
Abstract
Comparing the genetic structure of host populations with that of their parasites can shed light on the efficiency and independence of their respective dispersal mechanisms. The degree of congruence between host and parasite genetic structure should reflect to what extent they share dispersal mechanisms. Here, we contrast the genetic structure of the acanthocephalan parasite Profilicollis novaezelandensis with that of its intermediate host, the hairy-handed shore crab Hemigrapsus crenulatus, along the east coast of New Zealand’s South Island. We expected no congruence in their phylogeographic patterns because of the very different modes of dispersal used by the crabs (planktonic drift) and the acanthocephalans (bird-mediated dispersal). Based on analysis of cytochrome oxidase subunit I gene sequences, we found no significant genetic structure among isolated populations of the crab and those of their parasite, along a roughly 600 km stretch of coastline. Surprisingly, based on a distance-based co-evolutionary analysis statistical tool (PACo), we observed an overall significant level of congruence between host and parasite population-level phylogenies. The most parsimonious interpretation is that statistical significance does not translate into biological significance, with the result likely due to chance, possibly because bird movements that disperse parasites coincidentally match patterns of crab dispersal by ocean currents in parts of our study area. In this system, the connectivity among different localities and the apparent genetic mixing among populations may have implications for host–parasite co-evolution.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
At a time when the emergence, incidence and severity of parasitic diseases appear to be on the rise for many taxa in marine ecosystems (Ward and Lafferty 2004; Groner et al. 2016), it is crucial to understand how they spread among host populations. Comparative genetic structure, or comparative phylogeography, of hosts and their parasites provides a tool to address this, by assessing to what extent genetic differentiation among parasite populations is congruent with that among their respective host populations (e.g. Dybdahl and Lively 1996; Criscione and Blouin 2004; Keeney et al. 2009). In principle, parasites that spread independently of their hosts should have a spatial genetic structure distinct from that of the hosts, whereas parasites that depend on their hosts for dispersal should have a congruent genetic structure. Furthermore, comparing the genetic structure among host and parasite populations, and genetic diversity within populations, can provide insights into the spread of advantageous alleles (Morjan and Rieseberg 2004) and the evolution of local adaptation (Dybdahl and Lively 1996; Prugnolle et al. 2005), both key factors in host–parasite co-evolution.
Helminth parasites of coastal organisms typically use vertebrates such as fish or birds as definitive hosts and invertebrates as intermediate hosts. In general, we might expect strong congruence between the genetic structure of a helminth parasite and that of its definitive host, given that the definitive host’s movements are the main dispersal routes for the parasites (Blasco-Costa and Poulin 2013). Conversely, the parasite’s genetic structure is unlikely to match that of its intermediate host, as the two are dispersed via different mechanisms. However, multiple other factors affect genetic structure and host–parasite congruence, and it remains difficult to predict observed patterns based on host traits or the hosts’ roles in the parasite life cycle (Keeney et al. 2009; Mazé-Guilmo et al. 2016). Therefore, congruence (or lack thereof) between host and parasite genetic structure cannot be assumed, but must be tested. The recent application of methods originally developed to test for congruence between two phylogenetic trees, to compare the genetic structure of hosts and parasites across multiple populations (see Lagrue et al. 2016) allows for a novel and robust assessment of congruence in phylogeographic patterns.
In this study, we investigate the genetic structure of an acanthocephalan parasite and its crab intermediate host, as well as the level of congruence between their respective genetic structures, across several localities on the east coast of New Zealand’s South Island. The hairy-handed shore crab Hemigrapsus crenulatus Milne-Edwards, 1837 (Brachyura: Grapsidae) is abundant in sheltered intertidal or estuarine habitats (McLay 1988). They possess planktonic larval stages and are therefore dispersed by coastal currents. In our study area, the main current is the Southland Current, which flows northward along the east coast of the South Island (Sutton 2003) and shapes the genetic structure of several marine invertebrates (Wallis and Trewick 2009).
Along with two other crab species, H. crenulatus serves as the first intermediate host of the acanthocephalan Profilicollis novaezelandensis Brockerhoff and Smales 2002 (Polymorphidae). Crabs become infected through accidental ingestion of parasite eggs; inside a crab host, the parasites develop into juveniles, or cystacanths, and await transmission to avian definitive hosts. Suitable bird definitive hosts include gulls (Larus spp.) and oystercatchers (Haematopus spp.). Inside the bird intestine, the worms develop into adults, reproduce and pass their eggs outside via host faeces. Cystacanths have a relatively long life span, and over time large numbers can accumulate in individual crabs (over 100 in some individuals; Latham and Poulin 2002a). Moderate to heavy infections have been shown to induce crabs to spend more time away from their burrows and experience higher mortality, mostly due to bird predation (Latham and Poulin 2002a, b). In contrast, birds probably incur minimal costs from infection.
