
Abstract
Type 1 diabetes mellitus (T1DM) is an autoimmune condition driven by T lymphocytes that specifically declines the function of beta cells of pancreas. Immunological treatments aim to stop this decline in β-cell function thus preventing TIDM. Although TIDM occur at any age, it is one of the most common chronic disorders in children. T1DM accounts for 5 to 10% of all cases of diabetes amounting 21–42 million affected persons. Teplizumab is a novel drug recently approved by the US FDA for the treatment of T1DM. This drug reduces abnormal glucose tolerance who are at high risk for developing T1DM and have antibodies suggesting an immunological attack on their pancreas. A 14-day infusion of the drug prevents T cells’ attack of the insulin-producing cells of the pancreas. Adverse events due to teplizumab reported so far mild and of limited duration. This review gives an overview of the preclinical and clinical research on teplizumab for their role in new-onset T1DM.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Type 1 diabetes mellitus (T1DM) is an autoimmune disorder that results in the destruction of beta cells of the pancreas that produce insulin. T1DM patients need insulin replacement therapy. Diabetes-related ketoacidosis (DKA), a potentially fatal condition, can develop without insulin [1, 2]. T1DM has a genetic predisposition with a substantial association with specific HLA (DR and DQ) alleles, particularly DRB103-DQB10201 and DRB10401-DQB10302H.
Epidemiology
Although T1DM can occur at any age, it is one of the most common chronic disorders in children. T1DM accounts for 5–10% of all cases of diabetes mellitus amounting 21–42 million affected persons [3,4,5]. T1DM has a variable incidence and prevalence geographically [5, 6], and the incidence has changed over time [7, 8]. In the USA, T1DM currently affects an estimated 1.24 million, and this figure is projected to reach 5 million by 2050. In persons 20 years of age or less, the prevalence of T1DM has increased by 21% between 2001 and 2009. T1DM can affect a person at any age; however, the greatest occurrence is between the ages of 10 and 14 [9]. Thus, affecting from the young age, T1DM being a chronic incurable condition has significant impact on the quality as well as longevity of the affected person.
Natural history of T1DM
There is the autoimmune onslaught against beta cells at least 5 years prior to the clinical appearance of diabetes [10]. In the Diabetes Prevention Trial 1 (DPT1), loss of first-phase insulin secretion, verified islet autoantibodies, and the absence of the protective HLA allele DQB1*0602 are all linked to a greater than 50% chance of developing diabetes over the course of 5 years [11, 12]. The honeymoon phase characterized by a period of clinical remission following therapy for the acute presentation occurs even after the diagnosis of diabetes as there is still significant residual beta cell function [13]. Although it is frequently assumed that the reduction in beta cell loss is linear, this is probably not the case. The net decline represents the balance between cell regeneration and cell destruction [14]. It is likely that immune destruction waxes and wanes, and severe loss may be followed by a period of relative stability before further decline. The clinical remission following the initial presentation is most likely caused by the beta cells present at diagnosis regaining their ability to secrete insulin as opposed to fresh beta cell regeneration. Unfortunately, the remission is frequently brief, and because beta cell function decreases, many patients soon need full replacement dosages of insulin. To stop the loss of residual beta cell function, immunological treatments have been made both before the onset of TIDM and after diagnosis [15].
Current treatments
Data from the Diabetes Control and Complications Trial and the Epidemiology of Diabetes and its Complications study suggest that intensive insulin therapy along with strict glycaemic control can halt or slow the progression of microvascular complications. This also lowers the risk of developing macrovascular complications and lower mortality from all causes. T1D Exchange is a database of T1DM patients established in 2010 developed to a resource for fostering research on TIDM. Insulin therapy is the cornerstone of the care of T1DM. Multiple daily injections of insulin or subcutaneous insulin infusion using insulin pumps are two ways to give insulin subcutaneously. Effective control also requires the use of self-monitoring of blood glucose utilizing upgraded glucometer, continuous glucose monitors (CGM), and more recent insulin pumps with sensor-enhanced systems built in. Effective disease management of T1DM requires addressing the psychological elements of the condition [9, 16].
Need for new drug for TIDM
Few years before the beginning of clinical symptoms, immunologic signs of T1DM can be observed. Years prior to the onset of clinical symptoms of T1DM, there are CD4 + and CD8 + T lymphocytes that recognize autoantigens that kill majority of beta cells [17]. Targeting immune cells in immunotherapies has shown promising results in the management of T1DM. Immunosuppressive drugs such as cyclosporine, azathioprine, prednisone, and anti-thymocyte globulin were initially used, but their utility was limited due to their adverse effects and minor influence on the arrest of the disease development [18,19,20,21]. Rituximab, CTLA4Ig, and alefacept can delay the drop in C-peptide levels in some people in the first few years after the onset of the disease [22,23,24]. Not all patients respond to therapy, and effect of rituximab start to fade after 2 years [25]. Treatment with CTLA4-Ig can delay disease progression by 9.6 months in people with newly diagnosed T1DM [18, 22, 23]. Many medications, even those that were successful in treating other autoimmune diseases or in the non-obese diabetic (NOD) preclinical model, have fallen short, though. Anakinra and canakinumab, which inhibit the IL-1 pathway, neither substantially differ from placebo and drug-treated groups in those with recent-onset T1DM nor did immunization with GAD65 [26, 27]. Proinsulin-encoded vaccines or TNF-blocking therapies are still under development [28, 29]. Antibodies against anti-CD3 have been shown to prevent or even reverse the onset of T1D in preclinical animal models [30].
