
Abstract
A detailed characterisation of the organic composition of Arctica islandica (Linnaeus, 1767) shells with homogenous microstructure is compared with nacroprismatic shells of Pinctada fucata martensii (Dunker, 1872), Hyriopsis cumingii (Lea, 1852) and Diplodon chilensis patagonicus (d’Orbigny, 1835). Thermogravimetric analysis shows lowest total organic contents of 1.65 wt% for A. islandica shells, while all nacroprismatic shells are higher (3.14–4.13 wt%). Macromolecules extracted from the nacroprismatic shells are dominated by hydrophobic amino acids (~ 54%) in the acid extracts, while EDTA-extracts are moderately rich in aspartate and glutamate (16% in total) and in glycine–alanine (42%). In comparison, A. islandica shells have higher concentrations of proline, glycine and aspartate (ca 40%). Infrared spectroscopy shows some acidic protein bands in A. islandica, which cannot be found in the nacroprismatic shells. Alcian Blue and/or modified silver staining methods reveal many prominent bands. Protein bands at around 10, 14, 17, 21, 26, 31, 40 and 55 kDa are detected in A. islandica shells for the first time, thus may constitute a new set of proteins in mollusc shells. SDS-PAGE exhibits apparent molecular weights from 5 to 63 kDa in nacroprismatic shells. Distinct protein bands at around 17 and 26 kDa in A. islandica shells may correspond to a post-translational modification of proteins; these prominent bands, however, are absent in the nacroprismatic samples. Contrarily, SDS-PAGE of both, homogeneous and nacroprismatic shell microstructures show nonacidic-matrix-proteins.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Mollusc shell biomineralisation
Mollusk shells are natural biocomposites of calcium carbonate phases, such as calcite or aragonite, intimately intergrown with organic macromolecules (Weiner and Dove 2003). The organic moiety in bivalve shells contains a mixture of lipids, glycoproteins, proteins and polysaccharides-chitin (Farre and Dauphin 2009; Levi-Kalisman et al. 2001; Pereira-Mouriès et al. 2002), with variable ratios of proteins and polysaccharides depending on the species. The organic macromolecules are actively involved in shell formation and have been shown to control virtually all levels of mineralisation, including which mineral polymorph is formed (e.g. calcite, aragonite, vaterite), sizes and shapes of the grains, and the overall microstructure of the skeletal hard parts (Addadi and Weiner 2014).
Due to this crucial role in shell formation, the organic matrix in bivalve shells has received increased attention, resulting a number of proteins potentially involved in the control of bio-carbonate formation having been described to date (Arivalagan et al. 2016; Marin and Luquet 2005). These proteins contain carboxylates, sulphates, or phosphates as functional groups, which may bind to Ca2+ ions, thus lowering the interfacial energy between the solution and the macromolecular substrate and, in this way, being able to guide crystal nucleation and growth (Giuffre et al. 2013; Hamm et al. 2014).
Much of our knowledge about the composition and function of the organic matrix in mollusc shells is restricted to bivalves with nacroprismatic shells, whereas very little is known about the organic components in calcareous biominerals with different shell microstructures, such as crossed-lamellar or homogeneous structures (Agbaje et al. 2017; Carter 1990; Dauphin and Denis 2000). Arctica islandica (Linnaeus, 1767) (Bivalvia: Veneroida), an extremely long-lived bivalve (> 400 years) (Philipp et al. 2012) has a distinct and complex shell structure, referred to as ‘homogeneous’ (Ropes et al. 1984). The biogenic material has gained considerable importance for climate reconstruction in the northern hemisphere (Weidman and Jones 1994). Despite its importance, very little is known to date about the organic moieties in the calcareous biominerals of this species. In this study, we present the first detailed characterisation of the shell organic matrix of A. islandica and compare it to different bivalve shells with nacre-prism microstructure from marine and freshwater environments.
Shell microstructures
The homogeneous microstructure is widely distributed in the mollusc phylum. However, mollusc shell families consisting entirely of homogeneous layers are relatively rare in comparison to families with other shell microstructures such as the nacroprismatic structure (Taylor 1973). Arctica islandica shells are completely aragonitic and have a two-layered structure that consists of granular and irregularly arranged micrometre-sized aragonite prisms but lacks any readily visible architecture (Ropes et al. 1984). According to Taylor (1973), some of the mollusc shell families with almost entirely homogeneous layers descended from a nacroprismatic evolutionary ancestor.
The nacroprismatic structure is similarly widespread in the mollusc class (Carter 1990) and consists of an outer layer of prism-shaped aragonite or calcite grains in individual organic envelopes and an inner layer of aragonite tablets arranged in a brickwall-like way and individually separated by peripheral thin layers of organic matrix (Bevelander and Nakahara 1969). In marine bivalve shells as Pinctada fucata martensii, the outer prismatic layer is typically calcitic, whereas freshwater shells with this microstructure (Hyriopsis cumingii, Diplodon chilensis patagonicus) are entirely aragonitic.
A major question in understanding the parameters governing the formation of different types of shell microstructure is whether shells with different microstructures contain a similar population of matrix proteins, or not. A first step is to characterise and compare the bulk composition of soluble organic molecules in shells with different microstructures, which is what we are attempting here.
Organic macromolecules in bivalve shells
Generally, organic macromolecules in bivalve shells amount to less than 5 wt% and can be extracted by decalcification using water- (Bédouet et al. 2007; Ma et al. 2013; Pereira-Mouriès et al. 2002), EDTA- (Kono et al. 2000; Miyashita et al. 2000; Pereira-Mouriès et al. 2002) or acid-based methods (Dauphin 2003; Ma et al. 2013; Marie et al. 2007). A large part of these macromolecules are composed of hydrophobic components such as chitin or silk-fibroin. These insoluble molecules are often intimately associated with acidic proteins, forming an extensive three-dimensional architecture (Levi-Kalisman et al. 2001). In contrast, the soluble moiety is relatively low in content (< 2 wt%) and is composed of acidic proteins, glycoproteins, polysaccharides and polypeptides. Both, insoluble and soluble organic moieties are involved in carbonate crystal formation, and particularly the silk-like protein component of the shell macromolecules is thought to have an important function in the biomineralisation processes (Addadi and Weiner 2014).