In this system, hosts and parasites have seemingly efficient but completely independent modes of dispersal: the crabs rely on planktonic dispersal mediated by ocean currents, whereas the parasites are passively dispersed by their highly mobile definitive hosts. Crucially, planktonic crab juveniles are never infected and cannot contribute to parasite dispersal. These differences lead to three hypotheses. First, we hypothesise that there is weak genetic structure among populations of crabs along a north–south cline generated by the Southland current. Second, we hypothesise that there is no genetic structure among populations of parasites. Third, and more important, we hypothesise that the genetic structures of hosts and parasites, whether weak or strong, are completely independent of each other because of their distinct modes of dispersal. Here, we test these three hypotheses. Specifically, we use two genetic markers, the mitochondrial COI and nuclear 28S genes, to characterise the genetic diversity of H. crenulatus and P. novaezelandensis, explore the geographic population structure of both species and test whether they show congruent patterns.
Methods
Sample collection
Adult crabs, H. crenulatus, were collected from intertidal mudflats, river mouths and estuaries of eight locations along the East Coast of the South Island: Christchurch (CH, 43.550509°S, 172.703671°E), Kakanui (KA, 45.184492°S, 170.892708°E), Blueskin Bay (BB, 45.746889°S, 170.579611°E), Aramoana (AR, 45.784029°S, 170.681109°E), Portobello (PO, 45.839172°S, 170.648707°E), Taieri Mouth (TM, 46.050142°S, 170.190145°E), Papatowai (PA, 46.553866°S, 169.471249°E) and Invercargill (IN, 46.420003°S, 168.337081°E). All specimens were generally found underneath large rocks and caught by hand along the shoreline, within an area of approximately 50 m.
Crabs were killed by freezing after collection. The carapace width of each crab was measured to the nearest millimetre and the sex (determined by the shape of the abdomen) was recorded. Muscle tissue was removed from a walking leg and preserved in 99% ethanol for genetic analysis. The crabs were dissected and examined for acanthocephalan parasites. In infected crabs, the total number of Profilicollis sp. cystacanths was recorded; all cystacanths were collected, rinsed in distilled water to remove crab tissue and preserved in 99% ethanol for genetic analysis. Prevalence (proportion of infected hosts per location) and mean intensity (mean number of acanthocephalans per infected host) were calculated for each location. Acanthocephalan species identity was determined based on morphological characters, namely the number of hooks and rows in the proboscis as described by Brockerhoff and Smales (2002). One adult specimen identified as P. novaezelandensis, found in a royal spoonbill (Platalea regia Gould, 1838: Ciconiiformes: Threskiornithidae) from Otago Harbour (i.e. close to our AR and PO sampling sites) and kindly donated by Dr. B. Presswell, was also used for molecular analysis.
DNA extraction, PCR amplification and sequencing
A minimum of seven crabs and seven acanthocephalans per location (with the exception of Kakanui where no infected crabs were found) were selected for genetic analysis. Genomic DNA from all crabs and acanthocephalans was extracted using a modified fish buffer method (see Devlin et al. 2004). For each acanthocephalan, the whole individual was used for DNA extraction, while for the crab only a small section of muscle tissue (1–2 mm) was used.
For confirmation of Profilicollis species identity and respective phylogenetic position, a ~ 440 bp partial fragment of the 28S ribosomal DNA region (28S) was amplified; this marker is useful for phylogenetic classification and identification of acanthocephalans (Garcia-Varela et al. 2013). The 28S fragment was amplified using the primers T16 (5′-GAGACCGATAGCGAAACAAGTAC-3′) and T30 (5′-TGTTAGACTCCTTGGTCCGTG-3′) (Harper and Saunders 2001). Up to three specimens per locality were amplified as low genetic variability was expected and because this marker was only used to explore relationships between taxa. All polymerase chain reactions (PCRs) were performed in a final volume of 20 µL using an Eppendorf Mastercycler Pro thermal cycler. This consisted of 4 µL of 5× MyTaq Red Reaction buffer, 0.8 µL of each primer (10 mM), 0.2 µL bovine serum albumin (BSA, New England Biolabs), 0.1 µL MyTaq™ Red DNA Polymerase (Bioline, Bioline Pty Ltd, Alexandria, NSW, Australia) and 2 µL of the extracted DNA. Cycler conditions consisted of denaturation at 95 °C for 3 min, 35 cycles of 95 °C for 30 s, 50 °C for 30 s and 72 °C for 1 min for amplification, followed by a final extension at 72 °C for 10 min.
To explore population differences, a ~ 600 bp fragment of the mitochondrial cytochrome oxidase subunit I (CO1) gene was amplified for both the crab and acanthocephalan samples, using the universal invertebrate primers jgLCO1490 (5′-TITCIACIAAYCAYAARGAYATTGG-3′) and jgHCO2198 (5′-TAIACYTCIGGRTGICCRAARAAYCA-3′) (Geller et al. 2013). All PCRs were performed as described above, but with 1 µL of the extracted acanthocephalan DNA and 2–3 µL of the extracted crab DNA. Cycler conditions consisted of denaturation at 95 °C for 3 min, 35 cycles of 95 °C for 30 s, 46.2 °C for 30 s and 72 °C for 1 min for amplification, followed by a final extension at 72 °C for 10 min. Amplified products were sequenced for only the forward direction (unless otherwise stated), using the same primers as for amplification. PCR product purification and Sanger sequencing by capillary electrophoresis were performed by a commercial facility (Macrogen Inc., Seoul, Republic of Korea).
