
Abstract
This is a review of historical and modern literature data on the structure versus properties of wood lignin in view of the concepts developed by the authors based on their own research. Changes in the structure of lignin and related changes in its chemical reactivity during alkaline wood pulping are assessed based on the comparison of the structures of lignin at three kinetically distinct stages of delignification: initial, bulk and final. Lignin gradually moves from a solid to a liquid phase during the pulping process; therefore, structures of native, dissolved and residual lignin are elucidated and compared. The emphasis is on changes in the molecular weight distribution and content of alkylarylether bonds, and functional groups, in particular phenolic hydroxyls. For comparison, splitting rates for α- and β-alkylarylether bonds in both phenolic and non-phenolic lignin model compounds are analyzed. Based on the comparative analysis of the experimental data, it is suggested that native lignin in wood consists mainly of three distinct fractions that are different in chemical reactivities of alkylarylether bonds. This phenomenon results in three kinetically distinct stages of the pulping process. Wood delignification is essentially a process of lignin functionalization followed by its dissolution. The functionalization, i.e., formation of additional functional groups in the macromolecule, continues until it reaches the level sufficient for lignin dissolution under chosen conditions, and then, delignification occurs. At the bulk stage of pulping, the rate of delignification is directly proportional to the degree of functionalization. The data characterizing the effect of redox reactions on the structure and chemical reactivity of lignin in alkali–anthraquinone pulping are analyzed in detail in view of their general importance for our understanding of the chemical reactivity of lignin. Results of polarographic studies of numerous representative lignin model compounds (> 70 samples) and lignin samples, including chemically changed lignins, are compiled, and a diagram of reduction potentials of polarographically active functional groups in lignin is drawn. From a comparison of redox properties of lignin and 32 pulping additives, criteria for selection of potential alkaline pulping catalysts are derived. Solubility belongs to basic properties of polymers, lignin included, and all methods of delignification of plant materials can be essentially reduced to solid polymer functionalization followed by its dissolution. Factors contributing to lignin solubility are analyzed, including such characteristics as molecular weight, temperature, liquid–solid ratio and ionic strength. Based on the analyzed data, a uniform scale of solubility for different lignin types is proposed, and formulae for calculating lignin solubility in alkaline media are derived.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The chemical structure of lignin and its effect on chemical reactivity of lignin is one of the key issues in wood chemistry, first, due to practical implications of such knowledge in the pulp and paper industry. A variety of lignins in different plant species and a complex array of transformations that native lignins undergo during technological processes and natural transformation of lignocellulosic materials make explanation and prediction of chemical properties of lignin difficult even at the current level of development of analytical techniques.
Continuous progress in our understanding of lignin chemistry, specifically, in comparative analysis of molecular mass, functional groups, types of interunit bonds and, more generally, structures of lignins in wood, black liquor and pulp, nevertheless gives a clear idea of chemical processes during wood pulping (Brunow and Miksche 1976; Dimmel and Gellerstedt 2010; Fengel and Wegener 1989; Gellerstedt and Henriksson 2008; Gierer 1980, 1982a, b; Lindberg 1979; Santos et al. 2013; Sarkanen and Ludwig 1971; Shorygina et al. 1976). In this paper, focus is put on the studies that correlate structural changes in lignin with changes in its chemical reactivity and also discuss structural factors affecting its solubility in pulping liquors. This is a review of historical and modern literature data on the structure versus properties of wood lignin in view of the concepts developed based on the authors’ own research. The data on the structures of native and technical lignins are comparatively analyzed and related to lignin valorization, the latter issue attracting significant interest in recent years (Ragauskas et al. 2014; Rinaldi et al. 2016; Galkin and Samec 2016; Kärkäs et al. 2016).
Chemical reactivity of alkylaryl bonds in lignin and kinetics of delignification
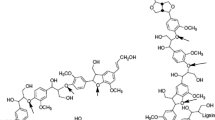
Lignin is an irregular polymer with a variety of bonds connecting its phenylpropane units (PPU), predominately α- and β-O-4 ether bonds (65/100 PPU in both hardwood and softwood lignin, see Tables 1 and 2). Typical chemical structures of softwood and hardwood lignin and typical products of their degradation in alkaline media are shown in Fig. 1.

