
Abstract
Purpose
To assess the efficacy and safety of empagliflozin (EMPA) as add-on to metformin (MET) in patients with type 2 diabetes mellitus (T2DM).
Methods
We searched PubMed, Embase, Medline, OVID, Cochrane Library and Web of Science. Randomized controlled trials of EMPA as add-on to MET for T2DM were included. Two investigators independently selected studies, extracted data and assessed the risk of bias. A meta-analysis was conducted by using RevMan 5.3 software and Stata 12 software.
Results
Seven trials including 4256 patients were analysed. Compared with placebo, two different doses of EMPA significantly reduced glycated haemoglobin (HbA1c) [10 mg: weighted mean difference (WMD) −0.57 %; 95 % confidence interval (CI) −0.65 to −0.49 %, P < 0.00001; 25 mg: WMD −0.65 %; 95 % CI −0.72 to −0.57 %, P < 0.00001]. Compared with active comparators (two sitagliptin, one linagliptin and one glimepiride), 10 mg of EMPA provided a similar reduction in HbA1c [WMD −0.10 %; 95 % CI −0.23 to 0.03 %, P = 0.13], while 25 mg of EMPA provided a significantly greater reduction in HbA1c [WMD −0.13 %; 95 % CI −0.20 to −0.06 %, P = 0.0005]. In addition, EMPA as add-on to MET also had a favourable effect on body weight and blood pressure. The risk of hypoglycaemia in the EMPA group was similar to the placebo group or active comparator group.
Conclusions
EMPA as add-on to MET was well tolerated and provided additional benefits beyond glucose lowering, such as weight loss and blood pressure reduction. However, high-quality trials with large samples are still needed in order to confirm their long-term safety.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Type 2 diabetes mellitus (T2DM) is a chronic progressive disease characterised by progressive decline in pancreatic beta-cell function and insulin resistance. Metformin (MET) was recommended as the optimal agent in pharmacotherapy treatment for T2DM due to its low cost, proven safety record, weight neutrality and possible benefits on cardiovascular (CV) outcomes [1]. However, as diabetes progresses, MET monotherapy often fails to achieve adequate glycaemic control, and we frequently consider other pharmacologic options combined with MET [1]. But many anti-diabetic agents (sulphonylurea, thiazolidinedione and insulin) are associated with adverse events (AEs) and tolerability issues, such as hypoglycaemia or weight gain, which are counter-productive effects and hamper adherence to treatment [2]. Therefore, there is a desperate need for novel agents to treat T2DM with improved safety and tolerability profiles. Empagliflozin (EMPA), a potent and highly selective inhibitor of sodium glucose co-transporter 2 (SGLT2), was approved in the European Union (EU) and the United States (US) in 2014 for the treatment of T2DM [3, 4]. EMPA lowers the plasma glucose concentration by inhibiting renal glucose reabsorption and increasing urinary glucose excretion [5]. As the mechanism is independent of insulin, EMPA is associated with a low risk of hypoglycaemia [6].
Currently, there is no consensus on the optimum second-line therapy when MET fails to achieve glycaemic control. The American Diabetes Association/European Association for the Study of Diabetes (ADA/EASD) guidelines encouraged patient choice and individualised treatment [1]. The drug selection of combination therapy for patients with T2DM must balance glucose-lowering efficacy, side-effect profiles, anticipation of additional benefits, costs, drug–drug interactions and patient compliance. There are potential benefits of EMPA as add-on to MET. First, EMPA shows excellent pharmacokinetic characteristics: a good oral bioavailability, a rather long elimination half-life (10–19 h) allowing once-daily administration and a negligible risk of drug–drug interactions [7, 8]. Second, SGLT2 inhibitor leads to a reduction in tissue glucose disposal and a rise in endogenous glucose production (EGP) [9, 10]. MET enhances the glucose-lowering actions of SGLT2 inhibitor by restraining EGP, which may provide long-term improvement of glycaemic control [11]. Given the complementary mechanisms of EMPA and MET, this combination may offer a promising treatment strategy for T2DM. Third, a series of studies have demonstrated that EMPA monotherapy significantly improved glycaemic control and reduced body weight and blood pressure.