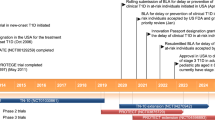
Teplizumab is a humanized IgG1 kappa CD3-directed monoclonal antibody that recognizes an epitope of the CD3-epsilon chain produced on mature cells. It is also known as MGA031 and hOKT3-g1 (Ala-Ala). Anti-CD3 medication has historically been used in organ transplantation to avoid graft-versus-host disease, but more recently, it has been investigated as a way to delay the development of T1DM in high-risk individuals. Antibodies against the CD3 portion of the T cell receptor (TCR) targets T cells. Although mechanism of action of teplizumab is still not entirely understood, however, it may involve deactivating pancreatic beta cell autoreactive T lymphocytes and partial agonistic signalling. Antigen-non-specifically, Fc-receptors can attach to the “tail-end” of anti-CD3 antibodies and cause serious side effects associated with cytokine release syndrome (CRS). Teplizumab is an Fc-non-binding antibody that was created to lower the prevalence of CRS [31]. Teplizumab got FDA approval in November 2022 to be the first medication that can postpone the onset of type 1 diabetes [32]. Here, we discuss the mechanism of action, safety, pharmacology, kinetics, and effect of teplizumab on T1DM and review the information from the clinical trials.
Development of teplizumab
P. Kung and G. Goldstein created the first murine monoclonal antibody (OKT3, Muromonab-CD3) that was specific for the human CD3 epsilon chain in 1979, and it was clinically tested for the treatment of patients with renal allografts in 1981 [33]. Later, acute rejection in recipients of liver and heart transplants was managed with OKT3 therapy [34]. However, the emergence of human anti-mouse antibody responses [35] and cytokine release syndrome (CRS) [36] has discouraged its therapeutic use. The Fc part of the antibody attaching to Fc receptors and cross-linking the CD3 molecule, the complement pathway activation, and the murine origin are all thought to be responsible for these unfavourable effects of the OKT3 [37,38,39]. Later research claimed that F(ab)2 fragments lacking the Fc region could reduce the release of inflammatory cytokines [40]. Thus, humanized anti-CD3 mAbs with decreased Fc receptor binding affinities was developed. One of the earliest humanized IgG2 antibodies, Nuvion (HuM291), was clinically tested for inflammatory bowel disease. With Nuvion, the incidences of CRS were decreased [41]. To develop teplizumab, amino acids 234 and 235 were substituted with alanine, which resulted in a 100–1000-fold reduction in the binding to Fc receptors and a reduction in T-cell activation, cytokine release, and complement activation [42, 43].
Pharmacokinetics
The pharmacokinetics of teplizumab following 14-day intravenous administration was best described by using a two-compartment model with saturable binding in central and peripheral compartments by Daifotis et al. At concentrations much higher than the binding capacity of the target (129 ng/mL), teplizumab pharmacokinetics were characterized by bi-exponential decay. The distribution and terminal half-lives for the bi-exponential decay were t1/2a = 0.19 day and t1/2b = 4.01 days. Teplizumab’s estimated daily clearance with the proven effective 14-day full-dose regimen was 2.3 l/day [44]. Anti-drug antibodies, which have been discovered in around 50% of patients treated with the treatment [45], speed up the clearance of teplizumab. The impact of the production of anti-drug antibodies on the clinical response is uncertain because a clear effect of repeated treatment has not been demonstrated in clinical studies and because it takes around 10 days for these antibodies to manifest. Teplizumab steady-state concentrations are not anticipated to be reached during the 14-day treatment duration. Teplizumab has a centre volume of distribution (Vd) of 2.27 L in a 60-kg patient. Teplizumab is a monoclonal antibody; thus, it is anticipated that proteases located throughout the body will break it down into minute peptides. Teplizumab-mzwv has a mean (SD) terminal elimination half-life and clearance in a 60-kg subject of 4.5 (0.2) days and 2.7 (0.8) L/day, respectively. Teplizumab has a mean terminal elimination half-life of 4.5 days in a 60-kg person. Teplizumab had a clearance of 2.7 L/day in a 60-kg patient [46].
Pharmacodynamics
Teplizumab is a monoclonal antibody that binds to CD3 molecules on the surface of CD4 + and CD8 + T lymphocytes, both of which are responsible for the destruction of pancreatic beta cells [46]. In early-onset T1DM patients receiving teplizumab, C-peptide levels have been found to rise, indicating enhanced beta cell function [47, 48]. Teplizumab’s pharmacodynamic time-response, safety, and exposure–response relationships have not been thoroughly understood [46]. Teplizumab usage in a 14-day course of therapy can result in lymphopenia if T cell depletion is not occurring. Teplizumab can also cause CRS from fifth day of onset of treatment. Teplizumab also carries the risk of hypersensitivity reactions and life-threatening infections [46].
