
Abstract
Each dietary analysis method has its own strengths and limitations and provides different perspectives for understanding the trophic biology of aquatic organisms. In this study, we used an integrative, multi-tool approach to comprehensively investigate the dietary differences of eight nominally herbivorous fishes from five feeding guilds in the Nansha Islands (N: 8°51′10°57', E: 112°50′115°35') of the South China Sea. The methods used in this study included 18S rDNA metabarcoding, standard gut content analysis, and stable isotope analysis. DNA metabarcoding revealed diverse algae categories (Ochrophyta, Rhodophyta, and Chlorophyta) in the gut contents of sampled fishes with high taxonomic resolution, and some microscopic and cryptic taxa, such as dinoflagellates and microinvertebrates, were precisely identified. The overlap of 95% prediction isotopic niche remained below 30% between different feeding guilds, and nearly 70% of the 107 genera detected in the gut contents were not shared by two or more fish species or feeding guilds. By combining stable isotope analysis and morphological identification with gut content metabarcoding, we found that the dietary overlap detected by metabarcodng between Ctenochaetus striatus and other fishes may be due to interference from debris of algae or other organisms. While each method has its own limitations in dietary analysis, our study suggests that an integrative, multi-tool approach can remedy the limitations of one method by utilizing the strengths of another. Our results indicate that a wide spectrum of resources on coral reefs could be utilized by different fish species in a complementary manner.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In recent decades, the significant development of various novel and traditional dietary analysis methods has greatly enhanced our understanding of the diets and trophic roles of aquatic organisms (Choat et al. 2002; Leal and Ferrier-Pages 2016; Clements et al. 2017; Pethybridge et al. 2018). Each method offers a distinct perspective that enhances our understanding of how aquatic organisms contribute to ecosystem functioning through their feeding processes. However, there is no single analytical method as they each have their own strengths and limitations and operate at specific scales of space and time (Nielsen et al. 2018). Therefore, there is a need for an integrative, multi-tool approach to comprehensively understand how species partition their diet and play different ecological roles in marine ecosystem.
Conventionally, dietary analysis of aquatic organisms is performed through morphological identification of gut contents and in situ observations (Choat and Bellwood 1985; Robertson and Gaines 1986; Montgomery et al. 1989; Choat et al. 2002; Rasher et al. 2013; Kelly et al. 2016). Both morphological identification of gut contents and in situ observations can provide precise taxonomic identities for dietary items. However, these methods can only provide a dietary snapshot, which represents feeding events that occur over limited time scales. Additionally, some particular food materials (e.g., liquid, gelatinous, or cryptic species) may be ignored or identified at a coarse taxonomic level (Leray et al. 2015), limiting the accuracy of the analysis. Despite these drawbacks, visual methods (morphological identification of gut contents and in situ observations) are still necessary and are applied in most dietary analysis studies. Stable isotope analysis is useful for determining diet composition and niche width over long timescales (Jackson et al. 2011). However, the use of stable isotope analysis should be based on knowledge of the primary producers (Pethybridge et al. 2018) and is also limited in its capacity to provide high-resolution taxonomic information. High-throughput sequencing (HTS) can yield high-resolution data on the taxonomic identity of the organisms in stomach, gut, faecal, or scat material (Leray et al. 2019; Brandl et al. 2020; Lin et al. 2021), which is paricularly useful for detecting rare, soft, or highly degraded items. However, it should be noted that the resolution of molecular approaches is dependent on accurate identification and appropriate reference collections (Pompanon et al. 2012; Nielsen et al. 2018). Furthermore, it is important to consider that the quantitative data generated through HTS can be affected by various factors (Pompanon et al. 2012; Leal and Ferrier-Pages 2016), and occurrence-based data have certain limitations, such as interference from environmental DNA and secondary predation (Deagle et al. 2013, 2019).
Herbivorous fishes are an important group in coral reefs, playing critical roles in herbivory and trophic dynamics within coral reef ecosystem (Pratchett et al. 2011; Adam et al. 2015; Kelly et al. 2016). Nominally herbivorous reef fishes are commonly classified into several groups based on their diets and foraging strategies (Choat et al. 2002; Crossman et al. 2005; Pratchett et al. 2011), including detritivore brushers (DB), scrapers/small excavators (SC), turf algae croppers (TC), macroalgal browsers (MB), and territorial farmers (TF). The diverse foraging patterns of herbivorous fishes may result in the ingestion of various types of algae or other components from coral reefs (Crossman et al. 2005; Rasher et al. 2013; Tebbett et al. 2017a). However, in some particular species or groups (such as detritivore brushers and scrapers/small excavators), the highly degraded and unidentified food items in their gut contents can result in a coarse taxonomic resolution of their diets. This may lead to an underestimation of their differences in resource use and ecological function. For instance, it was previously thought that scrapers/small excavators, detritivore brushers, and turf algal croppers all feed on the 'EAM' (Tebbett and Bellwood 2019). The 'epilithic algal matrix' is defined as an abundant algal matrix consisting of short turf algae (< 2 mm), detritus, sediments, microbes (including cyanobacteria), microalgae, and invertebrates (Wilson and Bellwood 1997; Wilson et al. 2003; Cheal et al. 2010; Tebbett and Bellwood 2019). The debate on the ability of croppers (Acanthurus) and detritivore brushers (Ctenochaetus) to remove the EAM has been ongoing for decades (Marshell and Mumby 2012; Tebbett et al. 2017a). However, recent studies have shown that Acanthurus only crop the upper portions of the turf algae, while Ctenochaetus brush detritus and microalgae from the substrate, leaving the algae untouched (Tebbett et al. 2017a; Tebbett and Bellwood 2019). Scrapers/small excavators are generally believed to remove the entire EAM (Tebbett and Bellwood 2019). Clements et al. (2017), using various lines of evidence such as feeding observations, trophic anatomy, and biochemical analyses, tissue composition, and digestive processes, have identified scrapers/small excavators as microphages. It is possible that they ingest cyanobacteria and other microbes from the substrata instead of the entire EAM (Nicholson and Clements 2020). Therefore, using multiple methods to analyse the dietary differences of these nominally herbivorous fishes will be helpful in clarifying their differences in resource use and ecological function.