Depending on the extraction method, the organic shell component shows somewhat differing composition: water-based extracts after decalcification tend to have higher percentages of silk-like gel (glycine and alanine), whereas EDTA extracts generally show higher of acidic amino acid concentrations (Pereira-Mouriès et al. 2002). Pereira-Mouriès et al. (2002) suggested that the water-based methods mobilise apolar proteins that are covalently attached to sugars, whereas these components are insoluble when using EDTA (Addadi et al. 2006; Pereira-Mouriès et al. 2002). Furthermore, decalcification using water-based methods is less aggressive compared to extraction methods using acid or EDTA, which can be partially destructive for the organic molecules (Bédouet et al. 2007). In this study, we present complementary results using both methods and aim at improving recovery.
Materials and methods
Materials
Arctica islandica (Arcticidae; Linnaeus, 1767), H. cumingii (Unionidae; Lea, 1852), D. chilensis patagonicus (Hyriidae; d’Orbigny, 1835) and P. fucata martensii (Pteriidae; Dunker, 1872) shells were used in this study. Samples are grouped by structure into nacre–prism structure (H. cumingii, D. chilensis patagonicus and P. fucata martensii) and homogeneous structure (A. islandica). All samples are entirely aragonitic except for P. fucata, which has a calcitic prism layer. H. cumingii and D. chilensis patagonicus are freshwater species, while P. fucata and A. islandica are marine bivalves.
Sample preparation and characterisation
All shells are covered by an organic periostracum (Carter and Aller 1975), which was mechanically removed with a DREMEL drill (Wisconsin, USA). Only ca. 2 mm of the outer shell containing the periostracum was removed. Mechanical removal of the periostracum also partially removed the prismatic layer, while complete removal of the prismatic layer is unlikely. The outer prismatic layer in the shells of all nacroprismatic bivalves studied here is of variable thickness, but is generally between 3 and 4 mm, while the nacreous layer is much thicker and generally makes up over 90% of the mineralised shell. Though small differences in compositions of the prismatic and nacreous layers in bivalves have been reported in the literature (Samata 1990), all our analyses are dominated by the composition of the nacreous shell layer. For homogeneous A. islandica shells and unlike for nacroprismatic shell, no significantly texturally different shell layers are known (Shirai et al. 2014).
After mechanical cleaning, the fragments were immersed in 30% H2O2 (Merck KGaA, 64271 Darmstadt; Germany) for 1 h at room temperature combined with ultrasonic treatment to remove any organic contaminants, washed extensively with Milli-Q water and rinsed with acetone to facilitate drying. After drying in air, the samples were powdered in a cleaned rock mill before Thermogravimetric Analysis (TGA) was conducted using a TGA 2050 Thermogravimetric Analyzer (TA Instruments, USA). About 30 mg of powdered sample was measured in a temperature interval from room temperature (21 °C) up to 1000 °C at 10 °C min−1 steps under nitrogen atmosphere.
Extraction of organic macromolecules
Powdered samples were decalcified for about 2 days either with a 10% ethylenediaminetetracetic acid disodium salt dihydrate (EDTA; Sigma-Aldrich) adjusted with KOH to pH ~ 8.0 or in 1 mM of HCl (pH > 5). After the addition of HCl solution to the shell material, pH of the shell solution was raised with Milli-Q water to the ~ 7. The extracts were centrifuged at 10,500 rpm (4 °C) for 20 min to separate the soluble and insoluble fractions. This procedure resulted in four different samples for A. islandica and H. cumingii shells, namely soluble fractions in EDTA (‘EDTA-Soluble Moiety’: ESM) and in dilute HCl (‘Acid-Soluble Organic Moiety’: ASM) as well as insoluble fractions (‘EDTA-Insoluble Moiety’ EIM and ‘Acid-Insoluble Organic Moiety’ AIM, Table 1). Diplodon chilensis patagonicus and P. fucata martensii samples were decalcified only in 1 mM HCl (pH > 5), not in EDTA, resulting in ASM and AIM fractions from these shells (Table 2).
In order to remove EDTA and HCl prior to lyophilisation, all insoluble fractions were washed thoroughly with Milli-Q water. Since EDTA can be removed by leaching from the matrices when soaked or flooded with water (Zaitoun and Lin 1997), the soluble fractions (EDTA and HCl based) were mixed with equal volumes of water and stored at 4 °C for about 1 month prior to concentration and lyophilisation. Fractionation followed the method described by Rahman et al. (2006) using Sep-Pak C18-2g (Waters Corporation Milford, MA, USA).
Additional organic matrix extracts were obtained without prior decalcification to study potential variations in ratios of charged to hydrophobic organic molecules and variations for Ca2+ binding polypeptides. For this purpose, coarsely fragmented samples (~ 500 mg each) were partly decalcified in 20% HCl for ca. 10 min at room temperature. The extracts were centrifuged and the soluble extracts were dried using SpeedVac dryer.
Fourier transform infrared spectroscopy and amino acid analysis
Fourier transform infrared (FTIR) spectra were measured from lyophilised samples of the soluble fractions (ESM, ASM) with a Thermo Nicolet iS10 ATR-FTIR spectrometer (Nicolet, MA, USA) equipped with a smart performer assessor in air. Spectra were acquired between 4000 and 500 cm−1 with 64 accumulations and a resolution of 4 cm−1. Backgrounds were recorded at the start of the analytical session and approximately after every half-hour.
For the analysis of amino acids, samples were acid hydrolysed with 6 M HCl for 24 h at 110 °C under nitrogen. Subsequently, hydrolysed samples were dissolved in Milli-Q water and an aliquot was derivatised with AQC reagent (Cohen and Michaud 1993) using AccQTag Ultra derivatisation kit (Waters Corporation, Milford, MA, USA). The separation and quantitation of the 16 acid hydrolysate amino acids was performed using a ACQUITY ultraperformance liquid chromatography (UPLC) system and BEH RP C18 1.7 µm column (Waters Corporation, Milford, MA, USA). A detailed protocol is described in Truong et al. (2015) The amino acid compositions expressed as mole per cent represent the average of duplicate results (Tables 1, 2). Due to the deamidation of glutamine to glutamic acid and asparagine to aspartic acid under acidic conditions the amounts reported for Glx and Asx are the sum of those respective components. Cysteine and tryptophan are partially destroyed by the acidic conditions and were not quantified.