Mechanism of action
T1DM is a chronic condition that progresses through three distinct stages, only stage 3 of which is characterized by outwardly visible symptoms [49]. The existence of at least two autoantibodies against pertinent antigens is the first sign of T1DM risk, indicating a crucial role for B cells in what has typically been thought to as a T cell-dominated illness [49, 50]. It is obvious that T and B cells contribute to T1DM when other findings from animal and human studies are considered, and treatment has centred on focusing on each of them individually as well as their interconnections [50]. The T cell receptor (TCR) is made up of six CD3 molecules, including two CD3 chains and TCR and chains. It is in charge of identifying antigens presented on other cells’ MHC complexes in order to evoke a response [51]. Teplizumab (also known as huOKT3ala-ala), a humanized IgG1 Fc-nonbinding adaptation of an existing mouse OKT3 antibody, is specific for the chain of CD3 and prevents T cell activation by steric inhibition of antigen recognition [42, 51, 52]. Teplizumab has recently proven effective in reducing the time it takes for people at high risk of developing T1D to receive a diagnosis [53]. The precise mechanism driving this action is still unknown, though. Teplizumab may function as a partial agonist at the TCR, boosting the proportion of worn-out T cells that are positive for KLRG1, TIGIT, and CD8. These worn-out T cells continue to exist but are unable to carry out effector duties; as a result, it is doubtful that they will help destroy further cells [49, 53, 54]. A rise in circulating CD8 + central memory (CD8CM) T cells is one shift in the T cell populations of clinical responders that has been observed in other investigations [55]. However, it is evident that individuals who have an active immune response and have not yet advanced to stage 3 benefit from treatment the most [49].
Preclinical studies
A low dosage of the CD3 monoclonal antibody (mAb) 145 2C11 restored self-tolerance to beta cell antigens (Ags) in adult overtly diabetic non-obese diabetic (NOD) mice in one of the earliest preclinical experiments. The entire and lasting remission of diabetes was shown within 2 to 4 weeks of treatment. Animals protected by CD3 Ab did not lose their autoreactive T cells. IFN-gamma production by activated spleen cells was markedly reduced in treated mice for 5 to 7 weeks following treatment. One distinctive characteristic was that the CD3 Ab-induced tolerance only developed as a result of treating obviously diabetic NOD mice. Treatment of mice with recently developed illness was the only method that consistently produced protection (14–20 weeks old) [56]. In a different experiment on animals, the causes of suppression in anti-CD3-treated and untreated NOD mice were investigated. In spontaneously diabetic NOD mice, a subpopulation of foxP3( +) cells inside a CD4( +)CD25(low) lymphocyte subset suppresses T cell immunity in a TGF-beta-dependent manner, a functional characteristic of “adaptive” regulatory T cells. It is possible that NOD mice produce these adaptive Tregs in an effort to control ongoing autoimmunity because this distinct Treg fraction is present in NOD mice but not in normal mice. Importantly, anti-CD3 immunotherapy can produce these TGF-beta-dependent adaptive CD4( +)CD25(low) T cells from peripheral CD4( +)CD25(-) T lymphocytes in two different in vivo models, which is associated with the recovery of self-tolerance [57]. You et al. conducted preclinical study to find out the evidence that CD4( +) regulatory T cells control progression of autoimmune insulitis in non-obese diabetic (NOD) mice. They studied the nature of these regulatory T cells and their mode of action in diabetes-prone NOD Rag(− / −) or severe combined immunodeficient (SCID) mice harbouring a transgenic T cell receptor derived from the diabetogenic T cell clone BDC2.5. It showed that diabetes onset is prevented in such mice by infusion of polyclonal CD4( +) T cells expressing L-selectin (CD62L) but not prevented or only marginally prevented by CD4( +)CD25( +) T cells. Similarly, they found with a cotransfer model that CD4( +)CD62L( +) T cells but not CD4( +)CD25( +) T cells inhibited diabetes transfer into NOD SCID recipients by transgenic NOD BDC2.5 SCID cells. Unexpectedly, cotransfer of transgenic NOD BDC2.5 SCID cells and spleen cells from WT diabetic NOD mice did not induce diabetes, whereas each individual population did so. Collectively, these data confirm the central role of CD4( +)CD62L( +) regulatory T cells in controlling disease onset in a well-defined transgenic model of autoimmune diabetes and suggest the intervention of homeostatic mechanisms as part of their mode of action [58]. In another study, CD3-specific antibodies’ unique capacity to restore self-tolerance in autoimmunity was established. They induce long-term remission of overt diabetes in non-obese diabetic (NOD) mice and