In this study, we used multi methods to analyse the diets of several common herbivorous fishes in the Nansha Islands of South China Sea (SCS). Eight common species from the five feeding guilds were selected for dietary analysis (Table 1). These species/feeding guilds were frequently observed feeding in the same area in SCS (Fig. 1) and were previously believed to rely on similar resources, such as EAM, as a food source (Wilson et al. 2003; Ng et al. 2021). Ctenochaetus striatus and surgeonfishes (Acanthurus japonicus, A. triostegus, and A. nigrofuscus) are the dominant species in the DB and TC, respectively. Parrotfishes (Scarus globiceps and S. schlegeli) in SC are selected to verify their possible overlapping on resource use with other fishes, when removing the entire ‘EAM’ (Tebbett and Bellwood 2019). Surgeonfish (Zebrasoma velifer, a concealed cropper) and damselfish (Plectroglyphidodon fasciolatus, a territorial farmer) were also sampled for dietary analysis to determine their potential dietary resources from EAM (Tebbett et al. 2022). The diets of eight fish species were evaluated using a combination of identification through gut content 18S rDNA metabarcoding, morphological analyses, and δ13C/δ15N stable isotope analysis. Although certain prokaryotes, such as cyanobacteria, serve as significant food sources for parrotfishes (Clements et al. 2017; Nicholson and Clements 2020), our gut content metabarcoding analysis only examined eukaryotic organisms ingested by herbivorous fishes. This decision was made to avoid potential interference from abundant non-target prokaryotes in both the fish gut and environment. We expected that an integrative approach to evaluating the dietary partitioning of these nominally herbivorous fishes would be beneficial, as the limitations of one method can be addressed by the strengths of another. Additionally, the presentation of trophic interactions at a fine taxonomic resolution would allow us to identify the taxa that contribute to the dietary partitioning of herbivores, thereby improving our understanding of how ecologically similar consumers coexist and exert varying effects on coral reefs. While the current dietary analysis is an important step in defining niche partitioning in nominally herbivorous fishes, a more comprehensive analysis is still required. It should consider incorporating additional fishes and dietary species, such as cyanobacteria, to further enhance our understanding of the resource use of herbivores in coral reef ecosystems.

Scrapers/small excavators (orange arrow), detritivore brusher (Ctenochaetus striatus, blue arrow), and turf algae croppers (Acanthurus japonicas and A. nigrofuscus, light green arrow) feeding in the same area at Nansha Islands, SCS. Photograph X. Lin
Methods
Sample collection
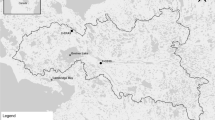
Herbivorous fishes were sampled from seven reefs in the Nansha Islands, SCS (Fig. 2a). These reefs were far from the mainland and at a certain distance (> 20 km) from one another (Fig. 2b). Fishes were collected from the crests and outer flats of reefs (Fig. 2c), which are the primary grazing areas of herbivorous/detritivorous fishes (Oakley-Cogan et al. 2020). Fish individuals were caught using a trammel net (10 m long × 1.5 m high, with maximum and minimum mesh size of 9.0 and 3.0 cm, respectively), and were removed from the trammel net within an hour of being trapped. Fishes were euthanized with an overdose of clove oil after collection and immediately placed on ice. The weight and body length of common fish species were measured, and the samples were stored at – 20 °C after capture. Body size is a crucial factor that contributes to dietary partitioning of herbivores (Kartzinel et al. 2015). To minimize the effect of body size on the dietary partitioning of fishes, individuals with similar body size from different species or feeding guilds were sampled for dietary analysis. The body mass of sampled individuals from SC, DB, and TF was mainly in the range of 40–60 g, and the body length was approximately 11 cm (Table 1). Concealed cropper (Z. velifer) was generally larger than other fishes, whereas territorial farmer (P. fasciolatus) was the smallest. Fish for dietary analysis were dissected within 2 months of capture. Fish gut contents were carefully collected and preserved in 75% ethanol at 4 °C. Muscles were collected and preserved at – 20 °C for δ13C/δ15N stable isotope analysis, but A. nigrofuscus samples were not sufficient for stable isotope analysis. Fish collection and euthanasia were conducted in accordance with the guidelines and approval of the Animal Research and Ethics Committee of the Chinese Academy of Sciences (SCSIO-IACUC-2019–000137).

Locations of surveyed sites on reefs in Nansha Islands of the South China Sea. a Geographic location of Nansha Islands. b Position of surveyed reefs and their distance from one another. c Map of survey sites (black filled circles) on each reef. Fish were collected from the crests or outer flats of each site. The fish species that were sampled from these sites were marked with different symbols on corresponding maps for easy identification. Number '2' inside the circles indicates that the fish species in that particular site was sampled twice for dietary analysis. Maps were prepared using Surfer 16
Gut content identification
Gut contents from three individuals of each species at each sampling sites were used as triplicates for DNA extraction and metabarcoding, resulting in 36 samples (a total of 3 × 36 = 108 individuals were used for gut content metabarcoding) (Table 1). Total DNA of the remaining gut contents was extracted using the FastDNA® Spin Kit for Faeces (MP Biomedicals, Santa Ana, USA), following the manufacturer’s instructions, and eluted in 30 μL of 10 mM Tris–HCl (pH 8.0). DNA quality was assessed using a Nanodrop® spectrophotometer (NanoDrop Technologies, Wilmington, Delaware, USA). To ensure data accuracy and reliability, negative control extractions were performed to identify potential contaminants. DNA samples were stored at − 20 °C until further use.
The DNA extracts were amplified using the universal TAReuk454FWD1-TAReukREV3 primer pair (TAReuk454FWD1:5-CCAGCASCYGCGGTAATTCC-3; TAReukREV3:5-ACTTTCGTTCTTGATYRA-3), which targets the V4 region (~ 380 bp) of eukaryotic 18S rDNA (Stoeck et al. 2010; Lin et al. 2018). PCRs were performed in triplicate in a 25 μL mixture containing 2.5 μL of TransStart Buffer, 2 μL of dNTPs, 1 μL of each primer, and 20 ng of template DNA. PCR was performed as described in a previous study (Lin et al. 2018). Indexed adapters were added to the ends of the amplicons using limited-cycle PCR. Finally, the library was purified using magnetic beads. PCRs of negative control extractions and no-template blanks were also performed. The concentration was detected using a microplate reader (Tecan, Infinite 200 Pro), and the fragment size was detected using 1.5% agarose gel electrophoresis. PCR product concentrations were validated with a Qubit 3.0 fluorometer. Libraries (10 nM) were multiplexed and loaded on an Illumina MiSeq, according to the manufacturer’s instructions (Illumina, San Diego, CA, USA). Sequencing was performed using an Illumina MiSeq platform (Illumina, San Diego, CA, USA).