Sequence processing
All sequence data editing and aligning was performed using Geneious v8.1.8 (http://www.geneious.com, Kearse et al. 2012). All sequences were uploaded to BLAST (Zhang et al. 2000) prior to analysis to confirm their identity and ensure no contamination had occurred. Sequences were trimmed in Geneious, using the trim function with default parameters. All sequences were inspected by eye for incorrect base calls and ambiguities and edited manually. If the identity of a base was not clear in a sequence, the reverse sequence was obtained to allow clarification (using the same reverse primer as for amplification). In this case, a contiguous sequence was assembled for each pair of forward and reverse sequences. Alignments were created using the MAFFT algorithm in Geneious using the Auto algorithm with default settings for COI fragments and E-INS-i algorithm for the 28S fragments (Katoh and Standley 2013). For the COI sequences, the reading frame was determined by translating the sequences specifying the appropriate gene code (invmtDNA) and by starting at different positions in the alignment and inspecting for stop codons. All sequences for this study are available on GenBank (Accession numbers MG602349–MG602475).
Phylogenetic analysis
To infer the phylogenetic relationships between the New Zealand acanthocephalan specimens and their closest relatives, and to assess the existence of different lineages within both crab and acanthocephalan taxa sampled in this study, Bayesian and maximum likelihood methods were used. Phylogenetic analyses were performed separately for each dataset and were performed remotely using the online phylogenetic tree tool CIPRES Science Gateway V. 3.3 (Miller et al. 2010). The parasite 28S sequences along with representative 28S sequences from other available acanthocephalans (see Garcia-Varela et al. 2013) were used to investigate the phylogenetic relationships of Profilicollis spp. Outgroups and all additional sequences for analyses were obtained from GenBank (http://www.ncbi.nlm.nih.gov) (Table S1, Supplementary Material). Model selection and optimal partitioning schemes were determined using PartitionFinder v2.1.1 (Lanfear et al. 2016) which implements the greedy algorithm (Lanfear et al. 2012) and utilises the software PhyML v3.0 (Guindon et al. 2010). For Bayesian analysis, model testing was restricted to those implemented in MrBayes and the Bayesian information criterion (BIC) was used for evaluating the likelihood of the proposed models and partitioning schemes. Bayesian analyses were performed in MrBayes v3.2.6 (Ronquist and Huelsenbeck 2003). For the acanthocephalan 28S phylogeny, the optimal model was identified as GTR+I+G. For COI Bayesian analysis, the best partitioning scheme consisted of each codon position being considered separately for both taxa. For the crab, this consisted of the models SYM+ I, F81+I and GTR+G, and for acanthocephalans GTR+I, F81+I and GTR, for the first, second and third codons, respectively. Two runs, each consisting of four Markov chains, were allowed to run for 10,000,000 generations and were sampled for every 1000. A heating chain parameter of 0.04 was used for all Bayesian analyses except for the host COI, where a temperature of 0.004 was used to allow better mixing of the chains (selected after preliminary analysis retrieved low acceptance rates for swaps between different chains). The first 2500 trees (25%) were discarded as burn-in and posterior probabilities were obtained from a majority-rule consensus. Mixing and convergence of each run were monitored by the standard deviation of statistics provided in MrBayes, and Tracer v1.6.0 (Drummond and Rambaut 2007) was used to assess the convergence and mixing of the two Bayesian runs and effective sample sizes (ESS).
For maximum likelihood methods, the RAxML default model of GTR was used for both markers. Bootstrap support values were obtained using the rapid bootstrap method with 1000 replicates. Pairwise uncorrected p distances were calculated within and between Profilicollis sp. obtained in this study and its closest related taxa in MEGA v.7 (Kumar et al. 2016).
Population genetics
To test for genetic structure between sampled localities, population genetic analyses were performed using the aligned COI sequence data for both crabs and acanthocephalans (without outgroups). PopART (Leigh and Bryant 2015) was used to create haplotype maps and networks for both the crabs and acanthocephalans. Networks were created using TCS network inference, a method to estimate genealogical relationships among sequences (Clement et al. 2000). Arlequin v3.5.2.2 (Excoffier and Lischer 2010) was used to calculate the number of haplotypes, number of polymorphic sites, haplotype diversity, nucleotide diversity, and perform analyses of molecular variance (AMOVAs). For the AMOVAs, genetic distance was determined using the Tamura–Nei model (Tamura and Nei 1993) which was determined by model selection in PartitionFinder. The results of the AMOVA analysis were tested for significance by 10,000 random permutations.
To further explore patterns of genetic diversity within the COI dataset for both acanthocephalans and crabs, the discriminant analysis of principal components (DAPC Jombart et al. 2010) was applied in R v3.4.1 (https://www.r-project.org). DAPC is a multivariate method designed to identify and describe clusters of genetically related individuals and provides graphical representation of between-group structures. Prior to analysis, aligned COI sequences without outgroups were imported in R using the Ape package (Paradis et al. 2004), and single nucleotide polymorphism (SNP) sites were extracted from sequences using the Adegenet package (Jombart 2008). A total of 96 SNPs from 50 specimens were obtained for acanthocephalans, and 87 SNPs from 60 specimens were obtained for the crabs. DAPC was implemented with the function dapc. For both the crab and acanthocephalan, 15 principal components of the PCA were retained in the preliminary data transformation, which contained 89% of the total genetic variation for the crab and 94% for the acanthocephalan. In the subsequent step, six discriminant functions were retained for the acanthocephalan, and seven for the crab, representing all eigenvalues. Basic scatter plots were then obtained using the function scatterplot.