in human type I diabetes. In this study, treatment with CD3 epsilon-specific antibodies induces transferable T-cell-mediated tolerance involving CD4 + CD25 + cells. However, these CD4 + CD25 + T cells are distinct from naturally occurring regulatory T cells that control physiological autoreactivity. CD3-specific antibody treatment induced remission in NOD Cd28 − / − mice that were devoid of such regulatory cells. Remission of diabetes was abrogated by coadministration of a neutralizing transforming growth factor (TGF)-beta-specific antibody. The central role of TGF-beta was further suggested by its increased, long-lasting production by CD4 + T cells from tolerant mice [59]. In autoimmune diabetes, short course treatment with FcR-nonbinding (FNB) anti-CD3 mAb in mice with recent onset of diabetes induces long-term disease remission. Induction of tolerogenic regulatory T cells (Tregs) has been implicated to be one of the mechanisms of action by FNB anti-CD3 mAb in these settings. In this study, Penaranda et al. examined the effect of FNB anti-CD3 mAb treatment on the homeostasis of naive, effector, and regulatory T cells in vivo. Anti-CD3 treatment induced a transient systemic rise in the percentage but not absolute number of CD4( +)Foxp3( +) Tregs due to selective depletion of CD4( +)Foxp3( −) conventional T cells. T cell depletion induced by FNB anti-CD3 mAb was independent of the proapoptotic proteins Fas, caspase-3, and Bim and was not inhibited by overexpression of the anti-apoptotic protein, Bcl-2. Tregs were not preferentially expanded, and we found no evidence of conversion of conventional T cells into Tregs, suggesting that the pre-existing Tregs are resistant to anti-CD3-induced cell death [60]. Interleukin (IL)-17-producing T helper cells (T(H)17) can drive antigen-specific autoimmune diseases and are considered the main population of pathogenic T cells driving experimental autoimmune encephalomyelitis (EAE) in the mouse model for multiple sclerosis developed by Esplugues et al. They reported that pro-inflammatory T(H)17 cells can be redirected to and controlled in the small intestine by using a model of tolerance induced by CD3-specific antibody and a model of sepsis and influenza A viral infection (H1N1). T(H)17-specific IL-17A secretion induced expression of the chemokine CCL20 in the small intestine, facilitating the migration of these cells specifically to the small intestine via the CCR6/CCL20 axis. These results identify mechanisms limiting T(H)17 cell pathogenicity and implicate the gastrointestinal tract as a site for control of T(H)17 cells. This study in mice, together with studies in NOD mice, humanized mice, and patients provide evidence for induction of regulatory T 34 cells with teplizumab [61]. Waldron-Lynch et al. used a humanized mouse reconstituted with human hematopoietic stem cells to study the mechanism of action of teplizumab, for the treatment of patients with type 1 diabetes mellitus. In this model, human gut-tropic CCR6( +) T cells exited the circulation and secondary lymph organs and migrated to the small intestine. These cells then produced interleukin-10 (IL-10), a regulatory cytokine, in quantities that could be detected in the peripheral circulation. Blocking T cell migration to the small intestine with natalizumab, which prevents cellular adhesion by inhibiting α(4) integrin binding, abolished the treatment effects of teplizumab. Moreover, IL-10 expression by CD4( +)CD25(high)CCR6( +)FoxP3 cells returning to the peripheral circulation was increased in patients with type 1 diabetes treated with teplizumab [62].
Clinical studies
In one of the first trial to show improvement in metabolic responses and delay of diabetes with immune therapy, Sims et al. performed a randomized controlled trial. They enrolled non-diabetic relatives at high-risk for T1DM to see the effect of a single 14-day course of teplizumab. In an extended follow-up (923-day median) of a previous report of teplizumab treatment, the median times to diagnosis were 59.6 and 27.1 months for teplizumab and placebo-treated participants, respectively (HR = 0.457, P = 0.01). Fifty percent of teplizumab-treated but only 22% of the placebo-treated remained diabetes free. Teplizumab treatment improved beta cell function, reflected by average on-study C-peptide AUC (1.94 vs. 1.72 pmol/mL; P = 0.006). Drug treatment reversed a decline in insulin secretion prior to enrolment followed by stabilization of the declining C-peptide AUC seen with placebo treatment. The teplizumab treatment was associated with increase in partially exhausted memory KLRG1 + TIGIT + CD8 + T cells (r = 0.44; P = 0.014) that showed reduced secretion of IFNγ and TNFα. A single course of teplizumab had lasting effects on delay of T1D diagnosis and improved beta cell function in high-risk individuals. Changes in CD8 + T cell subsets indicate that partially exhausted effector cells are associated with clinical response [47].