Raw sequencing data were subjected to the Illumina MiSeq/HiSeq platform pipeline (Bokulich et al. 2013), including quality filtering, sequence demultiplexing, and primer removal. Paired-end filtered reads were merged using Vsearch (1.9.6) and only sequences with lengths > 300 bp were obtained. Effective reads from the raw data were retained for further analysis. Sequences were clustered into molecular operational taxonomic units (OTUs) at a 97% threshold using Vsearch (1.9.6) with default parameters. Representative OTU sequences were aligned to the NCBI nucleotide database (https://www.ncbi.nlm.nih.gov/genbank, v2022.4.30) using the online BLAST function (Megablast v2.11.0) with default parameters, and taxonomic information was selected from the top best hit. A genus name was accepted only if the similarities of the five best hits were ≥ 98%. A family name was retained only if the similarities of all the best hits were ≥ 95%. A phylum or subphylum name was retained only if the similarities of all the best hits were ≥ 90%. Sequences with maximum similarities of < 90% were labelled NA (no account) (Lin et al. 2018). A feature table was obtained after OTU clustering with frequency information. To decrease the number of false positives, OTUs with a frequency < 10 were removed. Sequences identified as host (fish) or parasitic species were removed from all datasets. The filtered data matrix was then standardized by percentages.
The remaining gut content from each sample use for DNA metabarcoding was identified at a 4 × 10 magnification using a dissecting microscope with a transect-based method (Choat et al. 2002). Gut contents were classified into different components based on morphological characteristics, including turf algae, foliose algae, detritus (unrecognizable organic matter with irregular shape), microinvertebrates, and sands. The proportions of different components were estimated based on their volumes.
Stable isotope analysis
At least five individuals of each fish species were randomly selected and used for the stable isotope analysis, and a total of 66 individuals were used for gut content metabarcoding (Table 1). Fish muscles were freeze-dried for 48 h prior to homogenization using a mortar and pestle. δ13C and δ15N were measured using an elemental analyser (Flash2000, Thermo Fisher Scientific, Italy) connected to an isotope ratio mass spectrometer (Delta V advantage, Thermo Fisher Scientific, Germany). Stable isotope ratio (‰) was calculated as δ13C or δ15N = [Rsample/Rstandard – 1] × 103, where ‘δ’ is 13C or 15N and R is the ratio of 13C/12C or 15N/14N. The standard deviation in parts per thousand (‰) is relative to the conventional C (Vienna Pee Dee Belemnite) and N (atmospheric N2) standard reference materials, and ‘R’ represents heavier to lighter isotopic ratios (13C/12C or 15N/14N), indicating 15C- and 13N-enriched or -depleted items, respectively. A protein (casein) standard (Cat. No. B2155; Elemental Microanalysis, UK) was used as a certified reference material with analytical precisions of ± 0.13‰ and ± 0.08‰ for the δ13C and δ15N values, respectively.
Data analyses and visualization
Dietary niche width and overlapping area were calculated for each species and feeding guild based on both molecular and stable isotope data. A double-bit map of the δ13C–δ15N values of fish was constructed. Bivariate ellipses were fitted to stable isotope data using Bayesian inference with the aim of describing and comparing isotopic niches. A convex hull was plotted based on the variations in δ13C and a δ15N value of each fish species and the total polygon area (TA) was calculated. Bayesian multivariate normal distributions were fitted to the dataset for each species. The probability distributions of the standard ellipse areas (SEAs) were calculated (Jackson et al. 2011). TA and SEA were used as metrics to measure the isotopic niche width of fishes. The overlap between the corresponding 95% prediction ellipses was calculated using the maximum likelihood estimate to measure the degree of potential isotopic niche overlap between each pair of species. These analyses were performed using ‘SIBER’ R package (Jackson et al. 2011) for statistical analyses and the ‘ggplot2’ R package (Wickham 2016).
Both the relative read abundance and occurrence data (a presence/absence matrix of genera in each sample) generated from HTS were included for analysis of dietary similarity, and a minimum sequence percentage threshold of 1% in each sample was defined to determine occurrences that balanced the data, thereby maximizing the inclusion of real diet sequences and excluding background noise (secondary predation, environmental DNA, and other sequencing deficiencies). The dissimilarity matrix of dietary differences of the sampled fishes was constructed using an abundance‐based metric (Morista–Horn) and occurrence-based metric (Jaccard) with genus-level read data. Non-metric multi-dimensional scaling (NMDS) ordination was employed for graphical representation. Because C. striatus are not typical herbivores, they brush microalgae and detritus off turf algae rather than removing mature turf algae and macroalgae (Tebbett et al. 2017a), DNA derived from the debris of turf algae and macroalgae ingested by C. striatus may influence dietary discrimination against other fishes. Additional analyses are necessary to reduce this interference. Therefore, NMDS was conducted again without C. striatus. All analyses were performed using the ‘vegan’ package (Oksanen et al. 2020) for statistical analyses and the ‘ggplot2’ package (Wickham 2016) for graphical representation in R version 4.2.1 (R Core Team 2022). The dietary network was created within feeding guilds using Cytoscape (3.8.2) based on the average value of the genus-level read data from gut content sequencing.
Results
Diversity and composition of gut contents
A total of 108 individuals from eight fish species and five feeding guilds (Table 1) were sampled for gut content metabarcoding. After the quality control and filtering process, 1, 450, 316 reads were generated from DNA sequencing of gut content. Rarefaction curves showed that the generated reads were sufficient to cover the major gut contents of fishes (Supplemental S1). Algae were the most abundant and diverse reads, including Ochrophyta, Dinoflagellata, Rhodophyta, and Chlorophyta (Fig. 3a). There were also a considerable number of reads from microinvertebrates, including Arthropoda, Nematoda, and Platyhelminthes. Most algal and microinvertebrate phyla were detected in each gut content sample (Supplemental S2). Morphological identification revealed turf algae and detritus in the gut of all eight fish species from the five feeding guilds (Fig. 3b). Turf algae were abundant in the gut contents of the concealed cropper (Z. velifer) and turf algal croppers (A. japonicus, A. triostegus, A. nigrofuscus). As expected, detritus was abundant in the gut contents of C. striatus, whereas less detritus was found in other species. A considerable number of the microinvertebrates were found in the gut of territorial farmer (P. fasciolatus).

Diversity and composition of gut contents of sampled fishes. a Relative read abundance of gut content taxon in each gut sample; different colours represent the top 10 most abundant phyla, in the given order. b Dietary composition of fishes based on morphological identification
Dietary dissimilarity
The NMDS plot of dietary composition based on the Morista–Horn and Jaccard distance of the genus-level read proportion of gut contents revealed differences both across and within feeding guilds (Fig. 4a, b). Both the abundance and incidence read data showed that C. striatus was the most heterogeneous relative to other fishes (Fig. 4a, b). Samples from other species showed greater separation (Fig. 4c, d). Similar to the results based on molecular data, isotopic niches were distinct across feeding guilds, particularly for Z. velifer and P. fasciolatus (Fig. 4e). Scarus globiceps had the largest isotopic niche, followed by C. striatus (Fig. 4e and Supplemental S3). Isotopic niches (convex polygons) were much closer amongst scrapers, C. striatus, and turf algae croppers, with an obvious overlap on 95% prediction ellipses (Fig. 4e) between C. striatus and the two parrotfishes (overlapping area ranges 2.85 ~ 4.74‰2, Supplemental S3). In addition, the overlapping area of 95% prediction ellipses for turf algae croppers with scrapers and C. striatus ranges 0.92 ~ 1.53‰2 and 0.58 ~ 1.94‰2, respectively (Supplemental S3). Species within the same group showed high niche overlap, such as S. globiceps vs. S. schlegelii and A. japonicus vs. A. triostegus (Fig. 4e and Supplemental S3). However, except for the overlapping area between C. striatus and parrotfishes, the overlapping area of the prediction ellipse between the other group pairs was smaller than 30% of the total area.