To investigate if differences in prevalence among localities are associated with parasite genetic diversity (i.e. number of haplotypes, haplotype diversity and nucleotide diversity), we computed Pearson’s correlations coefficients (R function cor.test).
Co-phylogenetic analysis
To quantify the level of evolutionary congruence of each respective host–parasite association, the distance-based co-evolutionary analysis statistical tool PACo (Balbuena et al. 2013) was applied among individuals, rather than species. This approach allows one to determine whether the genetic differentiation among parasites tends to mirror that among hosts which could imply evolutionary dependence of the parasite in relation to its crab host. This analysis uses the null hypothesis that the arrangement of the parasite phylogeny does not depend on the host phylogeny. PACo analysis was also carried out in R v3.4.1, implementing the PACo script using the host and parasite Bayesian COI trees, and the respective binary matrix coding the host–parasite associations as the input. Prior to PACo analysis, host and parasite trees were pruned to include only 39 host–parasite pairs represented in the host–parasite binary matrix, using the function setdiff. This pruning procedure removes unnecessary individuals from the analysis such as outgroups, crab specimens found to harbour no parasites, as well as parasites where the respective host data are unavailable (i.e. where the corresponding host DNA failed to be extracted, amplified or sequenced successfully). Trees were then transformed into a matrix of patristic distances using the function cophenetic from the Ape package (Paradis et al. 2004) and transformed using a square root correction (function sqrt) to remove negative values (de Vienne et al. 2011). The level of co-phylogenetic congruence between host and parasite phylogenies was measured with the residual sum of squares using a Procrustean fit (Balbuena et al. 2013). The significance of this was established by 100,000 random permutations of the host–parasite association data. The contribution of each individual host–parasite association to the overall co-phylogenetic signal was measured by means of jackknife estimation of their respective squared residuals, together with a 95% confidence interval associated with each host–parasite link, where each link is removed in turn and the fit reassessed. A small residual value indicates a link that is more congruent with a concordant evolutionary histories scenario between parasites and crab hosts. A high residual value would indicate a link where the host is not driving the evolution of its parasite. To visualise these host–parasite associations, a tanglegram was manually created from the host and parasite Bayesian COI trees using the vector software Inkscape (https://www.inkscape.org).
Results
Collection data
A total of 263 Profilicollis sp. individuals were isolated from 239 H. crenulatus. Crabs infected with acanthocephalans were found at seven of the eight locations, with no infected crabs recovered from Kakanui. Among the sites at which infected crabs were found, there was a wide range of prevalence, from 28% at Invercargill to 90% at Portobello. Crab width ranged from 14 to 35 mm among all sites. Infection intensity (number of parasites per infected host) varied between 1 and 29 across all sites (Table 1). Based on morphological characteristics, all cystacanths were identified as P. novaezelandensis.
DNA sequencing and phylogenetics
In all phylogenetic analyses, Bayesian and maximum likelihood methods each yielded similar topologies for the 28S and COI datasets for both the crabs and acanthocephalans. For this reason, only Bayesian 50% majority-rule inference trees are displayed here, showing both posterior probabilities and bootstrap support values. In all Bayesian analyses, each separate run converged to an average deviation split of below 0.01, presenting good mixing and with ESS values for all parameters above 200.
Partial sequences of 28S were obtained for a total of 21 acanthocephalans (three per location). For the 28S marker, every sequence was found to be identical among all locations, indicating that only one species was sampled. For this reason, one sequence was selected as a representative and used in the phylogenetic inference. The 28S dataset used in phylogenetic analysis consisted of a final alignment length of 546 base pairs (including gaps). The 28S tree groups Profilicollis from this study with all other Profilicollis spp. for which sequences were available, being most closely related to P. bullocki (1.1% uncorrected p distance). This Profilicollis spp. clade is supported with 100% posterior probability and bootstrap support values (Fig. 1).

Bayesian 50% majority-rule inference tree for the acanthocephalan 28S nuclear gene. The tree shows the phylogenetic relationship of Profilicollis novaezelandensis with other representative acanthocephalans. Bootstrap support values (%, values below 50% are not shown) are indicated above the nodes, and the posterior probabilities (%, values below 75% are not shown) are below
Partial sequences of COI were obtained for 60 crab specimens (final alignment length of 598 bp, 7–9 per location) and 50 acanthocephalans (final alignment length of 587 bp, 7–8 per location). This marker was variable for both crabs (0.7% uncorrected p distance) and acanthocephalans (0.9% uncorrected p distance).
For this marker, all acanthocephalan sequences from this study form a well-supported monophyletic clade (98% bootstrap support and 100% posterior probability). Published P. antarcticus and P. bullocki sequences present a genetic divergence of more than 14% uncorrected p distance (15% and 14.6% respectively) from our clade. Cystacanth sequences from this study matched with 100% sequence similarity with the sequence of the adult P. novaezelandensis (see Figs. S1 and S2 in Supplementary Material).