Herold et al. conducted a phase 2, randomized, placebo-controlled, double-blind trial on teplizumab. They concluded that drug delayed the progression to clinical type 1 diabetes in high-risk participants. They also included relatives of patients with type 1 diabetes who did not have diabetes but were at high risk for development of clinical disease. Patients were randomly assigned to a single 14-day course of teplizumab or placebo. The progression to clinical type 1 diabetes was diagnosed by oral glucose-tolerance tests (OGTT) performed at 6-month intervals. The median time to the diagnosis of type 1 diabetes was 48.4 months in the teplizumab group and 24.4 months in the placebo group. The disease was diagnosed in 19 (43%) of the participants who received teplizumab and in 23 (72%) of those who received placebo. The hazard ratio for the diagnosis of type 1 diabetes (teplizumab vs. placebo) was 0.41 (95% confidence interval, 0.22 to 0.78; P = 0.006 by adjusted Cox proportional hazards model) [53]. Nourelden et al. conducted a metanalysis with the aim to assess the safety and efficacy of teplizumab in T1DM patients. Eight randomized clinical trials with 866 patients were selected. Teplizumab was associated with lower insulin use than placebo at 6 months (P < 0.001), 12 months (P < 0.001), 18 months (P < 0.001), and 24 months (P = 0.003). The area under the curve of C-peptide was significantly increased in the teplizumab group at 12 months (P = 0.03), 18 months (P = 0.03), and 24 months (P = 0.03). No significant effect of teplizumab on HbA1c levels was observed at any time point. Teplizumab was found to be associated with some side effects such as lymphopenia, skin, and subcutaneous tissue disorders [63]. In another open-label, randomized, controlled clinical trial, the immune therapies in new-onset type 1 diabetes (T1DM) have shown success. The main aim was to determine whether two courses of teplizumab reduce the decline in C-peptide levels in patients with T1DM, 2 years after disease onset. In the intent to treat analysis of the primary end point, patients treated with teplizumab had a reduced decline in C-peptide at 2 years when compared to control (P = 0.002). Thus, it was concluded that teplizumab treatment preserves insulin production and reduces the use of exogenous insulin in some patients with new-onset T1DM [64]. In one of the metanalysis to review the pharmacology, pharmacokinetics, safety, and efficacy of teplizumab, relevant studies from clinical trial registry, MEDLINE, International Pharmaceutical Abstracts, ClinicalTrials.gov, American Diabetes Association scientific posters, and Google Scholar (1966–May 2012) were included in this meta-analysis. While clinical data were limited, both phase 2 and phase 3 studies have demonstrated preserved C-peptide response as a measure of insulin production, decreased exogenous insulin use, and improved glycaemic control following a 12- to 14-day teplizumab infusion in patients diagnosed with T1DM within the previous 6 weeks. However, 1 phase 3 trial failed to find the same benefits in those diagnosed with T1DM within the previous 12 weeks when a lower cumulative teplizumab dose was used. Initial studies indicated that teplizumab is well tolerated, with a self-limiting rash as the most commonly reported adverse effect [65]. Protégé trial was a phase 3, randomized, double-blind, parallel, placebo-controlled study. In this, teplizumab was administered via intravenous route, daily for 14 days at baseline and again after 26 weeks, in new-onset T1DM. A 14-day treatment reduced the loss of C-peptide mean area under the curve (AUC), at 2 years versus placebo. Exogenous insulin needs were reduced as compared to placebo. Although anti-drug antibodies developed in some patients, but there was no apparent change in drug efficacy. No new safety or tolerability issues were observed over 2 years [45].
One of the only studies that were done for cost-effectiveness since the potentially high price may pose challenges for coverage and reimbursement for payers and policymakers. Mittal et al. used Markov microsimulation modelling, to compare the cost effectiveness of five options for choosing target individuals. The five options were all at-risk individuals, individuals without human leukocyte antigen (HLA)-DR3 or with HLA-DR4 allele, individuals without HLA-DR3 and with HLA-DR4 allele, individuals with anti-zinc transporter 8 (ZnT8) antibody negative, and no provision at all at different possible prices of teplizumab. Quality-adjusted life years were used to quantify effectiveness. Costs were calculated from the viewpoint of the health system. It was found to be cost-effective to treat only individuals without HLA-DR3 or with HLA-DR4 alleles. Thus, cost-effective provision of teplizumab to target individuals depends on the price of teplizumab and genetic and antibody characteristics of treated individuals [66]. Some important clinical trials are summarized in Table 1.
Conclusion
A 14-day infusion of the teplizumab, anti-CD3 immunotherapy, prevents T cells’ attack of the insulin-producing cells of the pancreas. Drug has been found to be effective in preclinical and clinical studies. It was shown to improve the metabolic responses and delay the onset of diabetes Adverse events due to teplizumab reported so far mild and of limited duration. Evidences from metanalysis and other RCT show that teplizumab treatment preserves insulin production and reduces the use of exogenous insulin in some patients with new-onset T1DM.
It is necessary to do more research to benefit from the knowledge gained from these initial studies.
Availability of data and materials
Not applicable.