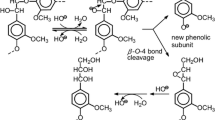
Structural fragments of lignin containing α- and β-O-4 bonds
The kinetics data in Tables 3 and 4 suggest certain types of structure–chemical reactivity relationships in lignin under the conditions of soda and kraft pulping. The α-O-4 linkages in phenolic units (I, III) are the most reactive, while in non-phenolic units (II, IV) they are stable (Ljunggren 1980). The β-O-4 linkages in phenolic (VI) and especially non-phenolic (XII) units are much less reactive. Thus, the ratio between the rate constants at 140 °C for units XII, VI and (I, III) is 1:24:2600 (Table 3). Unit XII is of special interest because β-O-4 bonds are the most common in lignin (Table 1), predominantly between non-phenolic structures because the content of phenolic hydroxyl groups in native lignin is just 10–15/100 PPU (Gellerstedt and Lindfors 1984a).
Chemical reactivity of β-O-4 bonds depends on the number and positions of substituents in the aromatic ring and side chain of the lignin fragment (Table 4). Rate constants in the series (V, VII, IX) and (VI, VIII, X) show how the susceptibility of β-O-4 bonds to splitting changes when going from softwood to hardwood lignin. It is known (Fengel and Wegener 1989; Sarkanen and Ludwig 1971) that guaiacyl units predominate in softwood lignin (see A and B in structures V and VI), while hardwood lignin is built from mixed guaiacyl and syringyl units (see A and B in structures IX and X).
Tables 4 and 5 illustrate the highest splitting rate of the β-O-4 bond in syringyl units as compared to other types of structures in lignin during kraft pulping. These data match published data on a higher delignification rate of hardwood versus softwood (Fengel and Wegener 1989; Sarkanen and Ludwig 1971; Shorygina et al. 1976).
Assessing the kinetics of the splitting reaction during soda pulping is more complicated. Under these conditions, a competing side reaction of enolization (formation of an enol ether structure XV) takes place, and the kinetics is affected by the ratio of the two reactions:

In Kondo et al. (1987), the rate constant k was measured based on the consumption of the initial model compound and k1 based on accumulation of one of the products of the β-O-4 linkage splitting. This product originating from the ring B in the initial model compound is either guaiacol (see G in Fig. 1) or 4-methylsiringol.
In kraft pulping, rate constants k and k1 are close, while k1 ≪ k in soda pulping. Thus, for a model compound of structure F, k/k1 = 8 at 130 °C. This explains why soda pulping is much slower than kraft pulping, and why deep delignification is not possible in soda pulping without substantial loss of carbohydrates.
Structures with shorter side chains (V, VII, IX) are more reactive in kraft pulping than related structures that carry γ-CH2OH groups (Table 4). It should be noted that the latter (VI, VIII, X) are the most typical of native lignin.
A comparative study of the effect of a substituent in A and B rings on β-O-4 linkage splitting in phenolic units of lignin (V) demonstrated that a substituent in ring B has a stronger effect under the conditions of soda pulping (Dimmel and Schuller 1986a, b). Consistent with other experimental data, it indicates that when the linkage splitting is a predominant reaction versus enolization (this is the case at high alkalinity), β-O-4 splitting is the rate-determining reaction. At lower alkalinity, formation of enol ether becomes a predominant reaction.
Under the conditions of kraft and anthraquinone (AQ) pulping, the rate of linkage splitting is affected by a substituent in the ring A only. This suggests quinone methide formation (1) that precedes the bond splitting being the rate-determining stage:

The effect of a side chain on the β-O-4 linkage splitting can be assessed based on the kinetic data published by Gierer and Ljunggren (1979, 1983) and Gierer et al. (1980). In the absence of hydroxyl groups in the side chain, this bond is stable toward alkali at elevated temperatures (see structure V′ in Table 6). In structures with α-hydroxyl substituents, it splits at a rather high rate in both phenolic and non-phenolic lignin structures (compare structures V and XI in Table 6). The non-phenolic units are less reactive, and the rate constant depends on the alkalinity but not concentration of sodium sulfide (see also Table 3).
The chemical reactivity of β-O-4 bond changes dramatically in structures combining γ-hydroxyl and α-carbonyl groups in a side chain (structure XIV in Table 6). It splits at room temperature at a rate exceeding that in any other lignin structure at 140–170 °C.
General analysis of the effect of a side chain structure on the kinetics of a β-O-4 bond splitting (Gierer and Ljunggren 1983) led to the identification of three major factors that determine how strong the effect of an adjacent group is:
-
a degree of ionization of the group;
-
nucleophilicity of the anion formed upon ionization;
-
a rate of formation of a product or intermediate involving the group.
The critical factor for the β-O-4 bond splitting in both phenolic and non-phenolic structures of lignin is the presence in the side chain of a functional group that can ionize in alkaline media.
Summing up the kinetic data on β-O-4 bond splitting in different structural fragments of lignin, the pulping reagents can be ranged in the following way:
-
phenolic units (V, VI)HS− > AHQ ≫ HO−
-
non-phenolic units (XI, XII)HO− > HS− ≫ AHQ
In actual wood pulping, many structures of lignin are involved in reactions with liquor components simultaneously, and it is often hard to conclude what reaction is rate-determining. Besides, non-phenolic units gradually convert into phenolic ones in the course of pulping, and concentrations of active reagents of the liquor decrease because of their reactions with both lignin and carbohydrates, and products of their degradation. Altogether, it results in a very complicated kinetics.
Considering structure–reactivity relationships in lignin, general descriptiveness of the proposed kinetic equations is less important than a correlation between the rate of splitting of α- and β-arylether bonds in lignin and rate of its dissolution. Such a correlation was established in a study (Ljunggren 1980) that was, however, criticized later for direct comparing of the rate constants in dimer models in a homogeneous solution and rate of dissolution of a lignin polymer in a heterogeneous system (Obst 1983).
A different approach to this problem was developed: α-O-4 and β-O-4 bond splitting in lignin releases free phenolic hydroxyl groups that leads to eventual dissolution of lignin in an alkaline medium; therefore, it makes sense to compare the rates of dissolution of lignin and of formation of new phenolic hydroxyls in lignin under the conditions of alkaline pulping.
Changes in molecular mass and rate of dissolution of lignin
In some studies (Evstigneyev 2001, 2003; Evstigneyev et al. 1987a, b, 1990), changes in the MMD through stages of pulping were followed using a GPC technique that eliminates polyelectrolyte interference (Spheron P-1000 gel, DMF eluent with LiBr and H3PO4).
In this study, unimodal gel chromatograms were obtained in lignins from soda, soda–AQ, kraft and kraft–AQ pulping in digesters. All GC curves were found to be unimodal, with the same position of the maximum, shape and σ values in all stages of pulping. It suggests that average MM of lignin remains the same through all stages of pulping. The GPC method, when distortion factors are suppressed, can be used reliably in comparative studies of lignin MMDs during pulping, while other methods are more suitable for absolute MM determination. Vapor osmometry demonstrates that Mn values of soda lignins are all close (Table 7). Compared to the GPC data, this suggests sodium sulfide and AQ having no special effect on lignin depolymerization as compared to sodium hydroxide.
In batch digesters, the liquor gradually becomes rich with dissolved lignin, and secondary reactions become possible that would affect MMD. Besides, some lignin is unavoidably lost during its separation by acidification of the liquor. To minimize these effects, pulping experiments in a flow-through reactor were conducted, with MMD characterization without separation of lignin from the liquor (Evstigneyev 2001). Sample gel chromatograms are presented in Fig. 2a, which can also be compared to a chromatogram of Björkman lignin of spruce so that the changes in lignin during pulping could be assessed.