Non-metric multi-dimensional scaling (NMDS) plot and dietary niche of eight common fish species from five feeding guilds. Non-metric multi-dimensional scaling ordination of the gut content of species derived from molecular identification were computed using the abundance‐based Morista–Horn metric a and incidence‐based Jaccard metric b. NMDS ordination excluding Ctenochaetus striatus using Morista–Horn c and Jaccard d metrics. e Isotopic niche breadth of fishes was measured based on the variations in δ13C and δ15N values and the total polygon area (dotted polygon); solid circle represents the 95% confidence interval, which indicates the potential isotopic niche. Different fish species are distinguished by different colours and symbols as shown in the upper right, and the same colour indicates the same feeding guild
Trophic interaction
Dietary networks were reconstructed between fishes and the 107 dietary genera (Fig. 5, Supplemental S4). However, nearly 70% of the genera could only be detected in a single species or feeding guild, and only one genus was shared among the five feeding guilds. Sphacelaria (#69) was the most common genus shared by all the feeding guilds (Fig. 5, Supplemental S4). Brown algae (Sphacelaria, #69), green algae (Parvocaulis, #35), microalgae (Gloeodinium, #45; Prorocentrum, #48; Symbiodinium, #50), and red algae (Jania, #91; Parviphycus, #94) were commonly detected in the gut contents of detritivorous brusher (C. striatus), scrapers, and turf algae croppers (Supplemental S4). The main genera in the gut contents of scrapers and C. striatus were microalgae, such as Prorocentrum and Symbiodinium. The gut contents of turf algae croppers were dominated by Ochrophyta genera, such as Sphacelaria. Both C. striatus and the territorial farmer (P. fasciolatus) showed a complex gut composition, including microinvertebrates and algae, but they shared few genera. The gut contents of the concealed cropper (Z. velifer) were dominated by Rhodophyta genera, which was the main taxon shared with other fishes.

Network diagram based on the sequencing data of fish gut contents. Nodes are either a genus of dietary items or a fish feeding guild. Genus node size is proportional to the relative read abundance, and edge thickness is weighted by the abundance of that genus found within feeding guilds. The abbreviations for the different guilds are as follows: scrapers (SC), turf algal croppers (TC), detritivore brusher (DB), territorial farmer (TF), and concealed cropper (CT). Colour for both panels represents food taxa. Taxonomic information and relative read abundance of gut content sequences are shown in Supplemental S4
Discussion
Nominally herbivorous reef fishes exhibit various foraging patterns (Choat and Bellwood 1985; Robertson and Gaines 1986; Montgomery et al. 1989; Rasher et al. 2013), suggesting that they may have divergent nutritional demands and ingest different food items from reefs (Choat et al. 2002; Crossman et al. 2005; Clements et al. 2017; Tebbett et al. 2017a, 2022; Nicholson et al. 2020). However, many previous studies have relied on a single dietary analysis method to investigate the diets of nominally herbivorous fishes, which may result in a limited understanding of the differences in their resource use. We utilized a combination of different techniques, including gut content DNA metabarcoding, morphological identification, and stable isotope analysis, to comprehensively assess the dietary differences of several fish species that are typically considered herbivorous. We found that different herbivorous reef fishes utilize a diverse range of food resources, and our study showcases the value of using multiple approaches to uncover these dietary differences.
Dietary targets of the studied fish species
Identifying the gut contents of C. striatus can be challenging due to the complex nature of their dietary sources and the high level of mechanical digestive processing (Choat et al. 2004). Previous studies have often classified the gut contents of C. striatus into broad morphological categories, such as turf algae, macroalgae, sediments, and other unidentified materials (which are mostly categorized as detritus) (Kelly et al. 2016; Tebbett et al. 2017a). Microbes and microalgae, which are often categorized as detritus, can also serve as a potential nutritional source for C. striatus (Tebbett et al. 2017a, b). However, most current studies have not identified the specific species of these cryptic organisms, instead grouping them together as detritus. With the advantages of high-throughput sequencing sensitivity, a wide range of organisms have been detected in the gut contents of C. striatus. However, some detected organisms may not represent the targeted food of a given fish but be occasionally ingested by the sampled fishes, and real food items should dominate in gut content sequencing (Deagle et al. 2019). Therefore, a threshold of 1% was used to exclude the background noise in the present study. Even so, gut content sequencing revealed considerable overlap between C. striatus and other fishes (Figs. 4a, b). Ctenochaetus striatus is likely to ingest debris or early algal settlers from the same substrates exploited by other fishes or algal material from the faeces of other fishes (Tebbett et al. 2022) and assimilate these debris. Overlaps in diet may also be mitigated by feeding on multiple species from different features within the same area (Adam et al. 2015).
Multiple lines of evidence suggest that most parrotfishes are considered to be 'microphages', and their capacity for bioerosion on reefs appears to be a result of targeting protein-rich autotrophic microorganisms that live on or within calcareous substrata (Clements et al. 2017; Nicholson and Clements 2023). Our results showed that the nitrogen values for parrotfishes (S. globiceps and S. schlegelii) are depleted. This supports the suggestions of Clements et al. (2017), who suggest that these fishes are possibly targeting cyanobacteria. The lower relative read abundance of macroscopic algae and higher read abundance of dinoflagellate in gut contents of parrotfishes also indicated that the dietary targets are microorganisms instead of the turf algae or EAM as a whole (Fig. 3a). Nicholson et al. (2020) found the only consistent feature across all parrotfishes biting cores was high density of filamentous cyanobacteria. Unfortunately, our molecular data do not include prokaryotes, especially for cyanobacteria. Therefore, we are unable to provide the exact species of cyanobacteria ingested by parrotfishes in the present study.