Polyacrylamide gel electrophoresis of proteins
The separation of matrix components was performed under denaturing conditions by mono-dimensional SDS-PAGE using Laemmli sample buffer. Soluble organic matrix fractions were used for electrophoresis analysis at 200 V. Proteins were separated on a pre-cast NuPAGE 4–12% Bis–Tris gel in MES running buffer according to protocols supplied by the manufacturer (Invitrogen; Carlsbad, CA, USA). After electrophoresis, the gels were stained with Coomassie Brilliant Blue G-250 and/or silver nitrate, Alcian Blue and/or modified silver nitrate. The Coomassie Brilliant Blue G-250 staining was followed by fixation in ammonium sulphate solution overnight. After recording the bands on the gel, it was destained in 10% glacial acetic acid overnight and stained with silver nitrate. For this step, a modified method was developed based on (Morrissey 1981). The modification entailed omission of the prefixed step, soaking of the gel in Milli-Q water for 3 h and use of 10 μg dithiothreitol for 20 min. The molecular weights were estimated using the pre-stained (Invitrogen) Novex Sharp standard MW-SDS marker kit for electrophoresis.
Gels were also studied for potential glycosylation in the ASM and ESM fractions. Saccharides were studied using Alcian Blue 8GX (Wall and Gyi 1988) at pH 1 for the very specific acidic sulfated sugars and subsequently stained with modified silver nitrate. The gel was washed with 50% ethanol twice to ensure clear backgrounds. The modification to the staining comprised fixation of the gel in 10 vol% methanol and glacial acetic acid, respectively, and sensitisation in 10 vol% glutaraldehyde for 30 min, prior to staining. After staining, the gel was soaked in 10 μg/ml dithiothreitol for 20 min and the impregnation step was performed by treatment with a solution containing 6 mM silver nitrate and formaldehyde for 30 min. Subsequently, the gel was developed in a solution containing 3 wt% sodium carbonate and formaldehyde. Staining was terminated by adding citric acid (44 wt% in milli-Q water) and rinsing in Milli-Q water before scanning.
Results
Total organic content
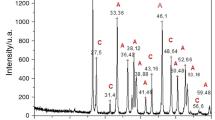
Thermogravimetric analysis coupled with differential thermal analysis (TGA/DTA) was used to investigate thermal stability and total organic contents of the shells. Peaks in the range 190–464 °C (Fig. 1a) are due to combustion of the complex mixture of proteins, glycoproteins and polysaccharides occluded in the shells and to transformation of aragonite to calcite. At 747–765 °C breakdown of calcium carbonate to calcium oxide and CO2 is recorded. The weight loss of 41–42% (Fig. 1b) at this temperature corresponds to the theoretical value of CO2 released (44%) from the calcium carbonate. As determined from these analyses, A. islandica shells have the lowest organic content of 1.65 wt% (Insert; Fig. 1b), while H. cumingii contains 3.14 wt%, similar to the D. chilensis shell 3.35 wt%. Pinctada fucata yielded the highest content in this sample suite of 4.13 wt%. Thus, the nacroprismatic samples generally contain higher organic contents than A. islandica with homogeneous shell structure.

Differential thermal analysis (a) and thermogravimetric analysis (b) show thermal stability and total organic matrix of homogeneous (AI: Arctica islandica) and nacroprismatic (HC: Hyriopsis cumingii, DC: Diplodon chilensis patagonicus and PF: Pinctada fucata martensii) shell structures. The bar chart insert in b represents the calculated total organic matrix for the range 150–500 °C
Bulk composition of the organic matrix
Acid-soluble organic matrix (ASM)
FTIR spectra of the lyophilised ASM extracts from all shell samples are presented in Fig. 2a. The organic moiety shows bands mainly in wavenumber areas characteristic for amide A (wavenumber area 3500–3000 cm−1), amide B (2990–2800 cm−1), amide I (1700–1600 cm−1), amide II (1590–1300 cm−1) and amide III (1290–1190 cm−1).

a FTIR spectra of ASM of A. islandica, P. fucata, D. chilensis and H. cumingii. The grey bar indicates the wavenumber region between 1800 and 1400 cm−1, which contains bands for amide I, II and the secondary structure of proteins. The shaded area is depicted in more detail in b. b Detailed FTIR spectra for the shaded area from a showing the different bands in the range 1800–1400 cm−1
Figure 2b details the wavenumber region between 1400 and 1800 cm−1, which contains bands for amide I, II and for the secondary structure of proteins. In this region, differences for the ASM between A. islandica and other samples are visible. Peaks at 1716 cm−1 (identified as aspartic acid, (Dauphin 2003), 1578 cm−1 for aspartate, 1559 cm−1 for glutamic acid, 1508 cm−1 for tyrosine, 1736 cm−1 for carbonyl of ester and the symmetric carboxylate absorption peak at 1419.4 cm−1 are present in the shells of A. islandica (Fig. 2b), while they are generally absent for the organic matrix in the nacroprismatic shells. Diplodon chilensis presents an exception with shoulder bands at 1714.5 cm−1 for glutamic acid (Fig. 2b) and 1418 cm−1 for symmetric carboxylate. Here again, the amide II peak near 1542 cm−1 is more distinct for A. islandica compared with the other samples. However, amide I peak at 1653 cm−1 for A. islandica is also prominent at 1652.5 cm−1 (α-helix conformation) for D. chilensis and 1646 cm−1 (random coil structure) for H. cumingii and P. fucata (Fig. 2a). The band at 1638 cm−1, present in all samples (however, as a shoulder in D. chilensis), could be assigned to the secondary protein (β-sheet) structure, which may originate from N-acetyl groups located on the polysaccharide. Similarly, lysine and C–H of aliphatic group peaks are present near 1522 and 1456 cm−1 in all samples. Amide A and B regions are also present in all samples, but are not characteristic for secondary structures (Fig. 2a).
It is possible that the strong amide A, I and II peaks from proteins are also due to the presence of sugars as shown by the spectra of chondroitin sulphate A (Dauphin 2003). Contrarily, the absorption bands at 1000–1150 cm−1, which are usually considered as characteristic for sugars, are absent from protein spectra. Strong peaks are present at 1050–1063 cm−1 (Fig. 2a) and are assigned to the sugar region. Peaks in this region are more prominent in the A. islandica spectrum compared with other samples and imply higher sugar contents in this shell compared with the nacre–prism samples. This is supported by the amino acid composition, which shows high concentrations of proline (15%; Supplementary Fig. 1S). In contrast, the peak at 1050–1063 cm−1 in ASM extracts may point to the presence of glucosaminoglycans (Pereira-Mouriès et al. 2002).