In five previous systematic reviews, Clar et al. [12], Vasilakou et al. [13] and Monami et al. [14] all assessed the efficacy and safety of SGLT2 inhibitor, but they did not include EMPA. Zhang et al. assessed the efficacy and safety of SGLT2 inhibitor as add-on to MET, but they focused primarily on dapagliflozin and canagliflozin, and only one trial of EMPA was included [15]. Liakos et al. examined the efficacy and safety of EMPA, in which only two trials of EMPA as add-on to MET were included [16].
To provide an up-to-date and more complete profile of the efficacy and safety of EMPA as add-on to MET, we conducted a systematic review and meta-analysis.
Methods
This systematic review was conducted in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement [17].
Data sources and search strategy
We searched PubMed, Embase, Medline, OVID, Cochrane Library and Web of Science up to September 4, 2015, without language restriction. Reference lists of eligible studies were hand-searched from relevant magazines. Potentially relevant unpublished data were searched from clinicaltrials.gov, the Food and Drug Administration (FDA) web site, ADA, European Union Drug Regulating Authorities Clinical Trials (EudraCT) and the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP).
We used the following terms: type 2 diabetes, T2DM, metformin, sodium glucose cotransporter 2 inhibitor, SGLT2, empagliflozin, BI-10773, Jardiance. These terms were adjusted to comply with the relevant rules in each database.
Study selection
Two investigators independently screened titles and abstracts, and subsequently examined potentially eligible studies in full-text form. Any discrepancies at each stage of selection were discussed by the third reviewer and resolved by consensus.
Inclusion criteria
We included randomised controlled trials if they met the following criteria: (1) patients aged ≥18 years with diagnosed T2DM [glycated haemoglobin (HbA1c) >7 to ≤10 %]; (2) comparison of EMPA and placebo or any other active comparator as add-on to MET, without other background therapy; (3) at least 12 weeks duration of intervention; (4) report at least one of the following outcomes: (a) HbA1c; (b) proportion of participants achieving HbA1c ≤ 7 %; (c) fasting plasma glucose (FPG); (d) body weight; (e) systolic blood pressure (SBP); (f) diastolic blood pressure (DBP); (g) estimated glomerular filtration rate (eGFR); (h) plasma lipids; (i) AEs.
We excluded studies with non-randomised designs, duplicate publications, EMPA monotherapy, healthy volunteers and animals.
Data extraction
Data were extracted independently by two investigators and checked by the third reviewer. For each study, we extracted information on the study characteristics, participants’ baseline characteristics, duration of treatment, interventions, efficacy outcomes and AEs.
We chose 10- and 25-mg doses of EMPA, which were approved by the FDA [4].
Risk of bias assessment
Two investigators assessed the quality of eligible studies by using the risk of bias tools [18]. The predefined key domains included: random sequence generation, allocation concealment, blinding and other items [i.e. intention-to-treat analysis (ITT), lost to follow-up].
Statistical analysis
We used RevMan 5.3 software and Stata 12 software for all statistical analyses. Separate analyses were performed according to the type of control group. Subgroup analyses were performed according to dose of EMPA and duration of treatment. For dichotomous data, the risk ratio (RR) was calculated, along with 95 % confidence intervals (CIs). In the case of continuous data, we used the weighted mean difference (WMD) with 95 % CIs. We calculated the I2 statistic to estimate heterogeneity, with values greater than 60 % being considered as high heterogeneity [19]. We also conducted a sensitivity analysis to assess the stability of results. Potential publication bias was assessed by Egger’s test.
Results
Search
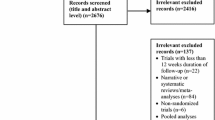
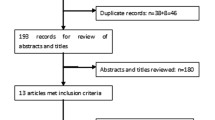
Figure 1 shows a flow diagram of the selected studies. We included seven trials [20–26], with a total of 4256 patients. Three trials [21, 24, 25] were of a double-blind design, one trial was a double-blind design for EMPA and placebo and one was of an open-label design for sitagliptin [20]. Two trials [22, 26] were an extension study. EMPA was compared with placebo in four trials [20–23] and with an active comparator in four trials [20, 24–26]. All eligible trials had no other background anti-diabetic therapy except MET. The duration of intervention ranged from 12 to 104 weeks. The characteristics of the included trials are summarised in Table 1.

