1937
de Groot BL, Grubmuller H (2001) Water permeation across biological membranes: mechanism and dynamics of aquaporin-1 and GlpF. Science 294:2353–2357
Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG (1995) A smooth particle mesh Ewald method. J Chem Phys 103:8577–8593
Fischer G, Kosinska-Eriksson U, Aponte-Santamaria C, Palmgren M, Geijer C, Hedfalk K, Hohmann S, de Groot BL, Neutze R, Lindkvist-Petersson K (2009) Crystal structure of a yeast aquaporin at 1.15 angstrom reveals a novel gating mechanism. PLoS Biol 7(6):e1000130
Fu D, Libson A, Miercke LJ, Weitzman C, Nollert P, Krucinski J, Stroud RM (2000) Structure of a glycerol-conducting channel and the basis for its selectivity. Science 290:481–486
Gonen T, Sliz P, Kistler J, Cheng Y, Walz T (2004) Aquaporin-0 membrane junctions reveal the structure of a closed water pore. Nature 429:193–197
Gonen T, Walz T (2006) The structure of aquaporins. Quart Rev Biophys 39:361–396
Gupta A, Sankararamakrishnan R (2018) dbSWEET: an integrated resource for SWEET superfamily to understand, analyze and predict the function of sugar transporters in prokaryotes and eukaryotes. J Mol Biol 430:2203–2211
Gupta AB, Sankararamakrishnan R (2009) Genome-wide analysis of major intrinsic proteins in the tree plant Populus trichocarpa: Characterization of XIP subfamily of aquaporins from evolutionary perspective. BMC Plant Biol 9:134
Gupta AB, Verma RK, Agarwal V, Vajpai M, Bansal V, Sankararamakrishnan R (2012) MIPModDB: a central resource for the superfamily of major intrinsic proteins. Nucleic Acids Res 40:D362–D369
Hashido M, Ikeguchi M, Kidera A (2005) Comparative simulations of aquaporin family: AQP1, AQPZ, AQP0 and GlpF. FEBS Lett 579:5549–5552
Hashido M, Kidera A, Ikeguchi M (2007) Water transport in aquaporins: osmatic permeability matrix analysis of molecular dynamics simulations. Biophys J 93:373–385
Hess B, Kutzner C, van der Spoel D, Lindahl E (2008) GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theoy Comput 4:435–447
Ho JD, Yeh R, Sandstrom A, Chorny I, Harries WEC, Robbins RA, Miercke LJW, Stroud RM (2009) Crystal structure of human aquaporin 4 at 1.8 angstrom and its mechansim of conductance. Proc Natl Acad Sci USA 106:7437–7442
Hu GD, Chen LY, Wang J (2012) Insights into the mechanisms of the selectivity filter of Escherichia coli aquaporin Z. J Mol Model 18:3731–3741
Hub JS, de Groot BL (2006) Does CO2 permeate through aquaporin-1? Biophys J 91:842–848
Hub JS, de Groot BL (2008) Mechanism of selectivity in aquaporins and aquaglyceroporins. Proc Natl Acad Sci USA 105:1198–1203
Janosi L, Ceccarelli M (2013) The gating mechanism of the human aquaporin 5 revealed by molecular dynamics simulations. PLoS ONE 8:e59897
Jensen MO, Dror RO, Xu HF, Borhani DW, Arkin IT, Eastwood MP, Shaw DE (2008) Dynamic control of slow water transport by aquaporin 0: Implications for hydration and junction stability in the eye lens. Proc Natl Acad Sci USA 105:14430–14435
Jiang JS, Daniels BV, Fu D (2006) Crystal structure of AqpZ tetramer reveals two distinct Arg-189 conformations associated with water permeation through the narrowest constriction of the water-conducting channel. J Biol Chem 281:454–460
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating water. J Chem Phys 79:926–935
Jorgensen WL, Maxwell DS, Tirado-Rives J (1996) Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc 118:11225–11236
Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22:2577–2637
Kandt C, Ash WL, Tieleman DP (2007) Setting up and running molecular dynamics simulations of membrane proteins. Methods 41:475–488