The territorial farmer (P. fasciolatus) ingested a certain amount of zooplankton and algal detritus (Figs. 3 and 5) with the highest δ15N values (Fig. 4e). Indeed, territorial farmers show limited herbivory on benthic algae (Ceccarelli et al. 2005), and their diets mostly constitute detritus rather than algae (Wilson and Bellwood 1997). As documented in previous studies (Choat et al. 2002; Tebbett et al. 2017b, 2022), turf algal croppers (Acanthurus spp.) mainly fed on turf algae, and also found that they ingest the brown algae Sphacelaria. Sphacelaria was the only genus detected in all the feeding guilds (Fig. 5, Supplemental S4), and can be the dominant algae on well-grazed turfs (Carpenter 1986). Sphacelaria is widely distributed in the southern part of the SCS (Draisma et al. 1997), possibly resulting in abundant living forms, detritus, or DNA in the studied reefs. Zebrasoma velifer has generally been viewed as a macroalgae browser due to the large quantities of macroalgae detected in its gut contents (Hiatt and Strasburg 1960; Robertson and Gaines 1986; Clements et al. 2017). Tebbett et al. (2022) classified Zebrasoma as concealed croppers due to their morphology and behaviour which suggested they may target different types of algae to macroalgal browsers. Indeed, in our study, the gut contents of Z. velifer predominantly contained turf algae rather than foliose algae (Fig. 3). In addition to morphological identification, DNA metabarcoding analysis indicates that red algae were the predominant type of algae consumed by Z. velifer (Fig. 5). This is not surprising, as red algae are a common group of algae that form algal turfs on reefs (Scott and Russ 1987; Harris et al. 2015).
New insights from a multi-method approach to dietary analysis
Using multiple different methods helped reveal the prevalence of dinoflagellates in the gut contents of parrotfishes and C. striatus (Fig. 3a). Parrotfishes and C. striatus overlapped both in gut content sequencing and isotopic niche (Fig. 4a, b, e), and the major shared species were from Symbiodinium and Prorocentrum (Fig. 5). Both the ‘brushing’ and ‘scrapping’ feeding modes can remove microalgae (e.g. diatoms, dinoflagellates, and cyanobacteria) from reefs (Tebbett et al. 2017a, 2017b). These microorganisms likely serve as nutrient sources for both C. striatus and parrotfishes (Marshell and Mumby 2012; Clements et al. 2017). A considerable overlap in isotopic niche between C. striatus and parrotfishes may be the result of both fishes consuming dinoflagellates (Fig. 4e and Supplemental S3). Moreover, Symbiodinium is a dinoflagellate commonly found in coral reef ecosystems as a symbiont (Hatcher, 1988). It is also found free living in sediments (Takabayashi et al. 2012; Quigley et al. 2017). The relatively high abundance of Symbiodinium in the gut contents of parrotfishes and Ctenochaetus may be attributed to the amount of sediment/sand these fishes consume when feeding (Fig. 3b). It is important to verify whether the Symbiodinium found in the fish gut contents is obtained from corals or from other natural sources. Prorocentrum often coexist with filamentous algae and may occasionally be consumed by surgeonfishes (Kohler and Kohler 1992). This dinoflagellate has been linked to Ciguatera (Faust 1991; Richlen and Lobel 2011). Ctenochaetus striatus is considered ‘high risk’ for Ciguatera poisoning (Mak et al. 2013; Rongo and van Woesik 2013) and a primary vector of toxins produced by benthic dinoflagellates (Yasumoto et al. 1976; Lewis 2006). Considering our limited understanding of Ciguatera in reef ecosystems, the presence of Prorocentrum in the gut contents of parrotfishes and C. striatus may help guide further research on the trophic transfer of Ciguatera in coral reef ecosystems.
Interestingly, a considerable proportion of microinvertebrates, such as crustaceans and nematodes, were also found in the fish gut contents (Figs. 3, 5). Microinvertebrates are abundant in EAM (Kramer et al. 2012). Consequently, when reef fishes ingest benthic algae or EAM from a reef base, they may also consume a significant number of benthic microinvertebrates (Kramer et al. 2013). However, their trophic utilization by herbivorous reef fishes has received limited attention, which means that their potential to be an energy resource for herbivorous fishes has been largely overlooked (Tebbett et al. 2023). The possibility of microinvertebrates being regularly consumed by nominally herbivorous fishes and their potential contribution to the nutritional ecology of herbivorous fishes needs to be explored further. Overall, our multi-method approach to dietary analysis provided new insights into the diets of these fishes and suggests that traditional classifications of fish as herbivorous or carnivorous may not accurately reflect the complexity of their diets.
Value in a multi-method approach to dietary analysis
Using DNA metabarcoding can enhance the resolution of taxonomic information on dietary targets, as suggested by Deagle et al. (2019), allowing identification of specific taxa involved in dietary partitioning with greater precision than conventional visual methods (Leray et al. 2015, 2019). Approximately, 70% of the genera found in the gut contents were only present in one species or feeding guild (Fig. 5, Supplemental S4), indicating a high level of dietary specialization. Similar results have also been reported for other reef fishes (Leray et al. 2015, 2019; Brandl et al. 2020). Leray et al. (2015) found that nearly 80% of prey items consumed by only one predator based on the results of gut content metabarcoding. Casey et al. (2019) demonstrated a detailed trophic network with a high degree of resolution, revealing fine-scale partitioning among various reef fish species. It is important to note that DNA sequencing may not be able to differentiate between living and dead organisms that are ingested by fishes. To ensure the accuracy of the results, it is necessary to verify the possible sources of the detected DNA sequences through other methods (Pompanon et al. 2012). By combining stable isotope analysis and gut content analysis, we can increase the confidence and reliability of our findings. By combining stable isotope analysis and morphological identification, the detected dietary overlap of C. striatus with other fishes through gut content metabarcoding may be due to interference from debris of algae or other organisms. In addition, the isotopic analysis showed a considerable overlap in isotopic niche between C. striatus and parrotfishes (Fig. 4e and Supplemental S3). DNA metabarcoding analysis detected a relatively high abundance of dinoflagellates in the gut contents of parrotfishes and C. striatus (Figs. 3a and 5), and standard gut content analysis revealed a significant presence of sand in the gut contents of both species (Fig. 3b). Therefore, it is likely that dinoflagellates or other microalgae associated with sediment/sand contribute to the isotopic overlap observed between C. striatus and parrotfishes. Our study highlights the benefits of combining metabarcoding (a high-resolution snapshot) and stable isotope analysis (long-term trophic position) to comprehensively capture the dietary differences among reef fishes. This approach can provide reliable results and improve our understanding of reef fish trophic ecology. It is worth noting that other methods may also provide valuable information. One such method is fatty acid analysis, which is primarily used to assess basal food sources (such as bacteria, detritus, diatoms versus dinoflagellates) (Crossman et al. 2005; Clements et al. 2017). Fatty acid analyses can provide information about the quality and nutritional value of different food sources (Pethybridge et al. 2018). Analysis of the short chain fatty acid composition in the gut fluid of nominally herbivorous fishes also indicates that they exhibit major differences in their primary targeted food resources (Crossman et al. 2005; Clements et al. 2017).