Population genetics
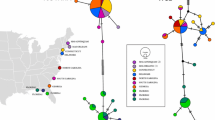
From the 60 H. crenulatus COI sequences, 43 polymorphic sites and 37 unique haplotypes were identified, while the 50 P. novaezelandensis COI sequences contained a total of 48 polymorphic sites and 27 unique haplotypes. Standard indices of genetic diversity for each species and across sampling sites are shown in Table 2. For H. crenulatus, most haplotypes differ by just one or two mutational steps (Fig. 2a). In contrast, in the acanthocephalan haplotype network, many haplotypes differ by multiple mutational steps, with some differing by up to six (Fig. 3a).

Haplotypic graphics for the crab Hemigrapsus crenulatus. a Statistical parsimony networks of COI haplotypes. Hatch marks indicate number of mutational steps and dark circles represent inferred, unsampled haplotypes. b Map of study area indicating the proportion of haplotype representation at each sampling location for H. crenulatus COI

Haplotypic graphics for the acanthocephalan Profilicollis novaezelandensis. a Statistical parsimony networks of COI haplotypes. Hatch marks indicate the number of mutational steps and dark circles represent inferred, unsampled haplotypes. b Map of study area indicating the proportion of haplotype representation at each sampling location for P. novaezelandensis COI
The distribution of haplotypes for H. crenulatus (Fig. 2b) illustrates a general lack of population structure. For example, the most abundant haplotype (haplotype 1) is found in Christchurch, Aramoana, Portobello, Taieri Mouth and Invercargill. All haplotypes are widespread and there is a high level of diversity driven by the many unique haplotypes.
In the haplotype network of P. novaezelandensis (Fig. 3b), despite a greater number of polymorphic sites, acanthocephalans have far fewer unique haplotypes compared to the diversity present in the crab populations. The haplotype map indicates some degree of population structure with haplotype 2 occurring only in the southern localities of Invercargill, Papatowai and Blueskin Bay. This contrasts with haplotype 7 which is found only at Aramoana and Christchurch, and not in any of the southern locations. However, there are also haplotypes that contribute to the lower degree of genetic structure within the COI such as haplotype 6, which is present in both Christchurch and Papatowai (Fig. 3b).
AMOVA tests for H. crenulatus and P. novaezelandensis COI datasets confirmed the lack of population structure among locations, with most of the variability being found within localities. Fixation indices were low and non-significant for both the crab and acanthocephalan (Table 3).
Discriminant analysis of principal component results further support this lack of distinct clustering. For H. crenulatus there is a very clear overlap of genetic variability among all localities, representing only one main genetic cluster (Fig. 4a). However, for P. novaezelandensis, the location Aramoana, which contains nearly all unique haplotypes, is separated from the others, though it still shows some degree of overlap with the main cluster (Fig. 4b).

Scatter plots of the DAPC analysis for Hemigrapsus crenulatus (a) and Profilicollis novaezelandensis (b) where each point represents an individual, and ellipses represent 95% confidence limits for locality-level groups. Ellipses and points are coloured according to the location, which were used as prior clusters; Christchurch (CH, maroon), Kakanui (KA, red), Blueskin Bay (BB, orange), Aramoana (AR, yellow), Portobello (PO, light green), Taieri Mouth (TM, dark green), Papatowai (PA, dark blue) and Invercargill (IN, light blue)
Overall, associations between the indices of parasite diversity and prevalence were positive, with a moderate to strong degree of association (nucleotide diversity: r = 0.38; haplotype diversity: r = 0.58; number of haplotypes: r = 0.63), but not statistically significant (all cases p > 0.1).
Co-phylogenetic analysis
The contribution of each host–parasite link to the global fit between host and parasite phylogenies is shown in Fig. 5. Individual host–parasite associations in the tanglegram appear very intertwined. Despite this, the PACo analysis gave a residual sum of squares (m 2 XY ) of 0.136 with a permutational value of p = 0.00111 for the overall fit of P. novaezelandensis with their host H. crenulatus. This significant p value indicates that the observed association is more congruent with both host and parasite evolutionary history than its random assembly.

Tanglegram of the co-phylogenetic relationships between the individual acanthocephalan parasites, Profilicollis novaezelandensis (a) and the respective intermediate host, Hemigrapsus crenulatus, from which they were obtained (b). Both the parasite and host trees are adapted from the COI gene data inference tree. c Jackknife residual bars resulting from PACo analysis for each respective host–parasite association represented in the tanglegram. The dotted line represents the mean residual value and the error bars are the upper 95% confidence intervals. Host–parasite links with bars below the mean residual value are considered indicative of congruent links. Jackknife bars are coloured according to the location; Christchurch (maroon), Kakanui (red), Blueskin Bay (orange), Aramoana (yellow), Portobello (light green), Taieri Mouth (dark green), Papatowai (dark blue) and Invercargill (light blue)
In the plot showing the associations between individual host–parasite pairs (Fig. 5c), bars below the mean residual value (dotted line) show associations that have low residual sum of squares and are therefore likely to represent co-evolutionary links. This allows us to identify which associations show the most support for the overall phylogenetic congruence. It is clear that the first 11 bars (in Fig. 5c) represent congruent links and are likely the main factors contributing to the overall significant p value. It is interesting to note that in the host–parasite relationships, nearly all specimens obtained from Invercargill have large squared residual values showing lack of support for a co-phylogenetic hypothesis driving the diversification of parasites in that locality.
