
Abstract
Solution-state nuclear magnetic resonance studies of membrane proteins are facilitated by the increased stability that trapping with amphipols confers to most of them as compared to detergent solutions. They have yielded information on the state of folding of the proteins, their areas of contact with the polymer, their dynamics, water accessibility, and the structure of protein-bound ligands. They benefit from the diversification of amphipol chemical structures and the availability of deuterated amphipols. The advantages and constraints of working with amphipols are discussed and compared to those associated with other non-conventional environments, such as bicelles and nanodiscs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Amphipols as an Alternative to Detergents for Membrane Protein Solution NMR Studies
Most structural studies of membrane proteins (MPs) are conducted in vitro on isolated proteins kept in artificial media. This requirement is due to the difficulty in identifying specific signals corresponding to the protein of interest in complex samples, even though outstanding progresses have been made to observe, for instance, NMR signals with atomic resolution inside living cells (see e.g., Renault et al. 2012). The choice of a suitable substitute to the native membrane that be compatible with various biochemical contexts and biophysical techniques is far from trivial (see e.g., Warschawski et al. 2011; Popot 2010; Raschle et al. 2010; Etzkorn et al. 2013). Above all, the artificial environment has to keep MPs stable and active. The vast majority of solution-state NMR studies of MPs to date have been performed in detergent solutions (http://www.drorlist.com/nmr/MPNMR.html; Kang and Li 2011). The limited stability of most MPs in the presence of detergents, compounded by the high concentrations, long durations, and relatively high temperatures required by solution NMR experiments, has led to the search for milder surfactants that remain compatible with the specific constraints of NMR. Among the most promising non-conventional media, nanometric lipid bilayers (hereafter ‘nanodiscs’; NDs) (Bayburt et al. 2002; Denisov et al. 2004; Ritchie et al. 2009), isotropic bicelles (Sanders and Landis 1995; Vold et al. 1997; Czerski and Sanders 2000; Poget and Girvin 2007), and amphipols (APols) (Tribet et al. 1996; Popot et al. 2011) represent powerful alternatives. APols are specially designed amphipathic polymers that can keep water-soluble in their native state MPs of all types and sizes. One of their major advantages over detergents, particularly relevant in the context of NMR, is their stabilizing properties, which hold for most MPs (reviewed in Popot et al. 2011). In addition to stabilizing target MPs, APols can also be used to fold them, either from a denatured state such as can be obtained from inclusion bodies (Pocanschi et al. 2006; Dahmane et al. 2009, 2011; Banères et al. 2011; Bazzacco et al. 2012) or in the course of cell-free expression (CFE) (Bazzacco et al. 2012). The very medium into which the protein has been folded can then be used to keep it soluble and stable for NMR experiments (Dahmane et al. 2011; Catoire et al. 2010a, b). Solution NMR studies in APols have been reported to date for three β-barrel MPs and two α-helical ones (Table 1). In this review, we try to delineate the advantages and constraints of the use of APols for solution-state NMR studies, particularly as regards sample preparation, handling, and NMR spectroscopic properties.
Preparing MP/APol Complexes for Solution NMR Studies
In general, MP/APol complexes derive from MP/detergent ones (Fig. 1). MPs expressed in their native environment or in a host membrane, or as inclusion bodies, are, in most cases, solubilized first using a detergent. This is due to the poor dissociating capacity of APols, which makes them inefficient at extracting MPs from a membrane or at dissolving inclusion bodies (Popot 2010; Popot et al. 2011). Once solubilized by detergents, MPs can be rapidly transferred to APols before they lose their activity. Replacing the detergent with APols at the protein hydrophobic surface can be achieved by supplementing the detergent solution with APols before (1) diluting the solution under the critical micellar concentration of the detergent (see e.g., Champeil et al. 2000; Martinez et al. 2002; Dahmane et al. 2013; Zoonens et al. 2014); or (2) adsorbing the detergent onto polystyrene beads, onto which APols do not adsorb (e.g., Zoonens et al. 2005, 2007, 2014); or (3) dialyzing it away (Dahmane et al. 2013; Zoonens et al. 2014); or (4), in the specific case of SDS, selectively precipitating it (see e.g., Pocanschi et al. 2006; Dahmane et al. 2009, 2013; Catoire et al. 2010b; Zoonens et al. 2014). Once the protein is complexed by APols, modifying buffer conditions for NMR becomes straightforward, whether by dialysis or using ultrafiltration devices. There is no need to add APols to the buffers used for dialysis or exchange. This is in contrast with detergent solutions, where the control of surfactant concentration is critical, whether in pure or mixed detergent solutions or in binary detergent/lipid preparations such as bicelles. Indeed, because of the large size of their particles (~40 kDa for the most commonly used APol, called A8-35; Gohon et al. 2006) and their very low critical aggregation concentration (~0.002 g L−1 for A8-35; Giusti et al. 2012), APols do not cross standard dialysis membranes. Furthermore, they do not dissociate spontaneously from MPs even at extreme dilutions (Zoonens et al. 2007). This has two practical consequences: it simplifies protocols, and it considerably limits the amount of APol consumed in any experiment, so that it seldom becomes an economical concern.

Various methods for preparing samples of MP/APol complexes for solution-state NMR studies. In the first NMR studies of MPs associated with APol A8-35, tOmpA and OmpX were overproduced as inclusion bodies and folded in detergent solution, after which the detergent was exchanged for A8-35 using polystyrene beads (Catoire et al. 2009, 2010a). Alternatively, the detergent can be removed by dilution under its critical micellar concentration, followed by a dialysis. APols can also assist MP folding (Pocanschi et al. 2006), which opened the way to folding MPs directly in APols from a urea solution (Dahmane et al. 2011), without ever using detergents, or from an SDS one (Etzkorn et al. 2013; Catoire et al. 2010b, 2011; Elter et al. 2014) (note: the references indicated in the figure concern NMR studies only). More references can be found in the text or in reviews (Popot 2010; Popot et al. 2011; Zoonens et al. 2014)
Indeed, most NMR studies require large amounts of material compared to other biophysical techniques, even though the advent of cryogenic probe technology has considerably lowered the detection threshold. Depending on the measurements to be carried out, a few tens to hundreds of nanomoles (0.1–1 mg) of protein are needed to achieve a good signal-to-noise ratio. Preparing such amounts of MPs is quite demanding in terms of biochemical work, and the quantities of surfactant that are needed to handle the protein from buffer to buffer until the NMR tube can become very costly. APols, beyond their stabilizing effects, have the added advantages of being chemically stable and cost-effective as compared to most other surfactants, which facilitates the handling of large amounts of MPs, especially when buffer exchanges are necessary in the course of sample preparation. Furthermore, some APols, such as A8-35, can be deuterated or otherwise labeled or tagged at an acceptable cost (see below).
NMR Studies of APol-Trapped MPs
The first NMR studies of APol-trapped MPs were carried out on two β-barrel MPs from the outer membrane of Escherichia coli, namely the transmembrane domain of OmpA (tOmpA) (Zoonens et al. 2005) and OmpX (Catoire et al. 2009, 2010a). Known to be highly stable in vitro, these proteins, which express very well, were chosen because of the extensive NMR data available from studies conducted in detergent solutions (Arora et al. 2001; Fernández et al. 2001), which had led to 3D structures very similar to those obtained by X-ray diffraction (Pautsch and Schulz 2000; Vogt and Schulz 1999). As no activity tests are available for these two proteins in solution, the fact that they had maintained their native conformation once trapped in APols was established by comparing their 1H and 15N NMR chemical shifts with those previously observed in detergent solutions, in addition to SDS-Page gel electrophoresis experiments, which can provide a first indication of the folding state of β-barrel MPs. In the case of full-length OmpA, it has also been shown that the protein, after refolding in A8-35 and transfer to black lipid membranes, induces the formation of native-like ion channels (Pocanschi et al. 2006). In early NMR studies (Zoonens et al. 2005; Catoire et al. 2009, 2010a), and more recently (Etzkorn et al. 2014), tOmpA and OmpX were overproduced as inclusion bodies and folded in detergent solution, after which the detergent was exchanged for A8-35 using polystyrene beads. In the meantime, the possibility of using APols to assist MP folding was demonstrated with one α-helical and two β-barrel MPs, one of them tOmpA (Pocanschi et al. 2006), opening the way to folding tOmpA directly in APols from a urea solution, without ever using detergents. This was achieved using sulfonated APols (SAPols), the folded state of the protein being checked by NMR (Dahmane et al. 2011).