Limitations and future work
It is important to note that while the multi-method approach used in this study allowed for a comprehensive analysis of dietary differences, the molecular data obtained did not include prokaryotes, particularly cyanobacteria. This is because the DNA metabarcoding approach used in this study focused on the identification of eukaryotic organisms. As a result, any cyanobacteria present in the gut contents would not have been detected using this method. It is possible that cyanobacteria may have been an important component of the diet of some of the herbivorous fishes in this study. It is important to carefully distinguish cyanobacteria ingested by fishes from other microbial taxa present in the fish gut and environment, as not all of these may be dietary targets. Therefore, future studies should consider using additional methods to specifically target cyanobacteria DNA in gut contents to obtain a more comprehensive understanding of the dietary composition of these fishes (Nicholson and Clements 2020, 2023). Few diatoms were detected in the present study, perhaps because their cell structures were destroyed during digestion(Choat et al. 2004) or because of the different DNA recovery rates between diatoms and dinoflagellates from frozen samples (Maki et al. 2017). Therefore, neglecting cyanobacteria and other autotrophic microorganisms may lead to an underestimation of the dietary niche of parrotfishes compared to other fish species. As a result, the actual differences in dietary niche between parrotfishes and other fish species may be higher than currently estimated in our study. Another limitation is the relatively low sample size for some species, which may have influenced our results and interpretation. In particular, there may be substantial variation in diets across sites for species sampled from multiple locations that are not captured in our analysis. This may result in a lower dietary diversity and narrower trophic niche being observed compared to the actual feeding habits of these fishes in the wild. This highlights the need for larger sample sizes and more extensive sampling across sites to fully understand dietary variation in these fishes. The size of individuals sampled, especially for parrotfishes, was also relatively small, averaging 10–11 cm. This may have shaped our results given that the diet of some fishes can change throughout ontogeny (Plass-Johnson et al. 2013). Future studies should consider sampling larger individuals to determine how the diet of these fishes changes over time. Lastly, the present study was restricted to a few fish species, which only represent the dominate species of herbivorous fishes in SCS. To enhance our comprehension of the utilization of resources by herbivorous fishes in coral reef ecosystems, it is important to include more fish species in future studies.
Conclusions
Through the integration of stable isotope analysis, morphological identification, and DNA metabarcoding, our study revealed substantial dietary partitioning among nominally herbivorous fishes. DNA metabarcoding revealed diverse algae categories (Ochrophyta, Rhodophyta, and Chlorophyta) in the gut contents of sampled fishes with high taxonomic resolution, as well as some microscopic and cryptic taxa, such as dinoflagellates and microinvertebrates. Our study found that the overlap of 95% prediction isotopic niches among feeding guilds was generally less than 30% of the total area, and nearly 70% of the genera were exclusively detected in the gut contents of a single species or feeding guild. While gut content metabarcoding has limitations in distinguishing between living organisms and detritus in diets, our study proposes a solution. By integrating stable isotope analysis and morphological identification with metabarcoding, we found that the observed overlap detected by metabarcoding between detritivore brusher C. striatus and other fishes may be attributed to interference from debris of algae or other organisms. This combined approach allows for a more accurate understanding of the dietary differences among nominally herbivorous fishes. While each method has its own limitations in dietary analysis, our study suggests that an integrative, multi-tool approach can remedy the limitations of one method by utilizing the strengths of another. Our results show a wide spectrum of resources on coral reefs could be utilized by different fish species in a complementary manner. However, our study also has important limitations that need to be addressed in future research, including the need for larger sample sizes and improved techniques for dietary analysis. By improving our techniques for dietary analysis and increasing sample sizes, we can gain a more complete understanding of the nutritional resources of these fishes and how they contribute to the functioning of coral reef ecosystems.
Data availability
The raw short-read DNA sequencing data was deposited in the NCBI Sequence Read Archive under the accession number SAMN26178891–SAMN26178918.
References
Adam TC, Kelley M, Ruttenberg BI, Burkepile DE (2015) Resource partitioning along multiple niche axes drives functional diversity in parrotfishes on Caribbean coral reefs. Oecologia 179:1173–1185. https://doi.org/10.1007/s00442-015-3406-3
Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, Mills DA, Caporaso JG (2013) Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods 10:57-U11. https://doi.org/10.1038/Nmeth.2276
Brandl SJ, Casey JM, Meyer CP (2020) Dietary and habitat niche partitioning in congeneric cryptobenthic reef fish species. Coral Reefs 39:305–317. https://doi.org/10.1007/s00338-020-01892-z
Carpenter RC (1986) Partitioning herbivory and its effects on coral reef algal communities. Ecol Monogr 56:345–363. https://doi.org/10.2307/1942551
Casey JM, Meyer CP, Morat F, Brandl SJ, Planes S, Parravicini V (2019) Reconstructing hyperdiverse food webs: gut content metabarcoding as a tool to disentangle trophic interactions on coral reefs. Methods Ecol Evol 10:1157–1170. https://doi.org/10.1111/2041-210X.13206
Ceccarelli DM, Jones GP, McCook LJ (2005) Foragers versus farmers: contrasting effects of two behavioural groups of herbivores on coral reefs. Oecologia 145:445–453. https://doi.org/10.1007/s00442-005-0144-y
Cheal AJ, MacNeil MA, Cripps E, Emslie MJ, Jonker M, Schaffelke B, Sweatman H (2010) Coral–macroalgal phase shifts or reef resilience: links with diversity and functional roles of herbivorous fishes on the Great Barrier Reef. Coral Reefs 29:1005–1015. https://doi.org/10.1007/s00338-010-0661-y