Nevertheless, it is worth mentioning that some of the confidence intervals associated with the bar plot are large and overlap with the mean residual value, notably in CH35C–CH35A, TM25C–TM25A, and PA7C–PA7A and so interpretations of their degree of congruence needs to be made cautiously.
Discussion
Comparing the genetic structure of host populations with that of their parasites is a powerful way to assess the potential for the spread of advantageous alleles and/or local adaptation (Dybdahl and Lively 1996; Morjan and Rieseberg 2004; Prugnolle et al. 2005), two important forces on the host–parasite co-evolutionary arms race. Here, we found no significant genetic structure among isolated populations of the crab H. crenulatus and populations of their acanthocephalan parasite P. novaezelandensis, along a roughly 600 km stretch of coastline. However, in contrast to our original hypothesis, we found significant congruence between the population-level phylogenies of hosts and parasites, despite their distinct and independent modes of dispersal.
The observed lack of genetic structure in both host and parasite fits with our original expectation for the parasite, but not for the host, where we expected to observe some weak genetic structuring. The absence of significant structure may have been influenced by small sample sizes, which in turn may have reduced the power of the analyses. To some extent, the observed lack of structure may also be due to the limited spatial scale of our study; however, the biological characteristics of both species argue against a mere scale effect. The crab H. crenulatus has a planktonic larval stage that can last several weeks (Wear 1970). Although this does not necessarily guarantee wide-ranging dispersal and connectivity among populations (Gilg and Hilbish 2003), most New Zealand marine invertebrates with planktonic larvae distributed by sea currents show weak genetic structuring on the scale of our study (Ross et al. 2009; Wallis and Trewick 2009). The main current in our study area is the Southland Current, flowing northward along the east coast of the South Island (Sutton 2003). In addition to potentially generating a north–south cline, the current encounters potential barriers in the form of the Otago and Banks peninsulas, which affect the structuring of some coastal marine species (e.g. Tobias et al. 2017). We detected neither a cline nor a subdivided structure resulting from dispersal barriers in our study, indicating that the crab populations along the east coast of the South Island form a well-mixed metapopulation. The lack of a north–south cline may indicate that lesser, more regional currents also play important dispersal roles, masking any larger role for the Southland Current.
Based on 28S sequences, the acanthocephalan cystacanths analysed here belong to a single species, P. novaezelandensis. A second species, P. antarcticus, occurs at very low prevalence in some of the localities we sampled (Latham and Poulin 2002a), but was not found among the sequenced specimens of our study. Nevertheless, as a limited number of acanthocephalan individuals were selected per location, we cannot completely rule out the presence of another species. Unlike the strong genetic structure and likelihood of cryptic species seen in freshwater acanthocephalans inhabiting isolated lakes and rivers, and using water-bound fish as definitive hosts (e.g. Steinauer et al. 2007; Martinez-Aquino et al. 2009), we found no significant genetic structure among the P. novaezelandensis sampled here.
This result is not surprising given that across a much larger area in North and South America, populations of P. altmani showed little population structure (Goulding and Cohen 2014). In both cases, shore bird definitive hosts appear to be efficient agents of dispersal for this parasite (Goulding and Cohen 2014; Rodriguez and D’Elia 2017). Indeed, the black-backed gull Larus dominicanus is a long-lived, widespread bird capable of long-distance dispersal, even between the North and South Islands (Fordham 1968; Rowe 2013). Their role in spreading P. novaezelandensis from one locality to another is evident from the strong correlation across localities between acanthocephalan infection levels in crabs and local average abundance of gulls; the more the gulls frequent a locality, the more do local crabs accumulate parasites (Latham and Poulin 2003). Our analysis does reveal some interesting trends, with some parasite haplotypes only occurring in adjacent localities, or some localities comprising mostly unique haplotypes. In fact, parasites showed a higher degree of genetic structure than their respective host. While it seems that dispersal of these acanthocephalan by birds is responsible for the lack of structure, one would assume that this would lead to lower genetic structure in the parasite. The relatively higher genetic structure suggests that local patterns of host movements and/or the composition of the local bird fauna are also important factors. However, there is no significant structure across all localities, and the acanthocephalan, like its crab host, exists as a well-mixed metapopulation.