These pioneering experiments were aimed at better characterizing MP/APol complexes and at exploring the resources and limitations of NMR to address their structure and dynamics when kept soluble by APols. tOmpA and OmpX from E. coli and KpOmpA from Klebsiella pneumoniae were shown by NMR to either retain or regain their native fold when associated with various APols (Dahmane et al. 2011; Zoonens et al. 2005; Catoire et al. 2009, 2010a, b; Etzkorn et al. 2014; Renault 2008) (Fig. 2). NMR spectroscopy was used to examine the organization of MP/A8-35 complexes at various levels, from a general description of the distribution of the alkyl chains of the surfactant (Zoonens et al. 2005; Catoire et al. 2009; Etzkorn et al. 2014) to the identification of hydrophobic contacts between specific amide protons of KpOmpA and octyl and isopropyl chains of A8-35 (Renault 2008) (Fig. 3). A consistent picture emerged from these studies, showing that, as anticipated, A8-35 adsorbs specifically onto the transmembrane region of the protein. This was also observed more recently by electron microscopy (Althoff et al. 2011) and in molecular dynamics (MD) calculations (Perlmutter et al. 2014).

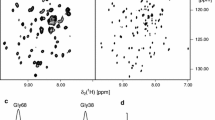
A comparison of the 2D 1H,15N NMR correlation spectra of three β-barrel membrane proteins kept water-soluble by either amphipols or detergents. Data from the following publications: ref. (Zoonens et al. 2005) (tOmpA/A8-35 and tOmpA/DHPC); ref. (Dahmane et al. 2011) (tOmpA/SAPol); ref. (Renault 2008) (KpOmpA/A8-35); ref. (Catoire et al. 2010a) (OmpX/A8-35 and OmpX/DHPC); and ref. (Bazzacco et al. 2012) (OmpX/NAPol)

Analysis of intermolecular contacts between KpOmpA transmembrane domain and amphipol A8-35. a Chemical structure of A8-35. b 1D 1H spectrum of [u-2H,13C,15N]-KpOmpA/A8-35 complexes (1:4, w/w) in 20 mM NaH2PO4 buffer (pD = 7.9) containing 100 mM NaCl, 10 mM EDTA, 10 % D2O. NMR assignments of A8-35 resonances (labels 1–8) are reported on the spectrum. 1H chemical shifts of side chain resonances are indicated in p.p.m. units. c Selected 2D [15N,1HN] planes from the 3D 15N-edited (1H,1H) HSQC-NOESY-TROSY spectra using 100 ms (red) and 200 ms (black) NOESY mixing times obtained on [u-2H,13C,15N]-KpOmpA/A8-35 complexes (1:4, w/w) in 20 mM NaH2PO4 buffer (pD = 7.9) containing 100 mM NaCl, 10 mM EDTA, 100 % D2O. 2D [15N,1HN] planes were extracted at the 1H frequencies indicated at the top of the spectra. d Distribution of intermolecular NOE signals throughout KpOmpA transmembrane domain (PDB accession code: 2K0L). Color code: Light gray, residue not detected; dark gray, residue for which intermolecular NOEs interactions could not be detected; light color, residues for which intermolecular NOEs are only detected in 3D NOESY using long mixing time (e.g., 200 ms); dark color, residues for which intermolecular NOE signals are detected in 3D NOESY using short mixing time (e.g., 100 ms). The colors correspond to the moieties shown in a. e Overview of intermolecular contacts with A8-35 molecules (red) plotted on the solution NMR structure of KpOmpA TM domain (blue). Side chains from aromatic residues located at the interface are indicated with a stick representation. f Distribution of micro- to milli-second molecular motions within KpOmpA transmembrane domain as revealed by chemical exchange experiments performed on [u-2H,13C,15N]-KpOmpA/A8-35 at 30, 35, 38 at 40 °C. Color code: gray, residue not detected; blue: residues with molecular motions slower than milli-second timescale; red residues with molecular motions occurring on micro- to milli-second timescales. All NMR experiments were performed on a Bruker AVANCE spectrometer operating at a 1H Larmor frequency of 700 MHz equipped with a triple-resonance QXI NMR probe and at 40 °C, unless indicated (Color figure online)
OmpX is the smallest member of the bacterial outer membrane protein family, and ground-state, low-energy structures describe a lumen cluttered by many amino acid side chains (Fernández et al. 2001; Vogt and Schulz 1999). In an apparent conflict, electrophysiological studies suggest that OmpX catalyzes the passage of small solutes through membranes (Dupont et al. 2004; Arnold et al. 2007). By the detection of hydrogen/deuterium exchange, NMR experiments conducted with OmpX in complex with A8-35 show that some amide protons of the membrane-spanning region exchange much more readily than others (Catoire et al. 2010a). These measurements, performed after extensive equilibration, show that the barrel is dynamic, suggesting that time-dependent conformational fluctuations may allow the transient formation of a channel.
Two α-helical MPs have been directly folded into APols in view of NMR studies, namely bacteriorhodopsin (BR) (Etzkorn et al. 2013) (see below) and the low-affinity leukotriene G protein-coupled receptor (GPCR) BLT2 (Yokomizo et al. 2000). The perdeuterated and uniformly 15N-labeled BLT2 receptor, folded and kept in partially deuterated A8-35 (DAPol), was then used to determine, from the distance constraints obtained from transferred nuclear Overhauser effect (NOE) signals, the structure of BLT2 agonists in their receptor-bound state (Catoire et al. 2010b, 2011). To this end, the receptor was overexpressed as inclusion bodies in E. coli cells (Banères et al. 2003; Mouillac and Banères 2010) grown in a 100 % D2O medium, solubilized and purified in sodium dodecylsulfate (SDS) solution, and folded by precipitating dodecylsulfate as its potassium salt in the presence of DAPol, according to a general protocol for APol-assisted folding of GPCRs (Dahmane et al. 2009; Banères et al. 2011). Perdeuteration of the receptor was mandatory to eliminate intermolecular spin diffusion effects between the protonated ligands and the receptor, which are more difficult to manage in structure calculations than intramolecular effects. Deuteration of the side chains of A8-35 (Gohon et al. 2004, 2006) limited overlaps between NOE signals originating from the ligand and those due to the APol. 15N labeling gave the opportunity to also observe, at low concentration (~10 μM) of receptor, protein spin resonances in non-crowded regions of a 2D 1H,15N CRINEPT (Riek et al. 1999) spectrum (cf. Supplementary Fig. 17 in ref. Catoire et al. 2010b).
More recently, following CFE and folding by transfer from SDS, a MP featuring seven transmembrane α-helices, BR, was studied in complex with A8-35. It gave rise to well-resolved NMR spectra, slightly better than those obtained in the detergent dodecyl-β-d-maltoside (DDM) (Raschle et al. 2010; Etzkorn et al. 2013; Elter et al. 2014) and significantly better than those in nanodiscs (NDs). In this study, it was noted that, whereas the transmembrane region of BR adopts the same structure in DDM, A8-35, and dimyristoylphosphatidylcholine-based NDs, some loop residues show chemical shift differences, indicating a different conformation or environment. In particular, in some loop regions, BR associated with APols displays NMR data that are similar to data observed by solid-state NMR of BR in native membrane, as opposed to BR associated with DDM or embedded in nanodiscs (Etzkorn et al. 2013).