Functional groups in the soluble organics and comparison of EDTA and acid extracts
Detailed infrared spectra were collected in the range 2000–500 cm−1 to identify the functional groups in both ESM and ASM extracts (Fig. 3a). In ESM, prominent asymmetric (1583 cm−1) and symmetric (1409–1408 cm−1) carboxylate bands, amide III band at 1330 cm−1, sulphate peaks, probably O-sulphate groups, near 1282.5, 1252, 927 and 854 cm−1 could be assigned (Fig. 3a). Similarly, a sulfated peak appeared at 1252 cm−1 for H. cumingii (ASM) and a broad shoulder in this area in A. islandica (ASM, see Fig. 3a) could be identified. Amide I carbonyl group with bands at 1646 cm−1 (H. cumingii) and 1653 cm−1 (A. islandica) in ASM can also be assigned as a weak band in ESM (Fig. 3a). Specific interpretations of amide I bands are difficult due to an overlap of the α-helical with random coil structures, which both have bands in this region (Figs. 2b, 3b). Depending on their source and particularly on the method of preparation (Brine and Austin 1981; Pereira-Mouriès et al. 2002), the occurrence of the absorption band in the high-frequency wing of the amide II band can be partly merged with carbonyl stretching group (Roeges 1994). The peak used for characterisation of amide II appears at 1542–1542.8 cm−1 in ASM and is also visible as a shoulder in ESM for both samples (see Fig. 3b).

a FTIR spectra of ASM and ESM of AI (Arctica islandica) and HC (Hyriopsis cumingii) showing bands in the range 2000–500 cm−1. The grey bar indicates the wavenumber region between 1800 and 1400 cm−1, which contains bands for amide I, II and the secondary structure of proteins. The shaded area is depicted in more detail in b. b Detailed FTIR spectra for the shaded area from a showing the range 1800–1400 cm−1 for ESM of H. cumingii and A. islandica
The region between 800 and 1200 cm−1 was characterised by well-resolved peaks in ESM as compared with ASM extracts and could be assigned to polysaccharide groups (Fig. 3a). The peaks at 1112 cm−1 (A. islandica) and 1109 cm−1 (H. cumingii), present in ESM, are usually attributed to the asymmetric in-phase ring stretching mode of C–O–C (Lavall et al. 2007). Furthermore, the peaks of prominent intensity in the range of 1063–1031 cm−1 (Fig. 3a) for ESM and ASM correspond to the C–O stretching bond in N-acetylglucosamine (Cárdenas et al. 2004; Stankiewicz et al. 1998). The small peaks near 963, 986 cm−1 (ESM) and a broad peak at 952 cm−1 (ASM) may be influenced by N-acetylglucosamine (Stankiewicz et al. 1998). Amine and/or alkene groups at 881 cm−1 (ESM) and 842–844 cm−1 (ASM) could also be identified (Stankiewicz et al. 1998). All spectra in the fingerprint region (754–500 cm−1) including a broad peak at 798 cm−1 (Fig. 3a) for both ESM and ASM are attributed to polysaccharide moieties, probably N-acetylglucosamine (Cárdenas et al. 2004).
The A. islandica spectra show some acidic protein bands, which cannot be found in nacroprismatic shells. Prominent peaks in the sugar region for the samples studied may correspond to the glucosaminoglycans.
Amino acid composition
Amino acid compositions are shown in Tables 1 and 2. Glycine, aspartate and proline residues make up ca 40% of the total amino acids in A. islandica followed by glutamate (ca 8.5%) (Bulk, AIM, EIM and ESM; Table 1). The charge to hydrophobic ratio in ASM is lower (C/HP = 0.59) than in ESM (C/HP = 0.85). In ASM, glycine and proline (31% of the total amino acids) are the main components followed by aspartate, alanine, threonine and glutamate (Supplementary Fig. 1S). These four residues constitute 34% of the total amino acid compositions in ASM (Table 1). Comparing the results with published data for Mercenaria stimpsoni shells with homogeneous microstructure (Samata 1990), the composition is similar to A. islandica with about 37% of the total amino acid composition consisting of aspartate, alanine, threonine and glutamate (Supplementary Fig. 1cS). The proportions of proline and glycine are higher in A. islandica than in M. stimpsoni shells; possibly these differences can be attributed to sampling preparation techniques and/or the different biological taxonomy of these calcareous biominerals. Nonetheless, other microstructures such as crossed lamellar reveal relatively higher proportion (40% in total; Samata 1990).
The nacroprismatic shells are generally characterised by high amounts of glycine and alanine, followed by polar amino acids (aspartate and glutamate) and serine (Tables 1, 2). The composition of the marine nacroprismatic sample (P. fucata) is close to that of the freshwater samples, except that the proportion of glycine is higher (33% compared to ~ 28%), while the proportion of hydrophobic amino acids (i.e. alanine) is lower (16% compared to ~ 21%). Comparing the results with published data for other shells from the same taxonomical orders studied here, Unionoida (Ma et al. 2013) and Pterioida (Pereira-Mouriès et al. 2002), about 50% of the total amino acid composition is in glycine–alanine form (Supplementary Fig. 1S).
Global amino acid compositions show some individual differences in the nacroprismatic shells (Supplementary Fig. 2S). The freshwater shell samples (H. cumingii and D. chilensis) have a C/HP ratio of 0.49, while the marine sample (P. fucata) yields a slightly higher ratio of 0.54. ASM extracts of the nacre–prism shells contain a large proportion of hydrophobic amino acids (C/HP = ~ 0.33) and are glycine–alanine rich (54% ± 1). These characteristics are significantly different to the results for A. islandica, which is rich in aspartate and glutamate (21%), has a higher C/HP of ca. 0.90 and a high proline content (ca 13%; Table 1).
Protein size determination
Various organic fractions of nacre–prism and homogeneous shell structures were analysed by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) and staining with CBB G-250 and/or silver nitrate (Supplementary Figs. 3S, 4S), and Alcian blue and/or modified silver nitrate (Figs. 4, 5). Staining with CBB G-250 did not reveal any protein bands in the samples and incubation of the polyacrylamide gel with traditional silver nitrate demonstrated only a few protein bands for the ASM and ESM extracts. The unsatisfactory response to routine staining methods (including combined silver nitrate and CBB G-250) led us to adapt and develop new fixing and staining methods (as described in “Materials and Methods”) to enhance the visibility of protein bands on the polyacrylamide gel.