An intriguing finding comes from the co-phylogenetic analysis, in which we applied the PACo approach within species and tested for congruence of evolutionary histories between the phylogeography of the crabs and that of their parasites. We expected no congruence because of the very different modes of dispersal used by the crabs (planktonic drift) and their acanthocephalan parasites (bird-mediated dispersal). In contrast, the analysis revealed an overall significant level of congruence. The most parsimonious and realistic interpretation for this result is that its statistical significance does not translate into biological significance. Firstly, the confidence intervals associated with the residual sums of squares are sometimes large (see Fig. 5c), casting some doubt over their accuracy. Secondly, given the modest number of localities sampled and small sample sizes, the result may simply be due to chance. The data indicate that host–parasite links from the southern-most location, i.e. Invercargill, contribute the least to the overall congruent pattern, whereas links indicating phylogenetic congruence consist of host–parasite pairs from multiple localities. One possible scenario is that in some places, bird movements that disperse parasites coincidentally happen to match patterns of crab dispersal by ocean currents, generating apparent congruence in genetic affinities. Several processes and biological traits of hosts or parasites can outweigh the role of the most mobile host in a parasite’s life cycle as dispersal route (Keeney et al. 2009; Blasco-Costa and Poulin 2013; Mazé-Guilmo et al. 2016); however, in this case, this seems unlikely, and the significant crab-acanthocephalan congruence in spatial genetics should be viewed with scepticism.
In summary, our findings indicate that populations of the crab H. crenulatus and populations of their acanthocephalan parasite P. novaezelandensis are genetically well mixed across the east coast of the South Island, but apparently independently of each other. If other genetic markers also support this pattern, there may be consequences for their co-evolution. In genetically structured host–parasite systems where the parasite induces a fitness cost to the host, adaptation to the local strains of the parasite is favoured by selection (Dybdahl and Lively 1996; Prugnolle et al. 2005). In our study system, the acanthocephalan interferes with the anti-predator behaviour of crabs and increases their mortality (Latham and Poulin 2002a, b). This impact on host fitness should exert strong selective pressures on the crab. During this and previous studies on this system (Latham and Poulin 2002a, 2003), we have never found acanthocephalans killed via encapsulation and melanisation within crabs, these being key immune defences of crustaceans against parasites known to vary through local adaptation (Bryan-Walker et al. 2007; Cornet et al. 2009). In our system, genetic mixing may give the parasite a small edge in the arms race. Analysis of several additional gene markers would be necessary to detect the genomic signature of local adaptation and determine whether it can explain the apparent lack of immune defence across all crab populations, regardless of local parasite prevalence.
References
Balbuena JA, Miguez-Lozano R, Blasco-Costa I (2013) PACo: a novel procrustes application to cophylogenetic analysis. PLoS One 8:e61048
Blasco-Costa I, Poulin R (2013) Host traits explain the genetic structure of parasites: a meta-analysis. Parasitology 140:1316–1322
Brockerhoff AM, Smales LR (2002) Profilicollis novaezelandensis n. sp (Polymorphidae) and two other acanthocephalan parasites from shore birds (Haematopodidae and Scolopacidae) in New Zealand, with records of two species in intertidal crabs (Decapoda: Grapsidae and Ocypodidae). Syst Parasitol 52:55–65
Bryan-Walker K, Leung TLF, Poulin R (2007) Local adaptation of immunity against a trematode parasite in marine amphipod populations. Mar Biol 152:687–695
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
Cornet S, Biard C, Moret Y (2009) Variation in immune defence among populations of Gammarus pulex (Crustacea: Amphipoda). Oecologia 159:257–269
Criscione CD, Blouin MS (2004) Life cycles shape parasite evolution: comparative population genetics of salmon trematodes. Evolution 58:198–202
de Vienne DM, Aguileta G, Ollier S (2011) Euclidean nature of phylogenetic distance matrices. Syst Biol 60:826–832
Devlin CM, Diamond AW, Saunders GW (2004) Sexing arctic terns in the field and laboratory. Waterbirds 27:314–320
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214
Dybdahl M, Lively CM (1996) The geography of coevolution: comparative population structures for a snail and its trematode parasite. Evolution 50:2264–2275
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Fordham R (1968) Dispersion and dispersal of the Dominican gull in Wellington, New Zealand. Proc N Z Ecol Soc 15:40–50
Garcia-Varela M, Pérez-Ponce de Léon G, Aznar FJ, Nadler SA (2013) Phylogenetic relationship among genera of Polymorphidae (Acanthocephala), inferred from nuclear and mitochondrial gene sequences. Mol Phylogenet Evol 68:176–184
Geller J, Meyer C, Parker M, Hawk H (2013) Redesign of PCR primers for mitochondrial cytochrome c oxidase subunit I for marine invertebrates and application in all-taxa biotic surveys. Mol Ecol Resour 13:851–861
Gilg MR, Hilbish TJ (2003) The geography of marine larval dispersal: coupling genetics with fine-scale physical oceanography. Ecology 84:2989–2998
Goulding TC, Cohen CS (2014) Phylogeography of a marine acanthocephalan: lack of cryptic diversity in a cosmopolitan parasite of mole crabs. J Biogeogr 41:965–976