Choat JH, Bellwood DR (1985) Interactions amongst herbivorous fishes on a coral-reef - influence of spatial variation. Mar Biol 89:221–234. https://doi.org/10.1007/Bf00393655
Choat JH, Clements KD, Robbins WD (2002) The trophic status of herbivorous fishes on coral reefs - I: Dietary analyses. Mar Biol 140:613–623. https://doi.org/10.1007/s00227-001-0715-3
Choat JH, Robbins WD, Clements KD (2004) The trophic status of herbivorous fishes on coral reefs - II. Food processing modes and trophodynamics. Mar Biol 145:445–454. https://doi.org/10.1007/s00227-004-1341-7
Clements KD, German DP, Piche J, Tribollet A, Choat JH (2017) Integrating ecological roles and trophic diversification on coral reefs: multiple lines of evidence identify parrotfishes as microphages. Biol J Linn Soc 120:729–751. https://doi.org/10.1111/bij.12914
Crossman DJ, Choat JH, Clements KD (2005) Nutritional ecology of nominally herbivorous fishes on coral reefs. Mar Ecol Prog Ser 296:129–142. https://doi.org/10.3354/Meps296129
Deagle BE, Thomas AC, Shaffer AK, Trites AW, Jarman SN (2013) Quantifying sequence proportions in a DNA-based diet study using ion torrent amplicon sequencing: which counts count? Mol Ecol Resour 13:620–633. https://doi.org/10.1111/1755-0998.12103
Deagle BE, Thomas AC, McInnes JC, Clarke LJ, Vesterinen EJ, Clare EL, Kartzinel TR, Eveson JP (2019) Counting with DNA in metabarcoding studies: How should we convert sequence reads to dietary data? Mol Ecol 28:391–406. https://doi.org/10.1111/mec.14734
Draisma SGA, Keum YS, Prud’homme van Reine WF, Lokhorst GM (1997) The species of Sphacelaria (Sphacelariales, Phaeophyceae) in China with a description of a new species. Phycologia 36:26–27
Faust MA (1991) Morphology of ciguatera-causing Prorocentrum lima (Pyrrophyta) from widely differing sites. J Phycol 27:642–648. https://doi.org/10.1111/j.0022-3646.1991.00642.x
Harris JL, Lewis LS, Smith JE (2015) Quantifying scales of spatial variability in algal turf assemblages on coral reefs. Mar Ecol Prog Ser 532:41–57. https://doi.org/10.3354/meps11344
Hiatt RW, Strasburg DW (1960) Ecological relationships of the fish fauna on coral reefs of the marshall islands. Ecol Monogr 30:65–127
Jackson AL, Inger R, Parnell AC, Bearhop S (2011) Comparing isotopic niche widths among and within communities: SIBER - Stable Isotope Bayesian Ellipses in R. J Anim Ecol 80:595–602. https://doi.org/10.1111/j.1365-2656.2011.01806.x
Kartzinel TR, Chen PA, Coverdale TC, Erickson DL, Kress WJ, Kuzmina ML, Rubenstein DI, Wang W, Pringle RM (2015) DNA metabarcoding illuminates dietary niche partitioning by African large herbivores. P Natl Acad Sci USA 112:8019–8024. https://doi.org/10.1073/pnas.1503283112
Kelly ELA, Eynaud Y, Clements SM, Gleason M, Sparks RT, Williams ID, Smith JE (2016) Investigating functional redundancy versus complementarity in Hawaiian herbivorous coral reef fishes. Oecologia 182:1151–1163. https://doi.org/10.1007/s00442-016-3724-0
Kohler ST, Kohler CC (1992) Dead bleached coral provides new surfaces for dinoflagellates implicated in ciguatera fish poisonings. Environ Biol Fish 35:413–416. https://doi.org/10.1007/Bf00004993
Kramer MJ, Bellwood DR, Bellwood O (2012) Cryptofauna of the epilithic algal matrix on an inshore coral reef, Great Barrier Reef. Coral Reefs 31:1007–1015. https://doi.org/10.1007/s00338-012-0924-x
Kramer MJ, Bellwood O, Bellwood DR (2013) The trophic importance of algal turfs for coral reef fishes: the crustacean link. Coral Reefs 32:575–583. https://doi.org/10.1007/s00338-013-1009-1
Leal MC, Ferrier-Pages C (2016) Molecular trophic markers in marine food webs and their potential use for coral ecology. Mar Genom 29:1–7. https://doi.org/10.1016/j.margen.2016.02.003
Leray M, Meyer CP, Mills SC (2015) Metabarcoding dietary analysis of coral dwelling predatory fish demonstrates the minor contribution of coral mutualists to their highly partitioned, generalist diet. PeerJ 3:e1047. https://doi.org/10.7717/Peerj.1047
Leray M, Alldredge AL, Yang JY, Meyer CP, Holbrook SJ, Schmitt RJ, Knowlton N, Brooks AJ (2019) Dietary partitioning promotes the coexistence of planktivorous species on coral reefs. Mol Ecol 28:2694–2710. https://doi.org/10.1111/mec.15090
Lewis RJ (2006) Ciguatera: Australian perspectives on a global problem. Toxicon 48:799–809. https://doi.org/10.1016/j.toxicon.2006.07.019
Lin XZ, Hu SM, Liu S, Huang H (2018) Unexpected prey of juvenile spotted scat (Scatophagus argus) near a wharf: The prevalence of fouling organisms in stomach contents. Ecol Evol 8:8547–8554. https://doi.org/10.1002/ece3.4380
Lin XZ, Hu SM, Liu Y, Zhang L, Huang H, Liu S (2021) Disturbance-mediated changes in coral reef habitat provoke a positive feeding response in a major coral reef detritivore Ctenochaetus striatus. Front Mar Sci 8:682697. https://doi.org/10.3389/Fmars.2021.682697
Mak YL, Wai TC, Murphy MB, Chan WH, Wu JJ, Lam JCW, Chan LL, Lam PKS (2013) Pacific ciguatoxins in food web components of coral reef systems in the Republic of Kiribati. Environ Sci Technol 47:14070–14079. https://doi.org/10.1021/es403175d
Maki A, Salmi P, Mikkonen A, Kremp A, Tiirola M (2017) Sample preservation, DNA or RNA extraction and data analysis for high-throughput phytoplankton community sequencing. Front Microbiol 8:1848. https://doi.org/10.3389/Fmicb.2017.01848
Marshell A, Mumby PJ (2012) Revisiting the functional roles of the surgeonfish Acanthurus nigrofuscus and Ctenochaetus striatus. Coral Reefs 31:1093–1101. https://doi.org/10.1007/s00338-012-0931-y
Montgomery WL, Myrberg AA, Fishelson L (1989) Feeding ecology of surgeonfishes (Acanthuridae) in the northern red-sea, with particular reference to Acanthurus-nigrofuscus (Forsskal). J Exp Mar Biol Ecol 132:179–207. https://doi.org/10.1016/0022-0981(89)90127-5
Ng D, Taira D, Heery EC, Todd PA (2021) Antagonistic effects of seawalls and urban sedimentation on epilithic algal matrix (EAM)-feeding fishes. Mar Pollut Bull 173:13098. https://doi.org/10.1016/j.marpolbul.2021.113098
Nicholson GM, Clements KD (2020) Resolving resource partitioning in parrotfishes (Scarini) using microhistology of feeding substrata. Coral Reefs 39:1313–1327. https://doi.org/10.1007/s00338-020-01964-0
Nicholson GM, Clements KD (2023) Micro-photoautotroph predation as a driver for trophic niche specialization in 12 syntopic Indo-Pacific parrotfish species. Biol J Linn Soc 139:91–114. https://doi.org/10.1093/biolinnean/blad005