Because BR can be expressed by CFE in the presence of non-ionic APols (NAPols; see below) (Bazzacco et al. 2012), and NMR spectra of NAPol-trapped OmpX are of a quality equivalent to those in A8-35 (Bazzacco et al. 2012), it seems likely that CFE in NAPols followed by purification can provide directly MP samples of NMR quality. A recombinant approach using heterologous expression in E. coli could also be considered, where BR would be expressed as a fusion protein to target it to inclusion bodies (cf. ref. Banères et al. 2011). SDS-denatured BR could then be folded to >90 % by transfer to A8-35 (Pocanschi et al. 2006; Dahmane et al. 2013) following the protocol mentioned above for BLT2, a procedure that is now routinely used to fold GPCRs (Banères et al. 2011; Bazzacco et al. 2012). Both CFE (Takeda and Kainosho 2012) and expression in E. coli (Plevin and Boisbouvier 2012) lend themselves to the sophisticated isotopic labeling schemes required for the study of large complexes by solution NMR. Overexpressing MPs as inclusion bodies allows the production of large amounts of labeled protein (tens of mg) at an affordable cost.
Which APols to Choose for NMR?
Over the years, various types of APols have been synthesized and studied either free or associated with MPs (Popot et al. 2011). Among them are: (1) A8-35, the first APol to have been developed and validated (Tribet et al. 1996; Gohon et al. 2006; Gohon et al. 2008); (2) phosphorylcholine-based APols (PC-APols) (Diab et al. 2007; Tribet et al. 2009); (3) sulfonated APols (SAPols) (Dahmane et al. 2011); and (4) glucose-based, non-ionic APols (NAPols) (Bazzacco et al. 2012; Sharma et al. 2012). The main chemical difference between them lies in the groups responsible for their solubility in aqueous solutions: A8-35, PC-APols, SAPols and NAPols carry, respectively, carboxylate, phosphorylcholine, sulfonate, and glucose groups (Fig. 4), which confer with them different behaviors at acidic pH and in the presence of multivalent cations, and different degrees of mildness toward MPs (Table 1) (reviewed in Popot et al. 2011). A8-35, SAPols and NAPols have all been validated for MP NMR studies (Dahmane et al. 2011; Bazzacco et al. 2012; Zoonens et al. 2005).

Compared 1H NMR spectra of various APols. The spectra were recorded either in deuterium oxide (A8-35 and DAPol, from Tribet et al. (1996) and Gohon et al. (2008), respectively), in deuterated methanol (perDAPol and SAPol, from Picard et al. (2006) and Dahmane et al. (2011), respectively), or in deuterated dimethylsulfoxide (DMSO) (NAPol) (from Bazzacco et al. 2012). The chemical structure and the corresponding NMR spectrum are presented on the same line. Each signal that has been formally identified and assigned is either individually related by an arrow to the corresponding chemical group, or more extensively by a brace to a category of functional groups
The solution and association properties of A8-35 have been thoroughly studied (Gohon et al. 2004, 2006; Giusti et al. 2012; Perlmutter et al. 2011). A8-35 is highly soluble in aqueous solutions (>200 g L−1), provided the pH is kept ≥7. This limitation is due to the fact that, at pH <7, some of the ionized carboxylate groups to which the polymer owes its solubility in water start to protonate, which increases its hydrophobicity and elicits aggregation both of the polymer itself and of MP/A8-35 complexes (Gohon et al. 2004, 2006). Above pH 7, 200 g L−1 is a comfortable value, which allows one to work, if necessary, with highly concentrated MP solutions. For instance, working with 1 mM of tOmpA (MW ≈ 20 kDa) requires ~20 g L−1 of protein and 80 g L−1 of A8-35. At ~100 g L−1 of A8-35, the viscosity of the solution is not affected (L. J. C., unpublished data), and there is therefore no impact on the NMR signal linewidths. However, the necessity to work above pH 7 can be detrimental for the detection of labile protons by NMR. 1H NMR observation of labile protons depends on their rate of exchange with the solvent (Wüthrich 1986). At the NMR chemical shift timescale, the exchange rate has to be slow enough to allow the detection of the signal; otherwise, the labile 1H signal is lost in the dominant signal from water protons. The rate of exchange of labile 1H in amino acids increases with the pH. For instance, the exchange rate of amide protons (1HN) of the polypeptide backbone is slowest at pH ~3. It is five orders of magnitude faster at pH 8 (Wüthrich 1986), rendering the observation of 1HN resonances problematic for those protons that are not protected from the solvent (note, however, that the higher the magnetic field, the easier the observation of labile protons becomes). The pH limitation inherent to the chemical structure of A8-35 has been a major incentive behind the development and validation of two types of pH-insensitive APols that have been shown to be compatible with high-resolution solution-state NMR studies, SAPols (Dahmane et al. 2011) and NAPols (Bazzacco et al. 2012; Sharma et al. 2012), both of which are highly soluble in the 0–14 pH range. They each have their own drawbacks, though: at variance with A8-35, NAPols are hard to synthesize and could not be perdeuterated at an affordable cost, whereas SAPols, which are easily amenable to deuteration, are time-consuming to purify in large amounts and, probably because of their higher charge density, appear to be less stabilizing toward MPs than A8-35 or NAPols (Table 1; reviewed in Popot et al. 2011).
The concentration of surfactant required to obtain monodisperse solutions of MPs can be quite high, which may cause signal overlap with the protein of interest. Studies of large proteins or protein complexes by NMR are generally conducted with isotope-labeled proteins along with an ad hoc methodology. Thanks to isotopic filters, 1H signals from the surfactant can be efficiently removed. However, in some cases, it is essential to work with deuterated surfactants, e.g., in the studies of interactions of organic molecules with a protein, especially when the cost of labeling isotopically the ligands of interest becomes extravagant. Two unsaturated fatty acid compounds, the eicosanoid acid leukotriene B4 (LTB4) and the heptadecanoid acid 12-HHT (12S-hydroxyheptadeca-5Z,8E,10E-trienoic acid), have been studied in interaction with BLT2 folded and stabilized in A8-35 (Catoire et al. 2010b, 2011). In both cases, the protein was trapped with DAPol. In Fig. 4 are compared the 1H resonances of hydrogenated A8-35 and DAPol. Despite the residual protonation, the resort to DAPol allowed the collection of enough intra-ligand 1H-1H interactions to perform structure calculations and competition experiments (Fig. 5). Of the three types of APols that have been validated for NMR to date, A8-35 and SAPol remain the most amenable to partial or total deuteration (Popot et al. 2011). A perdeuterated version of A8-35 (‘perDAPol’) has recently been synthesized, which will permit collecting more complete sets of distance constraints from NOE signals (Giusti et al. 2014) (Fig. 4).

Superimposed 2D 1H–1H NOESY spectra of a MP/DAPol/ligand complex and of hydrogenated and partially deuterated A8-35. In black, the perdeuterated BLT2 receptor complexed by DAPol, in the presence of the two protonated eicosanoids 12S-hydroxy-5Z,8Z,10E,14Z-eicosatetraenoic acid (12-HETE) and leukotriene B4 (LTB4) (from a competition experiment recorded at 950 MHz); in red and blue, spectra of pure A8-35 and DAPol, respectively, recorded at 400 MHz. The dashed lines on top and left of the spectra mark resonances from the two ligands. All samples were prepared in D2O solutions. Spectra were acquired at 25 °C. Thanks to the perdeuteration of the isopropyl and octyl side chains of DAPol, additional intra-ligand interactions could be observed (from Catoire et al. 2010a)
The availability of deuterated APols is also an asset in multidimensional heteronuclear 1H,13C NMR experiments. Because of the excess of APols over the protein and the large number of alkyl chains and repeating units in each APol chain, the natural abundance of 13C can give rise to correlation peaks with intensities close to or higher than signals from the protein (Fig. 6a). Methine, methylene and methyl groups, which are abundant in APols, are also common chemical groups in amino acids, creating signal overlaps. This was for instance observed in the study of the interaction of A8-35 with OmpX in a 2D heteronuclear 1H,13C Overhauser (HOESY) experiment (Catoire et al. 2009) (Fig. 6b). NMR methods, such as double-quantum approaches, would not eliminate all interference signals. Doing away with the contribution of natural abundance 13C in A8-35 and its deuterated derivatives would be readily possible by carrying out a complete synthesis, identical to that described for perDAPol (Giusti et al. 2014), starting from 13C-free acrylate, octylamine, and isopropylamine.