a SDS-PAGE of ESM soluble matrix (100 µg each) stained with silver nitrate + 50 µL formaldehyde. Lane 1: A. islandica; Lane 2: H. cumingii; Lane M: standard molecular weight (5 µL). The arrows in Lanes 1 and 2 indicate relative molecular weights at 10, 14, 17, 26, 40, 55 and above 160; and 36, 38 and 40 kDa. b SDS-PAGE of ESM, silver nitrate (silver nitrate + 50 µL formaldehyde) enhanced Alcian Blue. Lane 1: A. islandica (100 µg); Lane 2: H. cumingii (100 µg); Lane M: Standard molecular weight (5 µL). Bands around 17 and 26 kDa for A. islandica may be associated with the phosphorylated and/or sulfated structure of the carbohydrate-protein-linkage extracted

SDS-PAGE of ASM homogeneous (a) and nacroprismatic shells (b and b +) soluble matrix. a, b (20 µL, respectively) stained with silver nitrate + 50 µL formaldehyde, while b+ stained with Alcian Blue enhanced with silver nitrate + 50 µL formaldehyde. Lane M: standard molecular weight (5 µL); Lane 1A: ASM of A. islandica; Lane 1b and 1b + : P. fucata, 2b and 2b + : D. chilensis and, 3b and 3b + : H. cumingii. A distinct band at 40 kDa for A. islandica, P. fucata and H. cumingii (see Fig. 4; Fig. 4S Suppl.) agrees well with a similar band at 40 kDa found in nacre (Yan et al. 2007). The arrows in a indicate the relative molecular weights at around 21, 31, 40 and 55 kDa, while the arrows in b + designate the relative molecular weights at around 21, 31, 40, 45, 58, 63 and 110 kDa
For the A. islandica shell (ESM), modified silver nitrate staining visualised prominent protein bands at around 40, 26, 17, 14 and 10 kDa, and weaker bands above 160 and at around 55 kDa (Fig. 4a, Lane 1). The sugar moiety was confirmed by a specific glycoprotein staining in the A. islandica organic matrix (Fig. 4b, Lane 1). The very broad bands at around 26 and 17 kDa visible in all staining methods may correspond to post-translational modifications (glycosylation, phosphorylation or sulfatation) of proteins in A. islandica; however, no protein band was observed in shells of M. stimpsoni and M. lusoria (Samata 1990), which also have homogeneous microstructure.
For the ESM extract of H. cumingii, the fractionation by SDS-PAGE using modified silver nitrate staining (Fig. 4a; Lane 2) showed protein bands with molecular masses ranging from 5 to 50 kDa along with a smear from 15 to 30 kDa, probably related to the effect of glycosaminoglycans. Weaker bands with molecular weights of 36, 38 and 40 kDa could also be visualised. ESM extracts of A. islandica and H. cumingii (Fig. 4; Supplementary Fig. 4S) both showed similar bands at around 5–10 kDa and at 40 kDa.
Alcian Blue and or modified silver nitrate staining of ASM extracts (Fig. 5) revealed many bands in the range 5–110 kDa for all shells. Arctica islandica shows apparent bands at 21, 31, 40 and 55 kDa, respectively (Fig. 5a, Lane 1). Pinctada fucata revealed a prominent band at 40 kDa (Fig. 5bs, Lane 1), as well as H. cumingii (Fig. 5bs, Lane 3). Other bands for H. cumingii appear around 21, 31, 45, 58, 63 and 110 kDa, respectively, when silver nitrate-enhanced Alcian Blue is used (Fig. 5b+, Lane 3). Similarly, D. chilensis revealed prominent bands at around 10 and 14 kDa (Fig. 5bs, Lane 2). The band at 40 kDa, however, has not been observed in nacroprismatic samples before (Ma et al. 2013; Pereira-Mouriès et al. 2002).
The SDS-PAGE of soluble organic matrix extracts (ASM, ESM) in the studied shells revealed many discrete bands of proteins, some of which are reported here for the first time (e.g. proteins in A. islandica and D. chilensis shells). Two new glycoprotein bands at around 20 and 63 kDa (Fig. 4b; Lane 2) are found in ESM extracts of H. cumingii using combined Alcian Blue/silver nitrate staining and are attributed to a novel acidic sulfated glycoproteins for H. cumingii. Freshwater (H. cumingii and D chilensis) and marine shells (A. islandica) exhibit similar bands in the range 5–10 kDa. The band at 40 kDa is prominent in H. cumingii, A. islandica and P. fucata, respectively.
Comparison of EDTA and acid decalcification methods
Two complementary dissolution methods were used to optimise recovery, particularly of the soluble organic fraction (e.g. Bédouet et al. 2007). Our modification of the traditional EDTA decalcification method (see “Materials abd methods” section) was efficient in removing most residual EDTA from the extracted soluble organic matrix, which is otherwise a common problem. For instance, comparing the EDTA fractions with literature data, results for acid-extraction (Ma et al. 2013) match well with the amino acid composition of H. cumingii shells except for a difference in alanine, for which a higher concentration is reported in the literature (See Supplementary Fig. 1bS).
The EDTA extracts in our study yielded higher amounts of polar amino acid and higher C/HP ratios (Table 1) compared to the acid extract. Also, protein bands are more prominent in the soluble moiety extracted with EDTA, while the acid extracts generally have higher molecular weights (Fig. 5). This most likely indicates that some constituents, as for example silk-like constituent, which is rich in glycine and/or alanine (Addadi et al. 2006; Pereira-Mouriès et al. 2002), remain in the insoluble moiety when the shell is dissolved with EDTA (Tables 1, 2; Supplementary Fig. 1S).
Discussion
The soluble protein portion in A. islandica shells (ASM and ESM extracts) depicts eight major apparent molecular masses: of 10, 14, 17, 21, 26, 31, 40 and 55 kDa (Figs. 4 Lane 1, 5a Lane 1). CBB-G250 fails to stain these protein bands well and the protein bands ranging from 5 to 10 kDa show strong negative bands (Supplementary Fig. 4S). As CBB-G250 predominantly stains the acidic shell proteins, this suggests that a large portion of the organic macromolecules in A. islandica shells is non-acidic (Gotliv et al. 2003). CBB-G250 and Alcian Blue enhanced with silver nitrate stained three major bands at around 14, 26 and 17 kDa, respectively. We propose that these bands (26 and 17 kDa) are associated with sulfated polysaccharides. A band stained at around 55 kDa (ASM, Fig. 5a; Lane 1) may be a proteoglycan band, which are glycosaminoglycans associated to a core protein.
We first observed two discrete glycoprotein bands at 20 and 63 kDa amongst the soluble organic macromolecules in nacroprismatic H. cumingii shells (EDTA extract; Fig. 4b, Lane 2). Similar bands are reported for other molluscs: a 20-kDa protein band was observed in the bivalve Pinctada maxima (Bédouet et al. 2001; Pereira-Mouriès et al. 2002) and a 63-kDa acidic glycoprotein was found in the cephalopod Nautilus macromphalus (Marie et al. 2011). CBB-G250 and silver nitrate combined stained protein bands between 30 and 50 kDa in this extract (Supplementary Fig. 4S, Lane 2). Similar bands at 48 and 50 kDa were observed in freshwater cultured pearls (Natoli et al. 2010) and nacre (Ma et al. 2013) of H. cumingii, respectively. These findings are in accordance with the observation of a calcium-binding glycoprotein of an apparent molecular mass of 50 kDa in the bivalves Unio pictorum (Marie et al. 2007) and in Anodonta cygnea (Lopes et al. 2014) that belong to the same taxonomic order as H. cumingii (Unionida).