Nielsen JM, Clare EL, Hayden B, Brett MT, Kratina P (2018) Diet tracing in ecology: Method comparison and selection. Methods Ecol Evol 9:278–291. https://doi.org/10.1111/2041-210X.12869
Oakley-Cogan A, Tebbett SB, Bellwood DR (2020) Habitat zonation on coral reefs: Structural complexity nutritional resources and herbivorous fish distributions. PLoS ONE 15:e0233498. https://doi.org/10.1371/journal.pone.0233498
Oksanen JF, Blanchet G, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Henry M, Stevens H, Szoecs E, Wagner H (2020) vegan: Community ecology package. R package version 25–7
Pethybridge HR, Choy CA, Polovina JJ, Fulton EA (2018) Improving marine ecosystem models with biochemical tracers. Annu Rev Mar Sci 10:199–228. https://doi.org/10.1146/annurev-marine-121916-063256
Plass-Johnson JG, McQuaid CD, Hill JM (2013) Stable isotope analysis indicates a lack of inter- and intra-specific dietary redundancy among ecologically important coral reef fishes. Coral Reefs 32:429–440. https://doi.org/10.1007/s00338-012-0988-7
Pompanon F, Deagle BE, Symondson WOC, Brown DS, Jarman SN, Taberlet P (2012) Who is eating what: diet assessment using next generation sequencing. Mol Ecol 21:1931–1950. https://doi.org/10.1111/j.1365-294X.2011.05403.x
Pratchett MS, Hoey AS, Wilson SK, Messmer V, Graham NAJ (2011) Changes in biodiversity and functioning of reef fish assemblages following coral bleaching and coral loss. Diversity 3:424–452
Quigley KM, Bay LK, Willis BL (2017) Temperature and water quality-related patterns in sediment-associated Symbiodinium communities impact symbiont uptake and fitness of juveniles in the genus Acropora. Front Mar Sci. https://doi.org/10.3389/Fmars.2017.00401
R Core Team (2022) R: A language and environment for statistical computing. R Foundation for Statistical Computing
Rasher DB, Hoey AS, Hay ME (2013) Consumer diversity interacts with prey defenses to drive ecosystem function. Ecology 94:1347–1358. https://doi.org/10.1890/12-0389.1
Richlen ML, Lobel PS (2011) Effects of depth, habitat, and water motion on the abundance and distribution of ciguatera dinoflagellates at Johnston Atoll, Pacific Ocean. Mar Ecol Prog Ser 421:51–66. https://doi.org/10.3354/meps08854
Robertson DR, Gaines SD (1986) Interference competition structures habitat use in a local assemblage of coral-reef surgeonfishes. Ecology 67:1372–1383. https://doi.org/10.2307/1938693
Rongo T, van Woesik R (2013) The effects of natural disturbances, reef state, and herbivorous fish densities on ciguatera poisoning in Rarotonga, southern Cook Islands. Toxicon 64:87–95. https://doi.org/10.1016/j.toxicon.2012.12.018
Scott FJ, Russ GR (1987) Effects of grazing on species composition of the epilithic algal community on coral reefs of the central great-barrier-reef. Mar Ecol Prog Ser 39:293–304. https://doi.org/10.3354/Meps039293
Stoeck T, Bass D, Nebel M, Christen R, Jones MDM, Breiner HW, Richards TA (2010) Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol Ecol 19:21–31. https://doi.org/10.1111/j.1365-294X.2009.04480.x
Takabayashi M, Adams LM, Pochon X, Gates RD (2012) Genetic diversity of free-living Symbiodinium in surface water and sediment of Hawai’i and Florida. Coral Reefs 31:157–167. https://doi.org/10.1007/s00338-011-0832-5
Tebbett SB, Bellwood DR (2019) Algal turf sediments on coral reefs: what’s known and what’s next. Mar Pollut Bull 149:110542. https://doi.org/10.1016/j.marpolbul.2019.110542
Tebbett SB, Goatley CHR, Bellwood DR (2017a) Clarifying functional roles: algal removal by the surgeonfishes Ctenochaetus striatus and Acanthurus nigrofuscus. Coral Reefs 36:803–813. https://doi.org/10.1007/s00338-017-1571-z
Tebbett SB, Goatley CHR, Bellwood DR (2017b) The effects of algal turf sediments and organic loads on feeding by coral reef surgeonfishes. PLoS ONE 12:e0169479. https://doi.org/10.1371/journal.pone.0169479
Tebbett SB, Siqueira AC, Bellwood DR (2022) The functional roles of surgeonfishes on coral reefs: past, present and future. Rev Fish Biol Fisher 32:387–439. https://doi.org/10.1007/s11160-021-09692-6
Tebbett SB, Bennett S, Bellwood DR (2023) A functional perspective on the meaning of the term “herbivore”: patterns versus processes in coral reef fishes. Coral Reefs. https://doi.org/10.1007/s00338-023-02378-4
Wickham H (2016) ggplot2: elegant graphics for data analysis. Springer-Verlag, New York
Wilson S, Bellwood DR (1997) Cryptic dietary components of territorial damselfishes (Pomacentridae, Labroidei). Mar Ecol Prog Ser 153:299–310. https://doi.org/10.3354/meps153299
Wilson SK, Bellwood DR, Choat JH, Furnas MJ (2003) Detritus in the epilithic algal matrix and its use by coral reef fishes. Oceanogr Mar Biol: an Annual Review 41:279–309
Yasumoto TB, Raymond P, Vernoux JP (1976) Toxicity of the surgeonfishes II. Properties of the principal water soluble toxin. Bull Jpn Soc Sci Fish 42:359–365
Funding
This work was supported by the National Natural Science Foundation of China (Grant numbers 42106150 and 42176118), the Key Special Project for Introduced Talents Team of Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou) (Grant numbers GML2019ZD0405), the Innovation Academy of South China Sea Ecology and Environmental Engineering, Chinese Academy of Sciences (Grant number ISEE2018PY03), the Science, Technology Planning Project of Guangdong Province, China (Grant number 2020B1212060058).
Author information
Authors and Affiliations
Contributions
XL, SL, and LZ conceived the study; XL and SH designed the study and experimental protocols; XL performed the experiments, data analyses, and wrote the manuscript; YZ contributed the fish isotope data; HH, LZ, SL, and SH contributed significantly to the improvement of the manuscript and reviewed the final draft.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
The fish study was reviewed and approved by the Laboratory Animal Management and Ethics Committee, South China Sea Institute of Oceanology, Chinese Academy of Sciences.
Additional information
Responsible Editor: K. Clements .
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lin, X., Hu, S., Zhou, Y. et al. A multiple-methods approach to investigate dietary differences among nominally herbivorous fishes. Mar Biol 170, 134 (2023). https://doi.org/10.1007/s00227-023-04269-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00227-023-04269-2