Detection of APol 13C natural abundance in 2D heteronuclear 1H,13C experiments. a Chemical structure of A8-35. Circled numbers label the groups whose chemical shifts are shown in b and c. b Zoom on a selected region of two superimposed 2D 1H,13C heteronuclear Overhauser spectroscopy (HOESY) spectra, respectively, those of [u-2H,13C,15N]OmpX/A8-35 complexes (in black) and of A8-35 alone (in red). Circled black numbers refer to 1H (the corresponding 1H spectrum of A8-35 is displayed on the left) and 13C chemical shift resonances from the groups labeled on the chemical structure of A8-35 in a (from Catoire et al. 2009). c Superimposed 1D proton spectra (top) and 2D 1H,13C heteronuclear single quantum correlation (HSQC) spectra (bottom), recorded at room temperature at 400 MHz 1H Larmor frequency, of fully protonated A8-35 (HAPol, in black) and partially deuterated A8-35 (DAPol, in red). Groups labeled 1, 2, 3, 4, 7, and 8 in a are deuterated in DAPol
Should One Worry About the Size and Heterogeneity of MP/APol Complexes?
As described above, APols specifically adsorb onto the transmembrane, hydrophobic region of MPs (Zoonens et al. 2005; Catoire et al. 2009; Renault 2008; Althoff et al. 2011; Perlmutter et al. 2014). The thickness of the APol belt has been estimated to 1.5–2 nm (Althoff et al. 2011; Perlmutter et al. 2014; Gohon et al. 2008), which is slightly thicker than that of the thinnest detergent belts. Indeed, compared to MPs in complex with detergents commonly used for solution-state NMR studies, such as 1,2-hexanoyl-1-sn-glycero-3-phosphocholine (DHPC) or n-dodecylphosphocholine (DPC), the overall correlation times (τ c) of small A8-35-trapped MPs, tOmpA and OmpX, are ~30–50 % longer (Zoonens et al. 2005; Catoire et al. 2010a), consistent with the hydrodynamic radius of the complexes being somewhat larger. A limiting factor a decade ago, this range of τ c, a few tens of ns, does not, nowadays, hamper the observation of well-resolved peaks nor the acquisition of 3D NMR data within a reasonable time (Fig. 3) (Renault 2008), as long as there are not too many overlapping signals. This is mainly due to progress in instrumentation and NMR methodology, as well as in efficient isotope-labeling strategies designed for the study of large proteins and protein complexes (Plevin and Boisbouvier 2012). Striving to produce the smallest particles possible is therefore not as compelling today as it formerly was, and certainly should not be sought at the expense of distorting the protein’s native state and/or compromising its stability (see e.g., Poget and Girvin 2007).
APols being inherently polydisperse molecules (their mass distribution index is 1.5–2 (Sharma et al. 2012; Giusti et al. 2014)), a recurrent concern has been that they could form with MPs populations of complexes with a wide distribution of sizes or NMR chemical shifts due to variable electronic environments. This is both true and untrue. True, in the sense that complexes formed between a small MP, tOmpA, and A8-35 do migrate, upon size exclusion chromatography (SEC), as a broader band than tOmpA/octyltetraoxyethylene (tOmpA/C8E4) complexes (Zoonens et al. 2007). Untrue, for two reasons. First, this effect is probably due to the more discrete nature of APol versus detergent binding, A8-35 molecules (on average ~4.3 kDa; see Giusti et al. 2014) being ~10× bigger than detergent ones (482, 351, and 511 Da for DHPC, DPC, and DDM, respectively), and a less efficient thermodynamic pressure toward optimization of the bulk of the APol versus the detergent belt, rather than to APol heterogeneity. Second, it does not prevent NMR resonances from BR/A8-35 complexes from being at least as well resolved as those from BR/DDM ones (Etzkorn et al. 2013). Consistent with this interpretation, a version of A8-35 with restricted length polydispersity showed essentially the same behavior in SEC as the classical polydisperse mixture (F. G. & C. Tribet, unpublished results). It is interesting to note that trapping tOmpA with an A8-35/detergent mixture results in the formation of particles that migrate in SEC with the same apparent R S as pure tOmpA/A8-35 complexes, but are more homogeneous (Zoonens et al. 2007). A tentative interpretation of this observation is that detergent molecules provide the “small change” that permits optimization of the volume of the surfactant belt, and relaxation toward a more uniform size. Because APols have a stabilizing effect on MPs even when used in mixture with detergents (Champeil et al. 2000), using such mixtures could prove useful when particle homogeneity is a must.
Close attention must be paid to the fact that many factors can contribute to increasing the polydispersity of MP/APol complexes, which can lead to signal degradation. Among those factors that have been identified to date, one may cite: (1) deviations from the nominal composition of A8-35: a slightly higher hydrophobicity leads to aggregation of APol particles and MP/APol complexes (see Gohon et al. 2004, 2006); (2) incubation for a long time (days) at a pH too close to pH 7 (ibid.); (3) the presence of traces of calcium, which, presumably, bridge MP/A8-35 complexes (Picard et al. 2006) (adding EDTA improves the quality of solution NMR spectra; (Catoire et al. 2010b); (4) removal of the excess of free APol that is required for efficient MP trapping. This is a critical parameter. APols, as already mentioned, are not very dissociating, and, in the absence of a slight excess of them, MPs tend to form small oligomers (see Zoonens et al. 2007; Gohon et al. 2008). Solutions of A8-35-trapped tOmpA, for instance, appear monodisperse in SEC for an overall MP/APol mass ratio of 1:4, whereas the protein is estimated to bind only ~1.4 g A8-35 per g protein. Removing the excess APol by immobilized metal affinity chromatography leads to the formation of small oligomers, which redissolve if the polymer is added back (Zoonens et al. 2007). The presence of oligomers does not preclude solution-state NMR investigations, but it can degrade the intensity and quality of the signals. Checking for polydispersity can be done by SEC (Fig. 7a), radiation scattering (not shown), or analytical ultracentrifugation (AUC; Fig. 7b), as illustrated by a detailed analysis of BR/A8-35 complexes (Gohon et al. 2008), AUC being by far the most sensitive and quantitative technique.

Size exclusion chromatography (a) and analytical ultracentrifugation (b) analyses of two preparations of bacteriorhodopsin/A8-35 complexes. Sample BR/HAPol was trapped with unlabeled A8-35; sample BR/DAPol with A8-35 whose side chains was deuterated. Unrelated to the labeling of the polymer, their dispersity was slightly different. The BR/HAPol sample was almost perfectly monodisperse. In SEC (a), it migrated as a symmetrical peak, with a profile similar to that of a control globular protein, catalase. Analytical ultracentrifugation (AUC) (b) showed it to comprise ~98 % BR monomers. The SEC peak formed by sample BR/DAPol featured a slight shoulder. By AUC, this sample was shown to comprise a majority of monomers and a small proportion of small oligomers (~11 % dimers and ~2 % trimers). Conditions that may compromise the monodispersity of MP/APol complexes are discussed in the text. V 0 and V t: excluded and total volumes of the SEC column, respectively. J: detection by refractometry; A: free A8-35 particles; M, D, T: BR trapped in A8-35 as monomers, dimers, and trimers, respectively. Figures adapted from Gohon et al. (2008)
Studying MPs in vitro in their supposedly native homo- or hetero-oligomeric state is a difficult challenge. Once the problem of preserving the original oligomeric state is solved, which APols are good at, solution-state NMR is able to detect signals from large complexes, provided that a suitable isotope-labeling strategy in association with an appropriate NMR methodology be used (see e.g., Fiaux et al. 2002; Ruschak et al. 2010). Given that most MP/APols preparations derive from MPs solubilized in detergent solutions and that APols are less dissociating than detergents, the association state of the APol-trapped protein will essentially reflect that in detergent solution. APols are able to preserve fragile MP/MP interactions, as illustrated by the electron microscopy study of the I1III2IV1 supercomplex of the mitochondrial respiratory chain from bovine heart (transferred from digitonin) (Althoff et al. 2011) and by a recent biochemical study of photosynthetic supercomplexes (transferred from DDM). A detailed study of BR trapped by A8-35 revealed that each complex contained one BR monomer and lipids in a lipid/protein ratio similar to that in native purple membranes (Gohon et al. 2008). These examples indicate that the oligomeric state and bound lipids of the protein(s) in detergent solution tend to be preserved upon trapping with APols. Indeed, whereas APols are most unlipid-like molecules, their inability to effectively displace lipids from the hydrophobic surface of MPs probably accounts in part for their mildness (discussed in ref. (Popot et al. 2011)). It has also been noted that some MPs transferred from detergent solution to APols tend to recover functional properties more similar to those they exhibit when membrane-bound than to those observed in detergent solution. In the case of BR, this effect is clearly due to lipid rebinding upon transfer to APols (Dahmane et al. 2013), and such may also be the case for the nicotinic acetylcholine receptor (see Martinez et al. 2002) and, for a discussion, Popot et al. 2011). Mapping MP/lipid in APol-trapped complexes contacts by solution NMR techniques similar to those used for mapping MP/APol ones (see below) would be readily possible.