Flow diagram of the selected studies
Risk of bias assessment
Table 2 shows the risk bias assessment. In addition to three trials [22, 23, 26], the remaining trials were randomised, double-blind, placebo or active controlled trials and had clearly detailed randomisation and concealment procedures by using an interactive voice and web response system. Efficacy analyses of each trial were performed to give a full analysis set. The last observation carried forward (LOCF) approach was used to impute missing efficacy data. All eligible trials were funded by Boehringer Ingelheim and Eli Lilly and Company R.A.D.
Primary efficacy outcomes of EMPA as add-on to MET
Glycaemic levels
Compared with placebo, both doses of EMPA led to a greater reduction in HbA1c, with no significant between-study heterogeneity (10 mg: WMD −0.57 %; 95 % CI −0.65 to −0.49 %, P < 0.00001; 25 mg: WMD −0.65 %; 95 % CI −0.72 to −0.57 %, P < 0.00001) (Fig. 2). Compared with active comparator, 10 mg of EMPA had a similar reduction in HbA1c (WMD −0.10 %; 95 % CI −0.23 to 0.03 %, P = 0.13), while 25 mg of EMPA provided a significantly greater reduction in HbA1c (WMD −0.13 %; 95 % CI −0.20 to −0.06 %, P = 0.0005) (Fig. 3). There was no significant publication bias in this analysis (vs. PBO: P = 0.959; vs. active comparator: P = 0.853; Supplementary Figs. 1 and 2).

Meta-analysis for HbA1c change from baseline (EMPA group vs. placebo group)

Meta-analysis for HbA1c change from baseline (EMPA group vs. active control group)
With regards to FPG, both doses of EMPA were associated with a significant reduction versus placebo or active comparator (Table 3). Moreover, as compared to placebo, the proportion of participants achieving HbA1c ≤ 7 % was greater in the EMPA group, but compared to active comparator, this proportion showed no significant difference between the two groups (Table 3).
In order to compare EMPA with dipeptidyl peptidase-4 (DPP-4) inhibitor, we again analysed HbA1c and FPG by excluding the glimepiride-controlled trial [25]. Pooling the data (EMPA vs. DPP-4 inhibitor) showed that the overall estimate was not impacted by excluding the glimepiride-controlled trial (Supplementary Table 1).
Body weight
Compared with placebo, both doses of EMPA significantly reduced body weight (Table 3), with no significant heterogeneity (Table 3) and no publication bias (P = 0.251). Similarly, both doses of EMPA had a superior effect on body weight as compared to active comparator (Table 3), but the 25-mg regimen showed heterogeneity of 95 % and publication bias was detected (P = 0.016). When we performed a sensitivity analysis by excluding the glimepiride-controlled trial [25], the heterogeneity was reduced without affecting the overall estimate (WMD −2.52 kg; 95 % CI −3.41 to −1.62 kg, P < 0.00001; I2 = 57 %).
Blood pressure
Compared with placebo or active comparator, both doses of EMPA showed greater reductions in SBP and DBP (Table 3). There was no significant publication bias on Egger’s test in this analysis (SBP: vs. placebo, P = 0.967; vs. active comparator P = 0.291; DBP: vs. placebo, P = 0.855; vs. active comparator P = 0.553).
Since the therapeutic follow-up of eligible studies was divergent (12 to 104 weeks), we also performed a subgroup analysis on duration for primary efficacy outcomes, which is summarised in Supplementary Table 2.
Safety outcomes of EMPA as add-on to MET
Plasma lipids
Compared with placebo or active comparator, this combination increased low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C) and total cholesterol (TC), with statistically significant differences (Table 3).
Death
Compared with placebo, there were no reported cases of death in the two groups. Compared with active comparator, six deaths were reported in the EMPA group (N = 1515) and five in the active comparator group (N = 1290).
eGFR
Compared with placebo, 10 mg of EMPA did not decrease the eGFR and 25 mg of EMPA lowered the eGFR by 2.23 mL/min/1.73 m2 (Table 3). Compared with active comparator, DeFronzo et al. reported that, at 52 weeks, the mean eGFR changes from baseline for 10 mg of EMPA, 25 mg of EMPA and linagliptin were −0.1 mL/min/1.73 m2, 1.1 mL/min/1.73 m2 and −1.7 mL/min/1.73 m2, respectively [24]. At follow-up, the mean eGFR changes from baseline for 10 mg of EMPA, 25 mg of EMPA and linagliptin were 3.7 mL/min/1.73 m2, 4.8 mL/min/1.73 m2 and −0.5 mL/min/1.73 m2, respectively [24]. In the study by Ridderstråle et al. [25], after 104 weeks, the mean eGFR changes from baseline for 25 mg of EMPA and glimepiride were 1.7 mL/min/1.73 m2 and −1.8 mL/min/1.73 m2, respectively.