References
Saxby N, Beggs S, Kariyawasam N, Battersby M, Lawn S (2020) Do guidelines provide evidence-based guidance to health professionals on promoting developmentally appropriate chronic condition self-management in children? A Syst Rev Chronic Illn 16(4):239–252
Yue Y, Tang Y, Tang J, Shi J, Zhu T, Huang J et al (2018) Maternal infection during pregnancy and type 1 diabetes mellitus in offspring: a systematic review and meta-analysis. Epidemiol Infect 146(16):2131–2138
Lucier J, Weinstock RS (2022) Diabetes Mellitus Type 1. [Updated 2022 May 11]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. Available from: https://www.ncbi.nlm.nih.gov/books/NBK507713/
Zimmet P, Alberti KG, Shaw J (2001) Global and societal implications of the diabetes epidemic. Nature 414(6865):782–787
Chen L, Magliano DJ, Zimmet PZ (2011) The worldwide epidemiology of type 2 diabetes mellitus–present and future perspectives. Nat Rev Endocrinol 8(4):228–236
Greenbaum CJ, Speake C, Krischer J, Buckner J, Gottlieb PA, Schatz DA et al (2018) Strength in numbers: opportunities for enhancing the development of effective treatments for type 1 diabetes-the TrialNet experience. Diabetes 67:1216–1225
Atkinson MA, Eisenbarth GS, Michels AW (2014) Type 1 diabetes. Lancet 383(9911):69–82
Patterson CC, Gyürüs E, Rosenbauer J, Cinek O, Neu A, Schober E et al (2012) Trends in childhood type 1 diabetes incidence in Europe during 1989–2008: evidence of non-uniformity over time in rates of increase. Diabetologia 55(8):2142–2147
Indian Council of Medical Research (ICMR) [home page in Internet] ICMR Guidelines for management of type 1 diabetes. [Last Cited on 27th January 2023]. Available from: https://main.icmr.nic.in/sites/default/files/upload_documents/ICMR_Guidelines_for_Management_of_Type_1_Diabetes.pdf
Gianani R, Eisenbarth GS (2005) The stages of type 1A diabetes: 2005. Immunol Rev 204:232–249
Skyler JS, Krischer JP, Wolfsdorf J, Cowie C, Palmer JP, Greenbaum C et al (2005) Effects of oral insulin in relatives of patients with type 1 diabetes: the diabetes prevention trial–type 1. Diabetes Care 28(5):1068–1076
Skyler JS (2007) Prediction and prevention of type 1 diabetes: progress, problems, and prospects. Clin Pharmacol Ther 81:768–771
Martin S, Pawlowski B, Greulich B, Ziegler AG, Mandrup-Poulsen T, Mahon J (1992) Natural course of remission in IDDM during 1st yr after diagnosis. Diabetes Care 15(1):66–74
Pasquali L, Fan Y, Trucco M, Ringquist S (2006) Rehabilitation of adaptive immunity and regeneration of beta cells. Trends Biotechnol 24(11):516–522
Masharani UB, Becker J (2010) Teplizumab therapy for type 1 diabetes. Expert Opin Biol Ther 10(3):459–465
Subramanian S, Baidal D (2021) The management of type 1 diabetes. In: Feingold KR, Anawalt B, Boyce A, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279114/23
Eisenbarth GS, Srikanta S, Jackson R, Rabinowe S, Dolinar R, Aoki T et al (1985) Anti-thymocyte globulin and prednisone immunotherapy of recent onset type 1 diabetes mellitus. Diabetes Res 2(6):271–276
Bougnères PF, Landais P, Boisson C, Carel JC, Frament N, Boitard C et al (1990) Limited duration of remission of insulin dependency in children with recent overt type I diabetes treated with low-dose cyclosporin. Diabetes 39(10):1264–1272
Parving HH, Tarnow L, Nielsen FS, Rossing P, Mandrup-Poulsen T, Osterby R et al (1999) Cyclosporine nephrotoxicity in type 1 diabetic patients. A 7-year follow-up study. Diabetes Care 22(3):478–83
Silverstein J, Maclaren N, Riley W, Spillar R, Radjenovic D, Johnson S (1988) Immunosuppression with azathioprine and prednisone in recent-onset insulin-dependent diabetes mellitus. N Engl J Med 319(10):599–604
Chatenoud L (2010) Immune therapy for type 1 diabetes mellitus-what is unique about anti-CD3 antibodies? Nat Rev Endocrinol 6(3):149–157
Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R et al (2009) Type 1 Diabetes TrialNet Anti-CD20 Study Group. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med 361(22):2143–52
Orban T, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R et al (2011) Type 1 Diabetes TrialNet Abatacept Study Group. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet 378(9789):412–9
Rigby MR, DiMeglio LA, Rendell MS, Felner EI, Dostou JM, Gitelman SE et al (2013) T1DAL Study Team. Targeting of memory T cells with alefacept in new-onset type 1 diabetes (T1DAL study): 12 month results of a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Diabetes Endocrinol 1(4):284–94