The soluble organic fraction after acid extraction (ASM) of two nacroprismatic shells (D. chilensis patagonicus and of H. cumingii) show bands at around 14 and 58 kDa. The matrix proteins N14 (pearline) (Miyashita et al. 2000) and MSI60 (Sudo et al. 1997) have similar sizes, but detailed proteomics would be necessary to further clarify the identity of the bands observed here. Both protein bands have also been found in the nacreous layer of Pinctada shells (Kono et al. 2000), but we did not observe these bands in the P. fucata shells studied here (Fig. 5b; Lane 1). Instead, a prominent peak at 40 kDa stains in gels from P. fucata is also identified in A. islandica and H. cumingii shells. This band is inferred to be a conventional “insoluble matrix protein” (Yan et al. 2007).
Comparison of the organic macromolecules in A. islandica shells with nacroprismatic samples exhibits several important differences: The overall organic content is lower in A. islandica shells compared with the nacroprismatic samples (Fig. 1); it contains a higher C/HP ratio (Tables 1, 2), and hence more polar components (aspartate and glutamate) compared with the nacroprismatic shells. The composition of A. islandica shells is comparable to those of M. stimpsoni except that the total proportion of the polar amino acids as a group, namely aspartate, glutamate, threonine, serine and arginine, is relatively higher (42.8% in M. stimpsoni compared of 38.4% in A. islandica, see Supplementary Fig. 1cS). We further compared the composition of those five residues above with the shells of crossed lamellar microstructure (Samata 1990). The composition of crossed-lamellar bivalve shells is relatively close (36.0 vs 38.1%) compared to homogeneous A. islandica shell. However, unlike our study that reveals many prominent protein bands in A. islandica, shells of M. stimpsoni with homogeneous and other bivalve shells with crossed lamellar microstructure showed no protein band (Dauphin and Denis 2000; Samata 1990).
The nacroprismatic shells contain higher amounts of organic scaffolding macromolecules, including β-chitin lamellar sheets and silk-like fibroin gel (Levi-Kalisman et al. 2001) than A. islandica. This is supported by acidic bands of A. islandica in the FTIR spectra at 1736, 1716 cm−1 as well as by amide II bands (Fig. 2b), none of which are present in the nacroprismatic samples. Moreover, glycine and alanine are the main fractions in the nacroprismatic samples (Tables 1, 2), while A. islandica depicts lower amounts of these molecules (Table 1), even when considering all proline, glycine and aspartate in their glycine–alanine-rich forms. Furthermore, A. islandica demonstrates distinct polysaccharide bands in the FTIR spectra (Fig. 2a). These could be a result of higher contents of proline (15%; Table 1) in A. islandica, which by far exceed those in the nacroprismatic organic matrices (ca 3%; Gotliv et al. 2003; Ma et al. 2013).
The composition of the Ca-binding biomolecules in the soluble fractions of the nacroprismatic samples obtained by both decalcification methods are similar; they comprise glycine, threonine, serine, glutamate and aspartate (ca 54%; Supplementary Fig. 2S) with a large proportion of glycine, alanine and serine (ca 60%; Tables 1, 2) characteristic of silk fibroin-like proteins (Keith et al. 1993). These findings agree well with the composition of organic matrices in the nacre of other molluscs (Bédouet et al. 2007; Keith et al. 1993; Ma et al. 2013; Pereira-Mouriès et al. 2002), similar to the composition of insoluble spider silk (Lombardi and Kaplan 1990). Proteins with this amino acid composition are able to form a β-sheet structure, which has been proposed to be a key scaffolding constituent in the shell (Weiner and Traub 1980).
Previous studies on bivalve nacre established a major role for acidic amino acids, namely aspartic and glutamic acid in biomineralisation (Addadi and Weiner 2014; Gotliv et al. 2003) based on their Ca-binding function. However, compared to nacroprismatic shells, the amounts of aspartate and glutamate in A. islandica shells are lower (≤ 16% vs > 25%), suggesting that this role is not limited to acidic proteins, but extends to other compounds of the organic matrix. Glycosylated proteins with the potential to bind Ca2+ ions containing lower amounts of acidic properties (Asx + Glx < 22%), but high amounts of hydrophobic proteins (≤ 50%), were identified from Unio pictorium shells (Marie et al. 2007) and cultured pearls of H. cumingii (Ma et al. 2012; Natoli et al. 2010). Previous works (Ma et al. 2012; Yan et al. 2007; Zhang et al. 2006) suggested that aragonite crystal nucleation takes place at a preferential site on the surface of nonacidic matrix proteins and other non-proteinaceous macromolecules. This site is identified as rich in sulphur, probably in the form of sulphate groups (Marie et al. 2007).
Samata (1990) analysed the Ca-binding glycoproteins in molluscan shells with different microstructures and found the compositions similar between homogeneous and crossed lamellar structures. In our study both homogeneous- and nacroprismatic-shells are nonacidic, though glycine–alanine forms are prominent in nacroprismatic shells. We suggest that the composition of the Ca-binding glycoproteins may vary predominantly according to shell structure and/or species. It is obvious that no hydrophobic and/or acidic proteins appear to be specific to certain molluscan shell microstructures (Samata 1990).
Conclusions
We report the first analysis of shell-associated biopolymers for A. islandica shells and distinguish the homogeneous A. islandica shells from nacroprismatic shells. New staining methods for electrophoretic fractionations remove some major problems and improve the analysis of shell macromolecules. We show that impregnation of gels with silver nitrate solution containing formaldehyde (50 µl in 100 ml) before development can facilitate the visualisation of proteins on the gel. Using these newly adapted fixations and staining protocols we show that the choice of decalcification method significantly influences the sizes as well as the number of protein bands observed on the gels.
Many protein bands (e.g. around 20 and 63 kDa) are revealed in H. cumingii here for the first time. We show that shells with homogeneous and nacroprismatic microstructures have some protein band(s) in common: A. islandica, P. fucata and H. cumingii show a prominent protein band at 40 kDa. Pinctada fucata demonstrates just one band at 40 kDa, which is a nonacidic-matrix-protein (Yan et al. 2007). Moreover, the shell macromolecules of all studied samples are overall non-acidic, containing low amounts of aspartate and glutamate (21% for homogeneous and 16% for nacroprismatic shells).