Other adverse events
The incidence of total AEs, serious AEs, discontinuation due to AEs, hypoglycaemia and urinary tract infection did not differ between the EMPA group and the control group (Table 3). However, the EMPA group was associated with an increased risk of genital infection when compared to the comparator group (Table 3).
Discussion
Our systematic review showed that both doses of EMPA as add-on to MET significantly reduced HbA1c compared with the placebo group. Compared with active comparator, 10-mg dosing regimen had a similar reduction in HbA1c, while 25 mg of EMPA showed a significantly greater reduction in HbA1c. In addition, this combination also had a favourable effect on body weight and blood pressure.
In our subgroup analysis on duration, compared with placebo, EMPA had a short- and long-term favourable effect on HbA1c, which was not inconsistent with the findings of Zhang et al., who showed that SGLT2 inhibitor was non-inferior to placebo in the long term (1 and 2 years duration). However, further sensitivity analysis showed that all three durations had a favourable effect on HbA1c [15]. Compared with active comparator, our study indicated that 25 mg EMPA was superior in the long term (52–78 weeks and 104 weeks). In the future, subgroup analysis on the duration may deserve further study.
Overweight/obesity is a significant CV risk factor and increases insulin resistance [27, 28]. EMPA has shown therapeutic benefits in reducing body weight and blood pressure. Weight loss may be due to a loss of calories via increased urinary glucose excretion, accompanied by a reduction in body fat [10]. The reduction in blood pressure may be related to diuretic effects, weight loss and improved glycaemic control [29]. The EMPA CV outcome trial (EMPA-REG OUTCOME™: NCT01131676) indicated that EMPA was the only glucose-lowering agent that has demonstrated superiority in CV risk reduction [30]. In the present systematic review, EMPA as add-on to MET significantly improved glycaemic control, body weight and blood pressure. There is an expectation that this combination might reduce CV risk in patients with T2DM.
The safety profile of EMPA as add-on to MET was similar to its individual components. This combination did not increase the risk of hypoglycaemia. Genital infections (mild or moderate) were more common in the EMPA group than in the control group. Discontinuation due to AEs showed no significant difference between the two groups.
In the present systematic review, increases in LDL-C were observed in the EMPA group, which should be monitored and treated, as appropriate.
In our included trials, data on cancer and diabetic ketoacidosis (DKA) were not reported. However, in the authorisation process for EMPA, the European Medicines Agency (EMA) found two cases of bladder cancer in the EMPA group [31]. Therefore, further post-marketing surveillance is ongoing.
On May 15, 2015, the FDA warned that SGLT2 inhibitors might lead to DKA, based on 20 cases of acidosis reported in those treated with SGLT2 inhibitors, mainly in cases with T2DM and in a few cases with type 1 diabetes mellitus (T1DM) [32]. The FDA suggested: “Do not stop or change your diabetes medicines without first talking to your health care professional” [32]. In a recent study, Peters et al. advised that patients with T2DM taking SGLT2 inhibitors need not routinely monitor urine ketones [33]. However, they suggested being aware that DKA may occur in any individual with T1DM or T2DM and in the presence of nearly normal or moderately increased blood glucose levels, in contrast to the traditional view that DKA most commonly occurs in patients with T1DM and is usually accompanied by high blood sugar levels. So, both the FDA and Peters et al. recommended that all patients taking SGLT2 inhibitors who experience tachypnoea or hyperventilation, anorexia, abdominal pain, nausea, vomiting, lethargy or mental status changes should be promptly evaluated for acidosis, even if the glucose levels are nearly normal [32, 33].