Pescovitz MD, Greenbaum CJ, Bundy B, Becker DJ, Gitelman SE, Goland R et al (2014) Type 1 Diabetes TrialNet Anti-CD20 Study Group. B-lymphocyte depletion with rituximab and β-cell function: two-year results. Diabetes Care 37(2):453–9
Moran A, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R et al (2013) Interleukin-1 antagonism in type 1 diabetes of recent onset: two multicentre, randomised, double-blind, placebo-controlled trials. Lancet 381(9881):1905–1915
Agardh CD, Lynch KF, Palmér M, Link K, Lernmark A (2009) GAD65 vaccination: 5 years of follow-up in a randomised dose-escalating study in adult-onset autoimmune diabetes. Diabetologia 52(7):1363–1368
Mastrandrea L, Yu J, Behrens T, Buchlis J, Albini C, Fourtner S et al (2009) Etanercept treatment in children with new-onset type 1 diabetes: pilot randomized, placebo-controlled, double-blind study. Diabetes Care 32(7):1244–1249
Roep BO, Solvason N, Gottlieb PA, Abreu JRF, Harrison LC, Eisenbarth GS et al (2013) BHT-3021 Investigators; Steinman L. Plasmid-encoded proinsulin preserves C-peptide while specifically reducing proinsulin-specific CD8+ T cells in type 1 diabetes. Sci Transl Med 5(191):191ra82
Chatenoud L, Primo J, Bach JF (1997) CD3 antibody-induced dominant self-tolerance in overtly diabetic NOD mice. J Immunol 158(6):2947–2954
Nadeem A, Ashraf MR, Javed M, Hussain T, Tariq MS, Babar ME (2018) Review - microRNAs: a new paradigm towards mechanistic insight of diseases. Pak J Pharm Sci 31(5):2017–2026
Drugbank [Home page in Internet] Teclizumab. [Last cited on 27th January 2023] Available from: https://go.drugbank.com/drugs/DB06273
Cosimi AB, Colvin RB, Burton RC, Rubin RH, Goldstein G, Kung PC et al (1981) Use of monoclonal antibodies to T-cell subsets for immunologic monitoring and treatment in recipients of renal allografts. N Engl J Med 305(6):308–314
Hooks MA, Wade CS, Millikan WJ Jr (1991) Muromonab CD-3: a review of its pharmacology, pharmacokinetics, and 37. clinical use in transplantation. Pharmacotherapy 11(1):26–37
Chatenoud L, Baudrihaye MF, Chkoff N, Kreis H, Goldstein G, Bach JF (1986) Restriction of the human in vivo immune response against the mouse monoclonal antibody OKT3. J Immunol 137(3):830–838
Chatenoud L, Ferran C, Legendre C, Thouard I, Merite S, Reuter A et al (1990) In vivo cell activation following OKT3 administration. Systemic cytokine release and modulation by corticosteroids. Transplantation 49(4):697–702
Smith KG, Austyn JM, Hariri G, Beverley PC, Morris PJ (1986) T cell activation by anti-T3 antibodies: comparison of IgG1 and IgG2b switch variants and direct evidence for accessory function of macrophage Fc receptors. Eur J Immunol 16(5):478–486
Parren PW, Warmerdam PA, Boeije LC, Capel PJ, van de Winkel JG, Aarden LA (1992) Characterization of IgG FcR-mediated proliferation of human T cells induced by mouse and human anti-CD3 monoclonal antibodies. Identification of a functional polymorphism to human IgG2 anti-CD3. J Immunol 148(3):695–701
Vallhonrat H, Williams WW, Cosimi AB, Tolkoff-Rubin N, Ginns LC, Wain JC et al (1999) In vivo generation of C4d, Bb, iC3b, and SC5b-9 after OKT3 administration in kidney and lung transplant recipients. Transplantation 67(2):253–258
Hirsch R, Bluestone JA, DeNenno L, Gress RE (1990) Anti-CD3 F(ab’)2 fragments are immunosuppressive in vivo without evoking either the strong humoral response or morbidity associated with whole mAb. Transplantation 49(6):1117–1123
Cole MS, Stellrecht KE, Shi JD, Homola M, Hsu DH, Anasetti C et al (1999) HuM291, a humanized anti-CD3 antibody, is immunosuppressive to T cells while exhibiting reduced mitogenicity in vitro. Transplantation 68(4):563–571
Xu D, Alegre ML, Varga SS, Rothermel AL, Collins AM, Pulito VL et al (2000) In vitro characterization of five humanized OKT3 effector function variant antibodies. Cell Immunol 200(1):16–26
Vudattu NK, Herold KC (2014) Treatment of new onset type 1 diabetes with teplizumab: successes and pitfalls in development. Expert Opin Biol Ther 14(3):377–385
Daifotis AG, Koenig S, Chatenoud L, Herold KC (2013) Anti-CD3 clinical trials in type 1 diabetes mellitus. Clin Immunol 149(3):268–278
Hagopian W, Ferry RJ Jr, Sherry N, Carlin D, Bonvini E, Johnson S et al (2013) Protégé Trial Investigators. Teplizumab preserves C-peptide in recent-onset type 1 diabetes: two-year results from the randomized, placebo-controlled Protégé trial. Diabetes 62(11):3901–8