Groner ML, Maynard J, Breyta R et al (2016) Managing marine disease emergencies in an era of rapid change. Phil Trans R Soc B 371:20150364
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321
Harper JT, Saunders GW (2001) The application of sequences of the ribosomal cistron to the systematics and classification of the florideophyte red algae (Florideophyceae, Rhodophyta). Cah Biol Mar 42:25–38
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780
Kearse M et al (2012) Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinform 28:1647–1649
Keeney DB, King TM, Rowe DL, Poulin R (2009) Contrasting mtDNA diversity and population structure in a direct-developing marine gastropod and its trematode parasites. Mol Ecol 18:4591–4603
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Lagrue C, Joannes A, Poulin R, Blasco-Costa I (2016) Genetic structure and host–parasite co-divergence: evidence for trait-specific local adaptation. Biol J Linn Soc 118:344–358
Lanfear R, Calcott B, Ho SYW, Guindon S (2012) PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol 29:1695–1701
Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B (2016) PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol 34:772–773
Latham ADM, Poulin R (2002a) Field evidence of the impact of two acanthocephalan parasites on the mortality of three species of New Zealand shore crabs (Brachyura). Mar Biol 141:1131–1139
Latham ADM, Poulin R (2002b) Effect of acanthocephalan parasites on hiding behaviour in two species of shore crabs. J Helminthol 76:323–326
Latham ADM, Poulin R (2003) Spatiotemporal heterogeneity in recruitment of larval parasites to shore crab intermediate hosts: the influence of shorebird definitive hosts. Can J Zool 81:1282–1291
Leigh JW, Bryant D (2015) POPART: full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116
Martinez-Aquino A, Reyna-Fabian ME, Rosas-Valdez R, Razo-Mendivil U, Pérez-Ponce de León G, Garcia-Varela M (2009) Detecting a complex of cryptic species within Neoechinorhynchus golvani (Acanthocephala: Neoechinorhynchidae) inferred from ITSs and LSU rDNA gene sequences. J Parasitol 95:1040–1047
Mazé-Guilmo E, Blanchet S, McCoy KD, Loot G (2016) Host dispersal as the driver of parasite genetic structure: a paradigm lost? Ecol Lett 19:336–347
McLay CL (1988) Brachyura and crab-like Anomura of New Zealand. University of Auckland Marine Laboratory, Auckland
Miller MA, Pfeiffer W, Schwartz T (2010) Creating the CIPRES science gateway for inference of large phylogenetic trees. In: Proceedings of the gateway computing environments workshop, New Orleans, LA, pp 1–8
Morjan CL, Rieseberg LH (2004) How species evolve collectively: implications of gene flow and selection for the spread of advantageous alleles. Mol Ecol 13:1341–1356
Paradis E, Claude J, Strimmer K (2004) APE: analyses of phylogenetics and evolution in R language. Bioinform 20:289–290
Prugnolle F, Théron A, Pointier JP, Jabbour-Zahab R, Jarne P, Durand P, de Meeûs T (2005) Dispersal in a parasitic worm and its two hosts: consequences for local adaptation. Evolution 59:296–303
Rodriguez SM, D’Elia G (2017) Pan-American marine coastal distribution of the acanthocephalan Profilicollis altmani based on morphometric and phylogenetic analyses of cystacanths from the mole crab Emerita brasiliensis. J Helminthol 91:371–375
Ronquist F, Huelsenbeck JP (2003) MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinform 19:1572–1574
Ross PM, Hogg ID, Pilditch CA, Lundquist CJ (2009) Phylogeography of New Zealand’s coastal benthos. N Z J Mar Freshw Res 43:1009–1027
Rowe L (2013) Dispersal of southern black-backed gulls (Larus dominicanus dominicanus) banded in Canterbury, New Zealand, 1959–1993. Notornis 60:134–142
Steinauer ML, Nickol BB, Orti G (2007) Cryptic speciation and patterns of phenotypic variation of a highly variable acanthocephalan parasite. Mol Ecol 16:4097–4109
Sutton PJH (2003) The Southland current: a subantarctic current. N Z J Mar Freshw Res 37:645–652
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial-DNA in humans and chimpanzees. Mol Biol Evol 10:512–526
Tobias ZJC, Jorge F, Poulin R (2017) Life at the beach: comparative phylogeography of a sandhopper and its nematode parasite reveals extreme lack of parasite mtDNA variation. Biol J Linn Soc 122:113–132
Wallis GP, Trewick SA (2009) New Zealand phylogeography: evolution on a small continent. Mol Ecol 18:3548–3580
Ward JR, Lafferty KD (2004) The elusive baseline of marine disease: are diseases in ocean ecosystems increasing? PLoS Biol 2:e120
Wear RG (1970) Life-history studies on New Zealand Brachyura. N Z J Mar Freshw Res 4:3–35
Zhang Z, Schwartz S, Wagner L, Miller W (2000) A greedy algorithm for aligning DNA sequences. J Comput Biol 7:203–214
Acknowledgements
We thank Lynda Hay, Lance Hay, Jahmaine Hay and Kirby McKenzie for assistance with crab collection in the field, and Dr. Bronwen Presswell for providing an adult acanthocephalan specimen.
Funding
This research was funded internally by the Department of Zoology, University of Otago, and received no external funding from commercial or non-profit sectors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare having no conflict of interest.
Ethical statement
Collection and euthanasia of crabs were approved by the Otago University Animal Ethics Committee (Application no. ET 2/17).
Additional information
Responsible Editor: T. Reusch.
Reviewed by Undisclosed experts.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hay, E., Jorge, F. & Poulin, R. The comparative phylogeography of shore crabs and their acanthocephalan parasites. Mar Biol 165, 69 (2018). https://doi.org/10.1007/s00227-018-3326-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00227-018-3326-y