References
Addadi L, Weiner S (2014) Biomineralization: mineral formation by organisms. Phys Scr 89:098003
Addadi L, Joester D, Nudelman F, Weiner S (2006) Mollusk shell formation: a source of new concepts for understanding biomineralization processes. Chem Eur J 12:980–987. doi:10.1002/chem.200500980
Agbaje O, Wirth R, Morales L, Shirai K, Kosnik M, Watanabe T, Jacob D (2017) Architecture of crossed-lamellar bivalve shells: the southern giant clam (Tridacna derasa, Röding, 1798). R Soc Open Sci 4:170622. doi:10.1098/rsos.170622
Arivalagan J, Marie B, Sleight VA, Clark MS, Berland S, Marie A (2016) Shell matrix proteins of the clam, Mya truncata: roles beyond shell formation through proteomic study. Mar Genom 27:69–74. doi:10.1016/j.margen.2016.03.005
Bédouet L, Schuller MJ, Marin F, Milet C, Lopez E, Giraud M (2001) Soluble proteins of the nacre of the giant oyster Pinctada maxima and of the abalone Haliotis tuberculata: extraction and partial analysis of nacre proteins. Comp Biochem Physiol B: Biochem Mol Biol 128:389–400
Bédouet L, Marie A, Dubost L, Péduzzi J, Duplat D, Berland S, Puisségur M, Boulzaguet H, Rousseau M, Milet C (2007) Proteomics analysis of the nacre soluble and insoluble proteins from the oyster Pinctada margaritifera. Mar Biotechnol 9:638–649. doi:10.1007/s10126-007-9017-1
Bevelander G, Nakahara H (1969) An electron microscope study of the formation of the nacreous layer in the shell of certain bivalve molluscs. Calcif Tissue Res 3:84–92. doi:10.1007/BF02058648
Brine CJ, Austin PR (1981) Chitin variability with species and method of preparation. Comp Biochem Physiol B: Biochem Mol Biol 69:283–286
Cárdenas G, Cabrera G, Taboada E, Miranda SP (2004) Chitin characterization by SEM, FTIR, XRD, and 13C cross polarization/mass angle spinning NMR. J Appl Polym Sci 93:1876–1885. doi:10.1002/app.20647
Carter JG (1990) Skeletal biomineralization: patterns, processes and evolutionary trends. Wiley, New York
Carter JG, Aller RC (1975) Calcification in the bivalve periostracum. Lethaia 8:315–320
Cohen SA, Michaud DP (1993) Synthesis of a fluorescent derivatizing reagent, 6 aminoquinolyl-N-hydroxysuccinimidyl carbamate, and its application for the analysis of hydrolysate amino acids via high-performance liquid chromatography. Anal Biochem 211:279–287
Dauphin Y (2003) Soluble organic matrices of the calcitic prismatic shell layers of two pteriomorphid bivalves Pinna nobilis and Pinctada margaritifera. J Biol Chem 278:15168–15177. doi:10.1074/jbc.M204375200
Dauphin Y, Denis A (2000) Structure and composition of the aragonitic crossed lamellar layers in six species of Bivalvia and Gastropoda. Comp Biochem Physiol A: Mol Integr Physiol 126:367–377
Farre B, Dauphin Y (2009) Lipids from the nacreous and prismatic layers of two Pteriomorpha Mollusc shells. Comp Biochem Physiol B: Biochem Mol Biol 152:103–109
Giuffre AJ, Hamm LM, Han N, De Yoreo JJ, Dove PM (2013) Polysaccharide chemistry regulates kinetics of calcite nucleation through competition of interfacial energies. Proc Natl Acad Sci 110:9261–9266. doi:10.1073/pnas.1222162110
Gotliv BA, Addadi L, Weiner S (2003) Mollusk shell acidic proteins: in search of individual functions. ChemBioChem 4:522–529. doi:10.1002/cbic.200200548
Hamm LM, Giuffre AJ, Han N, Tao J, Wang D, De Yoreo JJ, Dove PM (2014) Reconciling disparate views of template-directed nucleation through measurement of calcite nucleation kinetics and binding energies. Proc Natl Acad Sci 111:1304–1309. doi:10.1073/pnas.1312369111
Keith J, Stockwell S, Ball D, Remillard K, Kaplan D, Thannhauser T, Sherwood R (1993) Comparative analysis of macromolecules in mollusc shells. Comp Biochem Physiol B: Biochem Mol Biol 105:487–496
Kono M, Hayashi N, Samata T (2000) Molecular mechanism of the nacreous layer formation in Pinctada maxima. Biochem Biophys Res Commun 269:213–218. doi:10.1006/bbrc.2000.2274
Lavall RL, Assis OB, Campana-Filho SP (2007) β-Chitin from the pens of Loligo sp.: extraction and characterization. Bioresour Technol 98:2465–2472
Levi-Kalisman Y, Falini G, Addadi L, Weiner S (2001) Structure of the nacreous organic matrix of a bivalve mollusk shell examined in the hydrated state using cryo-TEM. J Struct Biol 135:8–17
Lombardi SJ, Kaplan DL (1990) The amino acid composition of major ampullate gland silk (dragline) of Nephila clavipes (Araneae, Tetragnathidae). J Arachnol 18:297–306
Lopes A, Hinzmann M, Bobos I, Lopes-Lima M, Gonçalves JF, rgio Ferreira S, Domingues B, Machado J (2014) Functional studies on the shell soluble matrix of Anodonta cygnea (Bivalvia: Unionidae). Nautilus 128:105–113
Ma Y, Qiao L, Feng Q (2012) In-vitro study on calcium carbonate crystal growth mediated by organic matrix extracted from fresh water pearls. Mater Sci Eng, C 32:1963–1970
Ma Y, Berland S, Andrieu JP, Feng Q, Bédouet L (2013) What is the difference in organic matrix of aragonite vs. vaterite polymorph in natural shell and pearl? Study of the pearl forming freshwater bivalve mollusc Hyriopsis cumingii. Mater Sci Eng, C 33:1521–1529
Marie B, Luquet G, Pais De Barros JP, Guichard N, Morel S, Alcaraz G, Bollache L, Marin F (2007) The shell matrix of the freshwater mussel Unio pictorum (Paleoheterodonta, Unionoida). FEBS J 274:2933–2945. doi:10.1111/j.1742-4658.2007.05825.x
Marie B, Zanella-Cléon I, Corneillat M, Becchi M, Alcaraz G, Plasseraud L, Luquet G, Marin F (2011) Nautilin-63, a novel acidic glycoprotein from the shell nacre of Nautilus macromphalus. FEBS J 278:2117–2130. doi:10.1111/j.1742-4658.2011.08129.x
Marin F, Luquet G (2005) Molluscan biomineralization: the proteinaceous shell constituents of Pinna nobilis L. Mater Sci Eng, C 25:105–111