U.S Food and Drug Administration [Home page in Internet] FDA approved drug products: TZIELD (teplizumab-mzwv) injection for intravenous use. [Last Cited on 27th January 2023] Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-can-delay-onset-type-1-diabetes
Sims EK, Bundy BN, Stier K, Serti E, Lim N, Long SA et al (2021) Teplizumab improves and stabilizes beta cell function in antibody-positive high-risk individuals. Sci Transl Med 13(583):eabc8980
Smigoc Schweiger D (2022) Recent advances in immune-based therapies for type 1 diabetes. Horm Res Paediatr
Dayan CM, Korah M, Tatovic D, Bundy BN, Herold KC (2019) Changing the landscape for type 1 diabetes: the first step to prevention. Lancet 394(10205):1286–1296
Felton JL, Conway H, Bonami RH (2021) B Quiet: autoantigen-specific strategies to silence raucous B lymphocytes and halt cross-talk with t cells in type 1 diabetes. Biomedicines 9(1):42
Gaglia J, Kissler S (2019) Anti-CD3 antibody for the prevention of type 1 diabetes: a story of perseverance. Biochemistry 58(40):4107–4111
Kaplon H, Reichert JM (2021) Antibodies to watch in 2021. MAbs 13(1):1860476
Herold KC, Bundy BN, Long SA, Bluestone JA, DiMeglio LA, Dufort MJ et al (2019) An anti-CD3 antibody, teplizumab, in relatives at risk for type 1 diabetes. N Engl J Med 381(7):603–613
Linsley PS, Long SA (2019) Enforcing the checkpoints: harnessing T-cell exhaustion for therapy of T1D. Curr Opin Endocrinol Diabetes Obes 26(4):213–218
Tooley JE, Vudattu N, Choi J, Cotsapas C, Devine L, Raddassi K et al (2016) Changes in T-cell subsets identify responders to FcR-nonbinding anti-CD3 mAb (teplizumab) in patients with type 1 diabetes. Eur J Immunol 46(1):230–241
Chatenoud L, Primo J, Bach JF (1997) CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice. J Immunol 158(6):2947–2954
You S, Leforban B, Garcia C, Bach JF, Bluestone JA, Chatenoud L (2007) Adaptive TGF-beta-dependent regulatory T cells control autoimmune diabetes and are a privileged target of anti-CD3 antibody treatment. Proc Natl Acad Sci U S A 104(15):6335–6340
You S, Slehoffer G, Barriot S, Bach JF, Chatenoud L (2004) Unique role of CD4+CD62L+ regulatory T cells in the control of autoimmune diabetes in T cell receptor transgenic mice. Proc Natl Acad Sci USA 101(Suppl 2):14580–5
Belghith M, Bluestone JA, Barriot S, Mégret J, Bach JF, Chatenoud L (2003) TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med 9(9):1202–1208
Penaranda C, Tang Q, Bluestone JA (2011) Anti-CD3 therapy promotes tolerance by selectively depleting pathogenic cells while preserving regulatory T cells. J Immunol 187(4):2015–2022
Esplugues E, Huber S, Gagliani N, Hauser AE, Town T, Wan YY et al (2011) Control of TH17 cells occurs in the small intestine. Nature 475(7357):514–518
Waldron-Lynch F, Henegariu O, Deng S, Preston-Hurlburt P, Tooley J, Flavell R et al (2012) Teplizumab induces human gut-tropic regulatory cells in humanized mice and patients. Sci Transl Med 4(118):118ra12
Nourelden AZ, Elshanbary AA, El-Sherif L, Benmelouka AY, Rohim HI, Helmy SK et al (2021) Safety and efficacy of teplizumab for treatment of type one diabetes mellitus: a systematic review and meta-analysis. Endocr Metab Immune Disord Drug Targets 21(10):1895–1904
Herold KC, Gitelman SE, Ehlers MR, Gottlieb PA, Greenbaum CJ, Hagopian W, Boyle KD, Keyes-Elstein L, Aggarwal S, Phippard D, Sayre PH, McNamara J, Bluestone JA (2013) AbATE Study Team. Teplizumab (anti-CD3 mAb) treatment preserves C-peptide responses in patients with new-onset type 1 diabetes in a randomized controlled trial: metabolic and immunologic features at baseline identify a subgroup of responders. Diabetes 62(11):3766–74
Skelley JW, Elmore LK, Kyle JA (2012) Teplizumab for treatment of type 1 diabetes mellitus. Ann Pharmacother 46(10):1405–1412
Mital S, Nguyen HV (2020) Cost effectiveness of teplizumab for prevention of type 1 diabetes among different target patient groups. Pharmacoeconomics 38(12):1359–1372
Author information
Authors and Affiliations
Contributions
Concept or design of the article: Dr Saurav Misra, Dr Ajay Kumar Shukla. Drafted the article or revised: Dr Saurav Misra. Approved the version to be published: Dr Ajay Kumar Shukla.
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Misra, S., Shukla, A.K. Teplizumab: type 1 diabetes mellitus preventable?. Eur J Clin Pharmacol 79, 609–616 (2023). https://doi.org/10.1007/s00228-023-03474-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-023-03474-8