Khandelia H, Jensen MO, Mouritsen OG (2009) To gate or not to gate: using molecular dynamics simulations to morph gated plant aquaporins into constitutively open conformations. J Phys Chem B 113:5239–5244
Li T, Choi WG, Wallace IS, Baudry J, Roberts DM (2011) Arabidopsis thaliana NIP7;1: an anther-specific boric acid transporter of the aquaporin superfamily regulated by an unusual tyrosine in helix 2 of the transport pore. Biochemistry 50:6633–6641
Mitani-Ueno N, Yamaji N, Zhao FJ, Ma JF (2011) The aromatic/arginine selectivity filter of NIP aquaporins plays a critical role in substrate selectivity for silicon, boron, and arsenic. J Exp Botany 62:4391–4398
Parrinello M, Rahman A (1981) Polymorphic transitions in single-crystals—a new molecular dynamics method. J Appl Phys 52:7182–7190
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comp Chem 25:1605–1612
Smart OS, Neduvelil JG, Wang X, Wallace B, Sansom MSP (1996) HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J Mol Graph Model 14:354–360
Sui H, Han BG, Lee JK, Walian P, Jap BK (2001) Structural basis of water-specific transport through the AQP1 water channel. Nature 414:872–878
Tajkhorshid E, Nollert P, Jensen MO, Miercke LJ, O’Connell J, Stroud RM, Schulten K (2002) Control of the selectivity of the aquaporin water channel family by global orientational tuning. Science 296:525–530
Tieleman DP, Berendsen HJC (1998) A molecular dynamics study of the pores formed by Escherichia coli OmpF porin in a fully hydrated palmitoyloleoylphosphatidylcholine bilayer. Biophys J 74:2786–2801
Tornroth-Horsefield S, Wang Y, Hedfalk K, Johanson U, Karlsson M, Tajkhorshid E, Neutze R, Kjellbom P (2006) Structural mechanism of plant aquaporin gating. Nature 439:688–694
Verma RK, Gupta AB, Sankararamakrishnan R (2015a) Major intrinsic protein superfamily: channels with unique structural features and diverse selectivity filters. Methods Enzymol 557:485–520
Verma RK, Prabh ND, Sankararamakrishnan R (2014) New subfamilies of major intrinsic proteins in fungi suggest novel transport properties in fungal channels: Implications for the host-fungal interactions. BMC Evol Biol 14:Art. No. 173
Verma RK, Prabh ND, Sankararamakrishnan R (2015b) Intra-helical salt-bridge and helix destabilizing residues within the same helical turn: Role of functionally important loop E half-helix in channel regulation of major intrinsic proteins. Biochim Biophys Acta 1848:1436–1449
Wang Y, Schulten K, Tajkhorshid E (2005) What makes an aquaporin a glycerol channel? A comparative study of AqpZ and GlpF. Structure 13:1107–1118
Xin L, Su HB, Nielsen CH, Tang CY, Torres J, Mu YG (2011) Water permeation dynamics of AqpZ: a tale of two states. Biochim Biophys Acta 1808:1581–1586
Acknowledgements
We gratefully acknowledge Prof. Bert de Groot for sharing the Berger lipid parameters compatible with the OPLS-AA force-field. We thank the High Performance Computing facility of IIT-Kanpur supported by DST and MHRD, Government of India. RS is Pradeep Sindhu Chair Professor. RKV thanks Council of Scientific and Industrial Research (CSIR) for a Senior Research Fellowship. We thank all our lab members for suggestions and useful discussions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Alok Jain and Ravi Kumar Verma have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Jain, A., Verma, R.K. & Sankararamakrishnan, R. Presence of Intra-helical Salt-Bridge in Loop E Half-Helix Can Influence the Transport Properties of AQP1 and GlpF Channels: Molecular Dynamics Simulations of In Silico Mutants. J Membrane Biol 252, 17–29 (2019). https://doi.org/10.1007/s00232-018-0054-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00232-018-0054-7