Miyashita T, Takagi R, Okushima M, Nakano S, Miyamoto H, Nishikawa E, Matsushiro A (2000) Complementary DNA cloning and characterization of pearlin, a new class of matrix protein in the nacreous layer of oyster pearls. Mar Biotechnol 2:409–418. doi:10.1007/PL00021687
Morrissey JH (1981) Silver stain for proteins in polyacrylamide gels: a modified procedure with enhanced uniform sensitivity. Anal Biochem 117:307–310
Natoli A, Wiens M, Schröder HC, Stifanic M, Batel R, Soldati AL, Jacob DE, Müller WE (2010) Bio-vaterite formation by glycoproteins from freshwater pearls. Micron 41:359–366
Pereira-Mouriès L, Almeida MJ, Ribeiro C, Peduzzi J, Barthélemy M, Milet C, Lopez E (2002) Soluble silk-like organic matrix in the nacreous layer of the bivalve Pinctada maxima. Eur J Biochem 269:4994–5003. doi:10.1046/j.1432-1033.2002.03203.x
Philipp EE, Wessels W, Gruber H, Strahl J, Wagner AE, Ernst IM, Rimbach G, Kraemer L, Schreiber S, Abele D (2012) Gene expression and physiological changes of different populations of the long-lived bivalve Arctica islandica under low oxygen conditions. PLoS ONE 7:e44621
Rahman MA, Isa Y, Takemura A, Uehara T (2006) Analysis of proteinaceous components of the organic matrix of endoskeletal sclerites from the alcyonarian Lobophytum crassum. Calcif Tissue Int 78:178–185. doi:10.1007/s00223-005-0253-y
Roeges NP (1994) A guide to the complete interpretation of infrared spectra of organic structures. Wiley, New York
Ropes JW, Jones D, Murawski S, Serchuk F, Jearld A (1984) Documentation of annual growth lines in ocean quahogs, Arctica Islandica LINNE. Fish Bull 82:1–19
Samata T (1990) Ca-binding glycoproteins in molluscan shells with different types of ultrastructure. Veliger 33:190–201
Shirai K, Schöne BR, Miyaji T, Radarmacher P, Krause RA, Tanabe K (2014) Assessment of the mechanism of elemental incorporation into bivalve shells (Arctica islandica) based on elemental distribution at the microstructural scale. Geochim Cosmochim Acta 126:307–320. doi:10.1016/j.gca.2013.10.050
Stankiewicz BA, Mastalerz M, Hof CH, Bierstedt A, Flannery MB, Briggs DE, Evershed RP (1998) Biodegradation of the chitin-protein complex in crustacean cuticle. Org Geochem 28:67–76
Sudo S, Fujikawa T, Nagakura T, Ohkubo T, Sakaguchi K, Tanaka M, Nakashima K, Takahashi T (1997) Structures of mollusc shell framework proteins. Nature 387:563–564
Taylor JD (1973) The structural evolution of the bivalve shell. Palaeontology 16:519–534
Truong HH, Neilson KA, McInerney BV, Khoddami A, Roberts TH, Liu SY, Selle PH (2015) Performance of broiler chickens offered nutritionally-equivalent diets based on two red grain sorghums with quantified kafirin concentrations as intact pellets or re-ground mash following steam-pelleting at 65 or 97 °C conditioning temperatures. Anim Nutr 1:220–228
Wall RS, Gyi TJ (1988) Alcian blue staining of proteoglycans in polyacrylamide gels using the “critical electrolyte concentration” approach. Anal Biochem 175:298–299
Weidman CR, Jones GA (1994) The long-lived mollusc Arctica islandica: A new paleoceanographic tool for the reconstruction of bottom temperatures for the continental shelves of the northern North Atlantic Ocean. J Geophys Res Oceans 99:18305–18314. doi:10.1029/94JC01882
Weiner S, Dove PM (2003) An overview of biomineralization processes and the problem of the vital effect. Rev Mineral Geochem 54:1–29. doi:10.2113/0540001
Weiner S, Traub W (1980) X-ray diffraction study of the insoluble organic matrix of mollusk shells. FEBS Lett 111:311–316
Yan Z, Jing G, Gong N, Li C, Zhou Y, Xie L, Zhang R (2007) N40, a novel nonacidic matrix protein from pearl oyster nacre, facilitates nucleation of aragonite in vitro. Biomacromol 8:3597–3601. doi:10.1021/bm0701494
Zaitoun MA, Lin C (1997) Chelating behavior between metal ions and EDTA in sol–gel matrix. J Phys Chem B 101:1857–1860. doi:10.1021/jp963102d
Zhang C, Li S, Ma Z, Xie L, Zhang R (2006) A novel matrix protein p10 from the nacre of pearl oyster (Pinctada fucata) and its effects on both CaCO3 crystal formation and mineralogenic cells. Mar Biotechnol 8:624–633. doi:10.1007/s10126-006-6037-1
Acknowledgements
Mark Tran and Dr Christopher McRae are well-appreciated for their incessant technical supports. OBAA acknowledges Emily Gibson and Dr Bhumika Shah (Chemistry and Biomolecular Sciences, Macquarie University) for teaching him the basic techniques of gel electrophoresis. The authors are grateful to A. Checa and the anonymous reviewers for their valuable comments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded through an Australian Research Council (ARC) Future Fellowship to DEJ. The work was facilitated in part by the Australian Government’s National Collaborative Research Strategy (NCRIS) and its facilities at the Australian Proteome Analysis Facility (APAF).
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of mollusc shells were followed. Recently alive shells: Arctica islandica, Hyriopsis cumingii, Diplodon chilensis patagonicus and Pinctada fucata martensii were provided from the private collection of one of the authors (DEJ).
Additional information
Responsible Editor: A. G. Checa.
Reviewed by Undisclosed experts.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Agbaje, O.B.A., Thomas, D.E., Mclnerney, B.V. et al. Organic macromolecules in shells of Arctica islandica: comparison with nacroprismatic bivalve shells. Mar Biol 164, 208 (2017). https://doi.org/10.1007/s00227-017-3238-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00227-017-3238-2