When SGLT2 inhibitors were first released, there were concerns that their use would contribute to the deterioration of kidney function. However, several studies found that EMPA decreased the urinary albumin to creatinine ratio [34]. In the present systematic review, compared with placebo (study duration of three trials ≤24 weeks), 10 mg of EMPA did not show a decrease in the eGFR and 25 mg of EMPA lowered the eGFR by 2.23 mL/min/1.73 m2. Compared with active comparator, small eGFR changes from baseline were observed at week 52. After follow-up, renal function was preserved in the EMPA group [24, 25]. These findings seemed to support recent claims that SGLT2 inhibitors may have a potential renoprotective effect. In the future, more long-term studies are necessary to investigate this renoprotective potential. Currently, the prescribing information of EMPA states that, for GFR ≥45 to <60 mL/min/1.73 m2, the dose should be adjusted or maintained at 10 mg once daily; for eGFR <45 mL/min/1.73 m2, EMPA should be discontinued [35]. For MET, contraindication may be overly restrictive if serum creatinine ≥1.5 mg/dL (≥133 μmol/L) in men or 1.4 mg/dL (124 μmol/L) in women [1].
Cost also plays a major role in therapy selection. A 1-month supply of 10–25 mg of EMPA is about $355.7, of those of 100 mg sitagliptin, 5 mg linagliptin, 4 mg glimepiride and 4 mg rosiglitazone are about $343.37, $343.38, $6.73 and $132.07, respectively (http://www.goodrx.com). Like all newer agents not covered under insurance, EMPA is more expensive than older agents, which may influence physicians’ and patients’ treatment choices.
There were some limitations in this systematic review. First, in our eligible trials, an open-label design (three trials), the LOCF approach (seven trials) for handling missing data and industry funding (seven trials) may cause potential risks of bias. Second, EMPA had the highest selectivity over other SGLT2 inhibitors, including dapagliflozin and canagliflozin, in the vitro studies [36]. Mearns et al. [37] also showed that EMPA was significantly more efficacious compared to dapagliflozin. However, we did compare EMPA with other SGLT2 inhibitors due to the lack of head-to-head trials. Third, we included only four active-controlled trials (four sitagliptin, one linagliptin and one glimepiride). Hence, more study is needed in order to determine the differences between EMPA and other anti-diabetic agents. Finally, long-term data are needed to assess the safety of EMPA as add-on to MET, such as CV risk, eGFR, DKA and cancer.
In conclusion, EMPA as add-on to MET was well tolerated and provided additional benefits beyond glucose lowering, such as weight loss and reduction of blood pressure.
References
Inzucchi SE, Bergenstal RM, Buse JB, Diamant M, Ferrannini E, Nauck M, Peters AL, Tsapas A, Wender R, Matthews DR (2015) Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 38(1):140–149
Action to control cardiovascular risk in Diabetes Study Group, Gerstein HC, Miller ME, Byington RP, Goff DC Jr, Bigger JT, Buse JB, Cushman WC, Genuth S, Ismail-Beigi F, Grimm RH Jr, Probstfield JL, Simons-Morton DG, Friedewald WT (2008) Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 358(24):2545–2559
European Medicines Agency (EMA). EMEA/H/C/002677. Jardiance. EPAR. Product information. Available online at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002677/WC500168592.pdf. Accessed 18 June 2014
U.S. Food and Drug Administration. Jardiance® (empagliflozin) tablets, for oral use. Initial U.S. Approval: 2014 [prescribing information]. Ingelheim, Germany: Boehringer Ingelheim; 2014. Available online at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/204629s000lbl.pdf. Accessed 8 August 2014
DeFronzo RA, Davidson JA, Del Prato S (2012) The role of the kidneys in glucose homeostasis: a new path towards normalizing glycaemia. Diabetes Obes Metab 14(1):5–14