When to Favor Detergents, Amphipols, Bicelles, or Nanodiscs?
Pioneering structural studies in detergent solutions, carried out on rugged β-barrel MPs from the outer membrane of E. coli, led to very high-quality NMR data (e.g., Fernández et al. 2001). Nevertheless, in the absence of a functional test in vitro, high-quality spectroscopic signals do not prove that the protein is in its native conformation (Poget and Girvin 2007; Zhou and Cross 2013; Catoire et al. 2014). For instance, OmpX exhibits various backbone 15N/1HN chemical shifts depending on the surfactant used (Fernández et al. 2001; Lee et al. 2008; Hagn et al. 2013). These variations are unlikely to be due to changes in the transmembrane electronic environment, given that, whatever the surfactant used, the amino acids pointing toward the membrane face mostly CHn moieties. The chemical shifts differences must mainly reflect modifications in the structure of the protein. This being said, if the protein is stable and active in their presence, detergents remain a privileged medium for solution-state NMR measurements. This is primarily due to the fact that the overall correlation time of MP/detergent complexes is usually shorter than in other environments. Some detergents, such as DDM, one of the most widely used surfactants in structural biology, form with MPs relatively large complexes, but they remain smaller than MP/APol or MP/ND ones (e.g., Etzkorn et al. 2013). Mixed detergent/detergent or lipid/detergent micelles, as well as bicelles, represent a valuable alternative to mono-detergent solutions (Sanders and Landis 1995; Czerski and Sanders 2000; Poget and Girvin 2007; Catoire et al. 2014), even though short-chain detergents, which are mixed with lipids to generate bicelles (Triba et al. 2005), could be a destabilizing factor. In addition, the size of MP/bicelle complexes tends to be close to that of MP/APols ones (Catoire et al. 2010a; Lee et al. 2008). A new promising class of detergents, the maltose-neopentyl glycols (MNGs), has been described recently. MNGs appear to be less destabilizing to MPs than DDM (Chae et al. 2010). One advantage of MNGs and, more generally, of detergents over APols and NDs is their ability to directly extract MPs from their native or host membranes. The compatibility of MNGs with NMR investigations in solution remains to be tested. To study the interactions of protonated ligands with MPs, sugar-based detergents such as DDM or MNGs suffer from the prohibitive cost of perdeuteration.
Whereas other interesting alternatives to detergents have been proposed, among which lipopeptides (McGregor et al. 2003; Kelly et al. 2005; Privé 2009) and surfactant peptides (Zhao et al. 2006; Wang et al. 2011; Koutsopoulos et al. 2012), both of which appear as interesting tools for solution NMR, APols and NDs have emerged as particularly promising technologies. For very small MPs, the slower tumbling time of MP/APol complexes slightly reduces the resolution observed as compared to the smallest MP/detergent complexes. In many cases, this can probably be at least partially compensated by working at higher temperature, as MPs are much more thermostable in these environments (Fig. 8) (Etzkorn et al. 2013; Dahmane et al. 2009, 2013; Tifrea et al. 2011; reviewed in Popot 2010; Popot et al. 2011). It is likely that very fragile MPs will tend to be even more stable in NDs than in APols (for a discussion, see Popot 2010), as illustrated recently in an elegant study of BR (Etzkorn et al. 2013). In most cases, however, the stability afforded by APols is likely to suffice, in which case the greater simplicity of the biochemical preparation and the smaller size of the particles give APols an edge, at least as long as a bilayer-like environment is not essential to the measurements to be carried out. It is easier, however, to control the oligomeric state of MPs using NDs, by adjusting the size of the NDs and the ND/MP ratio at the reconstitution stage (Ritchie et al. 2009). With APols, this control is more difficult, because (1) the MP/APol ratio cannot be modified totally at will, as APols must be present in some excess at the trapping stage, (2) removing the excess of APol after trapping can induce artifactual associations, and (3) APols will not prevent monomers from associating if they have some affinity one for another (see Zoonens et al. 2007). If, as can be the case in some studies with GPCRs, it is essential to trap monomers and make sure that they do not associate, even in a transient manner, in the course of the experiment, NDs are definitely preferable to APols (for reviews, see Popot 2010; Nath et al. 2007).

Stability of APol-trapped versus detergent-solubilized BLT1 receptor. Top: Temperature-dependent stability of BLT1 kept in solution with the detergent fos-choline-16 in the presence of lipids (fos-choline-16/asolectin 2:1 mass ratio; D + L, in blue), or folded in pure A8-35 (AP, in red) or in A8-35 supplemented with asolectin in a 1:0.2 APol/lipid mass ratio (AP + L, in gray). Thermostability was assayed by heating the receptor at various temperatures for 30 min before performing an antagonist (LTB4) binding assay. Compared to the D + L sample, the thermostability is improved by ~11 °C in the AP + L one. Bottom: time-dependent stability of the BLT1 receptor at 25 °C. Same samples and color code as above. High-affinity LTB4 binding by the D + L sample dropped to ~50 % of its initial value after 20 days at 4 °C, whereas after folding in A8-35 or A8-35+ lipids, no significant loss was observed over the same period. Adapted from Dahmane et al. (2009)
A great advantage of APols remains their chemical versatility. APols carrying fluorophores (e.g., Zoonens et al. 2007; Giusti et al. 2012) or tags (e.g., Giusti et al. 2014; Charvolin et al. 2009; Le Bon et al. 2014a, b) are helpful in many biochemical and biophysical experiments, and they facilitate the characterization of samples (determination of the final MP/APol ratio, for example Zoonens et al. 2014). As already discussed, the use of deuterated (Gohon et al. 2004, 2006) or perdeuterated (Takeda and Kainosho 2012) APols is a great asset in the study of MP-bound hydrogenated ligands (Catoire et al. 2010b, 2011; Wüthrich 1986) and will also be for that of through-space interactions in solving MP structures (see below). It has also been exploited to map interactions between A8-35 and tOmpA, by taking advantage of the fact that 1HN lines of residues that are in contact with the surfactant become narrower if DAPol is substituted to HAPol (Zoonens et al. 2005). In other words, many complementary sets of NMR (and other) data can be collected on a MP experiencing chemically identical but differently labeled APol environments.
How to Solve a MP Structure Using APols?
As of now, no MP structure has been solved de novo by NMR using APols as the solubilizing medium. That is indeed possible is shown by (1) the high resolution of TROSY 1H,15N 2D spectra (Raschle et al. 2010; Etzkorn et al. 2013; Dahmane et al. 2011; Bazzacco et al. 2012; Zoonens et al. 2005; Catoire et al. 2010a) (Fig. 2) and (2) the fact that high-resolution 3D experiments can be carried out within a reasonable time span (Etzkorn et al 2014) (Fig. 3). All APols tested to date for use in solution NMR [A8-35 (Zoonens et al. 2005), SAPols (Dahmane et al. 2011), NAPols (Bazzacco et al. 2012)] form with MPs complexes of a similar size and can be used for backbone assignments. When studying large proteins or protein complexes by NMR, one of the best strategies is to take advantage of the favorable relaxation properties of methyl groups immersed in a perdeuterated environment (Plevin and Boisbouvier 2012) to either look at intra- or inter-molecular interactions or perform relaxation measurements. In this context, the 13C natural abundance of APols could mask some 13CH3 correlations and it is advantageous to work with APols that are either perdeuterated or, at least, carry perdeuterated side chains. This modification, which has been or can be easily achieved with A8-35 and SAPols, dramatically reduces interference signals arising from 13CHn residual APol groups (Fig. 6c). 13C-free A8-35 and SAPols could also be synthesized, provided 13C-depleted acrylic acid, octylamine, and either isopropylamine or taurine can be obtained at an affordable cost.
In the future, two further types of APols could conceivably be of help for NMR studies. First, it ought to be possible to develop a variant of SAPols with a lower density of charges along the chain, e.g., one in which the carboxylate groups currently present along with the sulfonate ones (Fig. 4a) would be replaced by isopropylamine. A SAPol whose charge density would be comparable to that of A8-35 would likely be as mild toward fragile MPs, while remaining soluble at all pH, and it could easily be obtained in perdeuterated form. Second, it might be interesting to examine the usefulness in NMR studies of APols carrying free radicals. It is relatively straightforward to functionalize APols with almost any desirable chemical moiety (reviewed in Le Bon et al. 2014a). Spin-labeled APols could possibly be of use to simplify NMR spectra by selectively suppressing, in a controlled and tunable manner, the resonance peaks from transmembrane residues. They could possibly also be used to improve the sensitivity of the measurements by dynamic nuclear polarization (magnetization transfer from the spin label to nuclei; see Griesinger et al. 2012).
Conclusion
APols, which are chemically highly stable, offer an excellent alternative to other surfactants for biophysical investigations of MPs, in particular by NMR. The improved stability of most MPs following trapping by APols makes them particularly attractive tools for structural and/or dynamics studies using this spectroscopy. Whereas APols are often criticized as being a highly artificial medium—which they undoubtedly are—one should keep in mind that they favor the retention of lipids, thus providing MPs with a more native-like environment than detergents do. It is one factor by which they stabilize MPs and, at least in some instances, bring them back to a functional behavior closer to that observed in the original membrane. APols are universal in terms of the MPs they can trap and extremely versatile in their uses. From a practical point of view, preparation and handling of MP/APols complexes until their final location in the magnet are both straightforward and easy to standardize. Current investigations indicate that, given good biochemistry and the use of efficient labeling strategies, NMR methodology and hardware, the resolution of NMR spectra collected on APol-trapped α-helical MPs is sufficient to obtain high-resolution information about proteins at least up to the size of BR.
It may be noted that solid-state NMR (ssNMR) is one of the rare techniques that has not been applied to date to the study of APol-trapped MPs. There is nothing to prevent it. The motivation to do so may seem limited, in the sense that one of the primary interests of ssNMR is to give access to MPs in an environment resembling the native one. Furthermore, MPs should not be expected to be more stable in APols than in a membrane environment. APols, however, could have the double advantage of making it possible to pack more MP in a given volume, increasing sensitivity—an important limitation in ssNMR—while keeping it associated with most of its bound lipids.
Abbreviations
- APol:
-
Amphipol
- A8-35:
-
Polyacrylate-based amphipol A8-35
- BLT2:
-
Low-affinity leukotriene receptor
- BR:
-
Bacteriorhodopsin
- C8E4 :
-
Octyltetraoxyethylene
- CFE:
-
Cell-free expression
- CRINEPT:
-
Cross-correlated relaxation-enhanced polarization transfer
- DHPC:
-
Dihexanoylphosphatidylcholine
- DAPol:
-
A8-35 with perdeuterated octyl and isopropyl chains and a hydrogenated polyacrylate backbone
- DPC:
-
n-Dodecylphosphocholine
- DDM:
-
n-Dodecyl-β-d-maltopyranoside
- GPCR:
-
G protein-coupled receptor
- 12-HHT:
-
12S-Hydroxyheptadeca-5Z,8E,10E-trienoic acid
- HOESY:
-
Hetero-nuclear Overhauser spectroscopy
- 12-HETE:
-
12S-Hydroxy-5Z,8Z,10E,14Z-eicosatetraenoic acid
- HSQC:
-
Hetero-single quantum correlation experiment
- KpOmpA:
-
Outer membrane protein A from Klebsiella pneumoniae
- LTB4 :
-
Leukotriene B4
- MD:
-
Molecular dynamics
- MP:
-
Membrane protein
- MW:
-
Molecular weight
- NAPol:
-
Non-ionic amphipol
- MNG:
-
Maltose neopentyl glycol
- NMR:
-
Nuclear magnetic resonance
- NOE:
-
Nuclear Overhauser effect
- ND:
-
Nanodisc
- OmpA:
-
Outer membrane protein A from Escherichia coli
- OmpX:
-
Outer membrane protein X from Escherichia coli
- perDAPol:
-
Perdeuterated A8-35
- PC-APol:
-
Phosphocholine amphipol
- R S :
-
Stokes radius
- SAPol:
-
Sulfonated amphipol
- SDS:
-
Sodium dodecylsulfate
- ssNMR:
-
Solid-state NMR
- SEC:
-
Size exclusion chromatography
- tOmpA:
-
Transmembrane domain of OmpA
- TROSY:
-
Transverse relaxation optimized spectroscopy
References
Althoff T, Mills DJ, Popot JL, Kühlbrandt W (2011) Arrangement of electron transport chain components in bovine mitochondrial supercomplex I1III2IV1. EMBO J 30:4652–4664
Arnold T, Poynor M, Nussberger S, Lupas AN, Linke D (2007) Gene duplication of the eight-stranded β-barrel OmpX produces a functional pore: a scenario for the evolution of transmembrane β-barrel. J Mol Biol 366:1174–1184
Arora A, Abildgaard F, Bushweller JH, Tamm LK (2001) Structure of outer membrane protein A transmembrane domain by NMR spectroscopy. Nat Struct Biol 8:334–338
Banères JL, Martin A, Hullot P, Girard JP, Rossi JC, Parello J (2003) Structure-based analysis of GPCR function: conformational adaptation of both agonist and receptor upon leukotriene B4 binding to recombinant BLT1. J Mol Biol 329:801–814
Banères JL, Popot JL, Mouillac B (2011) New advances in production and functional folding of G-protein-coupled receptors. Trends Biotechnol 29:314–322
Bayburt TH, Grinkova YV, Sligar SG (2002) Self-assembly of discoidal phospholipid bilayer nanoparticles with membrane scaffold proteins. Nano Lett 2:853–856