Brunton SA (2015) The potential role of sodium glucose co-transporter 2 inhibitors in the early treatment of type 2 diabetes mellitus. Int J Clin Pract 69(10):1071–1087. doi:10.1111/ijcp.12675
Macha S, Mattheus M, Pinnetti S, Woerle HJ, Broedl UC (2013) Effect of empagliflozin on the steady-state pharmacokinetics of ethinylestradiol and levonorgestrel in healthy female volunteers. Clin Drug Investig 33(5):351–357
Macha S, Mattheus M, Pinnetti S, Seman L, Woerle HJ (2012) Pharmacokinetics of empagliflozin, a sodium glucose cotransporter 2 inhibitor, and glimepiride following co-administration in healthy volunteers: a randomised, open-label, crossover study. J Diabetes Res Clin Metab. Available online at: http://www.hoajonline.com/journals/pdf/2050-0866-1-14.pdf. Accessed 4 February 2015
Merovci A, Solis-Herrera C, Daniele G, Eldor R, Fiorentino TV, Tripathy D, Xiong J, Perez Z, Norton L, Abdul-Ghani MA, DeFronzo RA (2014) Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J Clin Invest 124(2):509–514
Ferrannini E, Muscelli E, Frascerra S, Baldi S, Mari A, Heise T, Broedl UC, Woerle HJ (2014) Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J Clin Invest 124(2):499–508
Neschen S, Scheerer M, Seelig A, Huypens P, Schultheiss J, Wu M, Wurst W, Rathkolb B, Suhre K, Wolf E, Beckers J, Hrabé de Angelis M (2015) Metformin supports the antidiabetic effect of a sodium glucose cotransporter 2 inhibitor by suppressing endogenous glucose production in diabetic mice. Diabetes 64(1):284–290
Clar C, Gill JA, Court R, Waugh N (2012) Systematic review of SGLT2 receptor inhibitors in dual or triple therapy in type 2 diabetes. BMJ Open 2(5):1–12
Vasilakou D, Karagiannis T, Athanasiadou E, Mainou M, Liakos A, Bekiari E, Sarigianni M, Matthews DR, Tsapas A (2013) Sodium-glucose cotransporter 2 inhibitors for type 2 diabetes: a systematic review and meta-analysis. Ann Intern Med 159(4):262–274
Monami M, Nardini C, Mannucci E (2014) Efficacy and safety of sodium glucose co-transport-2 inhibitors in type 2 diabetes: a meta-analysis of randomized clinical trials. Diabetes Obes Metab 16(5):457–466
Zhang Q, Dou J, Lu J (2014) Combinational therapy with metformin and sodium-glucose cotransporter inhibitors in management of type 2 diabetes: systematic review and meta-analyses. Diabetes Res Clin Pract 105(3):313–321
Liakos A, Karagiannis T, Athanasiadou E, Sarigianni M, Mainou M, Papatheodorou K, Bekiari E, Tsapas A (2014) Efficacy and safety of empagliflozin for type 2 diabetes: a systematic review and meta-analysis. Diabetes Obes Metab 16(10):984–993
Knobloch K, Yoon U, Vogt PM (2011) Preferred reporting items for systematic reviews and meta-analyses (PRISMA) statement and publication bias. J Craniomaxillofac Surg 39(2):91–92
Higgins JP, Altman DG, Gøtzsche PC, Jüni P, Moher D, Oxman AD, Savovic J, Schulz KF, Weeks L, Sterne JA; Cochrane Bias Methods Group; Cochrane Statistical Methods Group (2011) The Cochrane collaboration’s tool for assessing risk of bias in randomised trials. BMJ 343:d5928
Guyatt GH, Oxman AD, Kunz R, Woodcock J, Brozek J, Helfand M, Alonso-Coello P, Glasziou P, Jaeschke R, Akl EA, Norris S, Vist G, Dahm P, Shukla VK, Higgins J, Falck-Ytter Y, Schünemann HJ; GRADE Working Group (2011) GRADE guidelines: 7. Rating the quality of evidence-inconsistency. J Clin Epidemiol 64(12):1294–1302
Rosenstock J, Seman LJ, Jelaska A, Hantel S, Pinnetti S, Hach T, Woerle HJ (2013) Efficacy and safety of empagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor, as add-on to metformin in type 2 diabetes with mild hyperglycaemia. Diabetes Obes Metab 15(12):1154–1160
Häring HU, Merker L, Seewaldt-Becker E, Weimer M, Meinicke T, Broedl UC, Woerle HJ; EMPA-REG MET Trial Investigators (2014) Empagliflozin as add-on to metformin in patients with type 2 diabetes: a 24-week, randomized, double-blind, placebo-controlled trial. Diabetes Care 37(6):1650–1659