Bazzacco P, Billon-Denis E, Sharma KS, Catoire LJ, Mary S, Le Bon C, Point E, Banères JL, Durand G, Zito F, Pucci B, Popot JL (2012) Nonionic homopolymeric amphipols: application to membrane protein folding, cell-free synthesis, and solution nuclear magnetic resonance. Biochemistry 51:1416–1430
Catoire LJ, Zoonens M, van Heijenoort C, Giusti F, Popot JL, Guittet E (2009) Inter- and intramolecular contacts in a membrane protein/surfactant complex observed by heteronuclear dipole-to-dipole cross-relaxation. J Magn Reson 197:91–95
Catoire LJ, Damian M, Giusti F, Martin A, van Heijenoort C, Popot JL, Guittet E, Banères JL (2010a) Structure of a GPCR ligand in its receptor–bound state: leukotriene B4 adopts a highly constrained conformation when associated to human BLT2. J Am Chem Soc 132:9049–9057
Catoire LJ, Zoonens M, van Heijenoort C, Giusti F, Guittet E, Popot JL (2010b) Solution NMR mapping of water-accessible residues in the transmembrane β-barrel of OmpX. Eur Biophys J 39:623–630
Catoire LJ, Damian M, Baaden M, Guittet E, Banères JL (2011) Electrostatically-driven fast association and perdeuteration allow detection of transferred cross-relaxation for G protein-coupled receptor ligands with equilibrium dissociation constants in the high-to-low nanomolar range. J Biomol NMR 50:191–195
Catoire LJ, Warnet XL, Warschawski DE (2014) Micelles, bicelles, amphipols, nanodiscs, liposomes or intact cells: the hitchhiker’s guide to the membrane protein study by NMR. In: Mus-Veteau I (ed) Membrane protein production for structural analysis. Springer, Berlin (in press)
Chae PS, Rasmussen SG, Rana RR, Gotfryd K, Chandra R, Goren MA, Kruse AC, Nurva S, Loland CJ, Pierre Y, Drew D, Popot JL, Picot D, Fox BG, Guan L, Gether U, Byrne B, Kobilka B, Gellman SH (2010) Maltose-neopentyl glycol (MNG) amphiphiles for solubilization, stabilization and crystallization of membrane proteins. Nat Methods 7:1003–1008
Champeil P, Menguy T, Tribet C, Popot JL, le Maire M (2000) Interaction of amphipols with the sarcoplasmic reticulum Ca2+-ATPase. J Biol Chem 275:18623–18637
Charvolin D, Perez JB, Rouvière F, Giusti F, Bazzacco P, Abdine A, Rappaport F, Martinez KL, Popot JL (2009) The use of amphipols as universal molecular adapters to immobilize membrane proteins onto solid supports. Proc Natl Acad Sci USA 106:405–410
Czerski L, Sanders CR (2000) Functionality of a membrane protein in bicelles. Anal Biochem 284:327–333
Dahmane T, Damian M, Mary S, Popot JL, Banères JL (2009) Amphipol-assisted in vitro folding of G protein-coupled receptors. Biochemistry 48:6516–6521
Dahmane T, Giusti F, Catoire LJ, Popot JL (2011) Sulfonated amphipols: synthesis, properties, and applications. Biopolymers 95:811–823
Dahmane T, Rappaport F, Popot JL (2013) Amphipol-assisted folding of bacteriorhodopsin in the presence and absence of lipids. Functional consequences. Eur Biophys J 42:85–101
Denisov IG, Grinkova YV, Lazarides AA, Sligar SG (2004) Directed self-assembly of monodisperse phospholipid bilayer nanodiscs with controlled size. J Am Chem Soc 126:3477–3478
Diab C, Tribet C, Gohon Y, Popot JL, Winnik FM (2007) Complexation of integral membrane proteins by phosphorylcholine-based amphipols. Biochim Biophys Acta 1768:2737–2747
Dupont M, De E, Chollet R, Chevalier J, Pages J-M (2004) Enterobacter aerogenes OmpX, a cation-selective channel mar- and osmo-regulated. FEBS Lett 569:27–30
Elter S, Raschle T, Arens S, Gelev V, Etzkorn M, Wagner G (2014) The use of amphipols for NMR structural characterization of 7-TM proteins. J Membr Biol (in press)
Etzkorn M, Raschle T, Hagn F, Gelev V, Rice AJ, Walz T, Wagner G (2013) Cell-free expressed bacteriorhodopsin in different soluble membrane mimetics: biophysical properties and NMR accessibility. Structure 21:394–401
Etzkorn M, Zoonens M, Catoire LJ, Popot JL, Hiller S (2014) How amphipols embed membrane proteins: Global solvent accessibility and interaction with a flexible protein terminus. J Membr Biol (in press)
Fernández C, Adeishvili K, Wüthrich K (2001) Transverse relaxation-optimized NMR spectroscopy with the outer membrane protein OmpX in dihexanoyl phosphatidylcholine micelles. Proc Natl Acad Sci USA 98:2358–2363
Fiaux J, Bertelsen EB, Horwich AL, Wüthrich K (2002) NMR analysis of a 900 K GroEL GroES complex. Nature 418:207–211
Giusti F, Popot JL, Tribet C (2012) Well-defined critical association concentration and rapid adsorption at the air/water interface of a short amphiphilic polymer, amphipol A8-35: a study by Förster resonance energy transfer and dynamic surface tension measurements. Langmuir 28:10372–10380
Giusti F, Rieger J, Catoire L, Qian, S, Calabrese AN, Watkinson TG, Radford S, Ashcroft A, Fradet A, Popot JL (2014) Synthesis, characterization and applications of a perdeuterated amphipol. J Membr Biol (in press)
Gohon Y, Pavlov G, Timmins P, Tribet C, Popot JL, Ebel C (2004) Partial specific volume and solvent interactions of amphipol A8-35. Anal Biochem 334:318–334
Gohon Y, Giusti F, Prata C, Charvolin D, Timmins P, Ebel C, Tribet C, Popot JL (2006) Well-defined nanoparticles formed by hydrophobic assembly of a short and polydisperse random terpolymer, amphipol A8-35. Langmuir 22:1281–1290
Gohon Y, Dahmane T, Ruigrok R, Schuck P, Charvolin D, Rappaport F, Timmins P, Engelman DM, Tribet C, Popot JL, Ebel C (2008) Bacteriorhodopsin/amphipol complexes: structural and functional properties. Biophys J 94:3523–3537
Griesinger C, Bennati M, Vieth HM, Luchinat C, Parigi G, Höfer P, Engelke F, Glaser SJ, Denysenkov V, Prisner TF (2012) Dynamic nuclear polarization at high magnetic fields in liquids. Prog Nucl Magn Reson Spectrosc 64:4–28
Hagn F, Etzkorn M, Raschle T, Wagner G (2013) Optimized phospholipid bilayer nanodiscs facilitate high-resolution structure determination of membrane proteins. J Am Chem Soc 135:1919–1925
Kang CB, Li Q (2011) Solution NMR study of integral membrane proteins. Curr Opin Struct Biol 15:560–569
Kelly E, Privé GG, Tieleman PD (2005) Molecular models of lipopeptide detergents: large coiled-coils with hydrocarbon interiors. J Am Chem Soc 127:13446–13447
Koutsopoulos S, Kaiser L, Eriksson HM, Zhang S (2012) Designer peptide surfactants stabilize diverse functional membrane proteins. Chem Soc Rev 41:1721–1728
Le Bon C, Popot JL, Giusti F (2014a) Labeling and functionalizing amphipols for biological applications. J Membr Biol (in press)
Le Bon C, Della Pia EA, Giusti F, Lloret N, Zoonens M, Martinez KL, Popot JL (2014b) Synthesis of an oligonucleotide-derivatized amphipol and its use to trap and immobilize membrane proteins. Nucleic Acids Res (in press)
Lee D, Walter KF, Brückner AK, Hilty C, Becker S, Griesinger C (2008) Bilayer in small bicelles revealed by lipid-protein interactions using NMR spectroscopy. J Am Chem Soc 130:13822–13823
Martinez KL, Gohon Y, Corringer PJ, Tribet C, Mérola F, Changeux JP, Popot JL (2002) Allosteric transitions of Torpedo acetylcholine receptor in lipids, detergent and amphipols: molecular interactions vs. physical constraints. FEBS Lett 528:251–256
McGregor CL, Chen L, Pomroy NC, Hwang P, Go S, Chakrabartty A, Privé GG (2003) Lipopeptide detergents designed for the structural study of membrane proteins. Nat Biotechnol 21:171–176
Mouillac B, Banères JL (2010) Mammalian membrane receptors expression as inclusion bodies in Escherichia coli. Methods Mol Biol 601:39–48
Nath A, Atkins WM, Sligar SG (2007) Applications of phospholipid bilayer nanodiscs in the study of membranes and membrane proteins. Biochemistry 46:2059–2069
Pautsch A, Schulz GE (2000) High-resolution structure of the OmpA membrane domain. J Mol Biol 298:273–282
Perlmutter JD, Drasler WJ, Xie W, Gao J, Popot JL, Sachs JN (2011) All-atom and coarse-grained molecular dynamics simulations of a membrane protein stabilizing polymer. Langmuir 27:10523–10537
Perlmutter JD, Popot J-L, Sachs, JN (2014) Molecular dynamics simulations of a membrane protein/amphipol complex. Submitted for publication
Picard M, Dahmane T, Garrigos M, Gauron C, Giusti F, le Maire M, Popot JL, Champeil P (2006) Protective and inhibitory effects of various types of amphipols on the Ca2+-ATPase from sarcoplasmic reticulum: a comparative study. Biochemistry 45:1861–1869
Plevin MJ, Boisbouvier J (2012) Isotope-labelling of methyl groups for NMR studies of large proteins In: Clore M, Potts J (eds) Recent developments in biomolecular NMR, RSC biomolecular sciences no. 25. Royal Society of Chemistry. doi:10.1039/9781849735391
Pocanschi CL, Dahmane T, Gohon Y et al (2006) Amphipathic polymers: tools to fold integral membrane proteins to their active form. Biochemistry 45:13954–13961
Poget SF, Girvin ME (2007) Solution NMR of membrane proteins in bilayer mimics: small is beautiful, but sometimes bigger is better. Biochim Biophys Acta 68:3098–3106
Popot JL (2010) Amphipols, nanodiscs, and fluorinated surfactants: three nonconventional approaches to studying membrane proteins in aqueous solutions. Ann Rev Biochem 79:737–775
Popot JL, Althoff T, Bagnard D et al (2011) Amphipols from A to Z. Ann Rev Biophys 40:379–408
Privé G (2009) Lipopeptide detergents for membrane protein studies. Curr Opin Struct Biol 19:1–7
Raschle T, Hiller S, Etzkorn M, Wagner G (2010) Nonmicellar systems for solution NMR spectroscopy of membrane proteins. Curr Opin Struct Biol 20:471–479
Renault M (2008) Etudes structurales et dynamiques de la protéine membranaire KpOmpA par RMN en phase liquide et solide. Ph. D. Thesis Université Paul Sabatier, Toulouse, France
Renault M, Tommassen-van Boxtel R, Bos MP, Post JA, Tommassen J, Baldus M (2012) Cellular solid-state nuclear magnetic resonance spectroscopy. Proc Natl Acad Sci USA 109:4863–4868
Riek R, Wider G, Pervushin K, Wüthrich K (1999) Polarization transfer by cross-correlated relaxation in solution NMR with very large molecules. Proc Natl Acad Sci USA 96:4918–4923
Ritchie TK, Grinkova YV, Bayburt TH, Denisov IG, Zolnerciks JK, Atkins WM, Sligar SG (2009) Chapter 11—reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol 464:211–213
Ruschak AM, Religa TL, Breuer S, Witt S, Kay LE (2010) The proteasome antechamber maintains substrates in an unfolded state. Nature 467:868–871
Sanders CR 2nd, Landis GC (1995) Reconstitution of membrane proteins into lipid-rich bilayered mixed micelles for NMR studies. Biochemistry 34:4030–4040
Sharma KS, Durand G, Gabel F, Bazzacco P, Le Bon C, Billon-Denis E, Catoire LJ, Popot JL, Ebel C, Pucci B (2012) Non–ionic amphiphilic homopolymers: synthesis, solution properties, and biochemical validation. Langmuir 28:4625–4639
Takeda M, Kainosho M (2012) Cell-free protein synthesis using E. coli cell extract for NMR studies. Adv Exp Med Biol 992:167–177
Tifrea DF, Sun G, Pal S, Zardeneta G, Cocco MJ, Popot JL, de la Maza LM (2011) Amphipols stabilize the Chlamydia major outer membrane protein and enhance its protective ability as a vaccine. Vaccine 29:4623–4631
Triba MN, Warschawski DE, Devaux PF (2005) Reinvestigation by phosphorus NMR of lipid distribution in bicelles. Biophys J 88:1887–1901
Tribet C, Audebert R, Popot JL (1996) Amphipols: polymers that keep membrane proteins soluble in aqueous solutions. Proc Natl Acad Sci USA 93:15047–15050
Tribet C, Diab C, Dahmane T, Zoonens M, Popot JL, Winnik FM (2009) Thermodynamic characterization of the exchange of detergents and amphipols at the surfaces of integral membrane proteins. Langmuir 25:12623–12634
Vogt J, Schulz GE (1999) The structure of the outer membrane protein OmpX from Escherichia coli reveals possible mechanisms of virulence. Structure 7:1301–1309
Vold RR, Prosser RS, Deese AJ (1997) Isotropic solutions of phospholipid bicelles: a new membrane mimetic for high-resolution NMR studies of polypeptides. J Biomol NMR 9:329–335
Wang X, Corin K, Baaske P, Wienken CJ, Jerabek-Willemsen M, Duhr S, Braun D, Zhang S (2011) Peptide surfactants for cell-free production of functional G protein-coupled receptors. Proc Natl Acad Sci USA 108:9049–9054
Warschawski DE, Arnold AA, Beaugrand M, Gravel A, Chartrand E, Marcotte I (2011) Choosing membrane mimetics for NMR structural studies of transmembrane proteins. Biochim Biophys Acta 1808:1957–1974
Wüthrich K (1986) NMR of proteins and nucleic acids. Wiley, New York
Yokomizo T, Kako K, Terawaki K, Izumi T, Shimizu T (2000) A second leukotriene B(4) receptor, BLT2. A new therapeutic target in inflammation and immunological disorders. J Exp Med 192:421–432
Zhao X, Nagai Y, Reeves PJ, Kiley P, Khorana HG, Zhang S (2006) Designer short peptide surfactants stabilize G protein-coupled receptor bovine rhodopsin. Proc Natl Acad Sci USA 103:17707–17712
Zhou HX, Cross TA (2013) Influences of membrane mimetic environments on membrane protein structures. Annu Rev Biophys 42:361–392
Zoonens M, Catoire LJ, Giusti F, Popot JL (2005) NMR study of a membrane protein in detergent–free aqueous solution. Proc Natl Acad Sci USA 102:8893–8898
Zoonens M, Giusti F, Zito F, Popot JL (2007) Dynamics of membrane protein/amphipol association studied by Förster resonance energy transfer. Implications for in vitro studies of amphipol-stabilized membrane proteins. Biochemistry 46:10392–10404
Zoonens M, Zito F, Martinez KL, Popot JL (2014) Amphipols: a general introduction and some protocols. In: Mus-Veteau I (ed) Membrane protein production for structural analysis. Springer, Berlin
Acknowledgments
We are extremely grateful to Sophie Walmé and Marie-Noëlle Rager (ChimieParisTech, Paris), and Carine van Heijenoort (ICSN, Gif/Yvette) for assistance with NMR experiments. We express our gratitude to B. Pucci (Université d’Avignon et des Pays de Vaucluse) for his long-term involvement in the development of non-ionic amphipols. This work was supported by the Centre National de la Recherche Scientifique (CNRS), Paris-7 University (Sorbonne Paris Cité), the “Initiative d’Excellence” program from the French State (Grant “DYNAMO,” ANR-11-LABX-0011-01), Human Frontier Science Program Organization Grant RG00223/2000-M, from the Agence Nationale pour la Recherche ANR-07-BLAN-0092 “Amphipol-assisted folding of GPCRs,” and E.U. Specific Targeted Research Project “Innovative tools for membrane protein structural proteomics.” LJC is a recipient of Projects Exploratoires/Premier Soutien (PEPS, LeukomotiVe project) from the CNRS.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Planchard, N., Point, É., Dahmane, T. et al. The Use of Amphipols for Solution NMR Studies of Membrane Proteins: Advantages and Constraints as Compared to Other Solubilizing Media. J Membrane Biol 247, 827–842 (2014). https://doi.org/10.1007/s00232-014-9654-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00232-014-9654-z