Merker L, Häring HU, Christiansen AV, Roux F, Salsali A, Kim G, Meinicke T, Woerle HJ, Broedl UC; EMPA-REG EXTEND™ MET investigators (2015) Empagliflozin as add-on to metformin in people with Type 2 diabetes. Diabet Med 32(12):1555–1567. doi:10.1111/dme.12814
Ross S, Thamer C, Cescutti J, Meinicke T, Woerle HJ, Broedl UC (2015) Efficacy and safety of empagliflozin twice daily versus once daily in patients with type 2 diabetes inadequately controlled on metformin: a 16-week, randomized, placebo-controlled trial. Diabetes Obes Metab 17(7):699–702
DeFronzo RA, Lewin A, Patel S, Liu D, Kaste R, Woerle HJ, Broedl UC (2015) Combination of empagliflozin and linagliptin as second-line therapy in subjects with type 2 diabetes inadequately controlled on metformin. Diabetes Care 38(3):384–393
Ridderstråle M, Andersen KR, Zeller C, Kim G, Woerle HJ, Broedl UC; EMPA-REG H2H-SU trial investigators (2014) Comparison of empagliflozin and glimepiride as add-on to metformin in patients with type 2 diabetes: a 104-week randomised, active-controlled, double-blind, phase 3 trial. Lancet Diabetes Endocrinol 2(9):691–700
Ferrannini E, Berk A, Hantel S, Pinnetti S, Hach T, Woerle HJ, Broedl UC (2013) Long-term safety and efficacy of empagliflozin, sitagliptin, and metformin: an active-controlled, parallel-group, randomized, 78-week open-label extension study in patients with type 2 diabetes. Diabetes Care 36(12):4015–4021
Fox CS, Pencina MJ, Wilson PW, Paynter NP, Vasan RS, D’Agostino RB Sr (2008) Lifetime risk of cardiovascular disease among individuals with and without diabetes stratified by obesity status in the Framingham heart study. Diabetes Care 31(8):1582–1584
Båvenholm PN, Kuhl J, Pigon J, Saha AK, Ruderman NB, Efendic S (2003) Insulin resistance in type 2 diabetes: association with truncal obesity, impaired fitness, and atypical malonyl coenzyme A regulation. J Clin Endocrinol Metab 88(1):82–87
Baker WL, Smyth LR, Riche DM, Bourret EM, Chamberlin KW, White WB (2014) Effects of sodium-glucose co-transporter 2 inhibitors on blood pressure: a systematic review and meta-analysis. J Am Soc Hypertens 8(4):262–275.e9
Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE (2015) Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 373(22):2117–2128
European Medicines Agency (EMA) (2014) Jardiance®. EPAR. Public assessment report (EAM). Available online at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002677/WC500168594.pdf. Accessed 24 September 2014
FDA warns that SGLT2 inhibitors for diabetes may result in a serious condition of too much acid in the blood. Available online at: http://www.fda.gov/drugs/drugsafety/ucm446845.htm
Peters AL, Buschur EO, Buse JB, Cohan P, Diner JC, Hirsch IB (2015) Euglycemic diabetic ketoacidosis: a potential complication of treatment with sodium-glucose cotransporter 2 inhibition. Diabetes Care 38(9):1687–1693
Shubrook JH, Bokaie BB, Adkins SE (2015) Empagliflozin in the treatment of type 2 diabetes: evidence to date. Drug Des Devel Ther 9:5793–5803
Jardiance®. Summary of product characteristics. Available online at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002677/WC500168592.pdf. Accessed 29 September 2014
Scott LJ (2014) Empagliflozin: a review of its use in patients with type 2 diabetes mellitus. Drugs 74(15):1769–1784
Mearns ES, Sobieraj DM, White CM, Saulsberry WJ, Kohn CG, Doleh Y, Zaccaro E, Coleman CI (2015) Comparative efficacy and safety of antidiabetic drug regimens added to metformin monotherapy in patients with type 2 diabetes: a network meta-analysis. PLoS One 10(4):e0125879. doi:10.1371/journal.pone.0125879
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 94 kb)
Rights and permissions
About this article

Cite this article
Zhong, X., Lai, D., Ye, Y. et al. Efficacy and safety of empagliflozin as add-on to metformin for type 2 diabetes: a systematic review and meta-analysis. Eur J Clin Pharmacol 72, 655–663 (2016). https://doi.org/10.1007/s00228-016-2010-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-016-2010-8