Normalized gel chromatograms of lignin from a soda and b kraft pulping of spruce wood in a flow-through digester. BL Björkman lignin (Evstigneyev 2001)
Figure 2 is consistent with increasing MM during pulping; however, this change is not dramatic and is observed only at the ramping stage (first 80–100 min; no changes at 120–180 min). It means that MM of dissolved lignin does not change at the bulk and final stages of soda pulping.
Comparable results were obtained for kraft pulping (Fig. 2b). A comparison of VR values in the peak maximum shows that the values for the initial, bulk and final stages of kraft pulping are the same as in soda pulping within an experimental error.
Changes in MMD during alkaline pulping can also be elucidated in experiments on Björkman lignin, which can condense under these conditions. The sample was cooked in glass ampules and analyzed directly, without lignin separation from the liquor (Fig. 3). In the course of pulping, the MMD curves moved to the lower molecular mass region. Quantification here can be done by comparison of Mn and VR values for Björkman lignin and soda lignins (Table 7). It can be seen that soda pulping causes a decrease in MM of Björkman lignin of spruce from ~ 2100 to ~ 1600, while in kraft pulping the lower limit of MM changes is less than 1600 that suggests more intensive lignin degradation. No increase in MM was observed in both processes.

Normalized gel chromatograms of spruce Björkman lignin a soda and b kraft pulping (Evstigneyev 2001)
A general conclusion here is that sodium sulfide and AQ do not significantly affect lignin depolymerization during alkaline delignification of wood. Further, there are no indications of lignin condensation in soda pulping, at least, not in the form of intermolecular aggregation. Why is it that delignification is faster in the presence of sodium sulfide or AQ? They indeed accelerate splitting of β-O-4 bonds that results in a higher content of phenolic hydroxyl groups in kraft lignin and soda–AQ lignin (see below), but this does not result in more intense degradation of lignin macromolecule.
In the authors’ opinion, the explanation is due to the fact that in lignin the units are connected by both ether and carbon–carbon bonds, the latter being resistant to the pulping conditions. Table 1 shows that the total content of carbon–carbon bonds is close to that of ether bonds (α-O-4 and β-O-4). Thus, it is suggested that most lignin units are connected to each other by both splittable and resistant bonds. In this case, breaking α-O-4 and β-O-4 bonds does not necessarily lead to depolymerization of lignin. On the other hand, it leads to enrichment of lignin with phenolic hydroxyl groups that improves solubility. This could be the mechanism of delignification acceleration by sodium sulfide and AQ. Schematically, it can be illustrated using a phenylcoumarane structure as an example:

A small fraction of units connected to the macromolecule exclusively by labile ether bonds (likely located on a periphery of the macromolecule) detaches during pulping and manifests itself in black liquor as monophenols.
A method of assessment of the theoretical yield of monomers after catalytic hydrogenolysis of native and technical lignins has recently been developed based on these considerations (Evsnigneyev 2018).
As demonstrated, differences in the rates of soda, soda–AQ, kraft and kraft–AQ pulping cannot be explained based on the changes in macromolecular properties of lignin affected by different liquor reagents. Therefore, it is more likely that the effect of sodium sulfide and AQ on alkaline delignification lies in the functionalization, changes in the chemical structure of lignin that are different from degradation and condensation reactions.
Lignin functionalization and the alkaline delignification rate
A rate curve for wood delignification in kraft pulping normally has three distinct parts that differ in the rate of lignin dissolution: initial (fast), bulk (slow) and final (very slow) (Sjöström 1981). The curves, generally, have a typical S-shape that can be illustrated by the data on soda, soda–AQ, kraft and kraft–AQ pulping of spruce (Evstigneyev et al. 1992a; Fig. 4), kraft pulping of aspen and turpentine pine (Gullichsen 1999), and kraft and sulfite pulping of Western hemlock (Chiang et al. 1989). It is important from the technological point of view to understand why delignification slows down so significantly in the end of pulping. We can answer this question based on the literature data on the structures of native, dissolved and residual lignins.

Delignification curves of soda (\(\bullet\)), soda–AQ (+), kraft (o) and kraft–AQ (\(\oplus\)) pulping of spruce (Evstigneyev et al. 1992a)
To assess structural changes in lignin leading to its dissolution during pulping, correlations were studied between, on the one hand, the content of functional groups and alkylaryl bonds in dissolved lignins and, on the other hand, rate and degree of delignification (Evstigneev 2003; Evstigneyev and Rusakov 1990; Evstigneyev et al. 1990, 1992a). It is proven that fast dissolution of lignin begins upon accumulation of sufficient phenolic groups, usually functionalization of about 50% PPU.
At the bulk stage, the rate of lignin dissolution is proportional to the rate of release of free phenolic hydroxyl groups (Fig. 5). Adding 3.7 mM OHphen is needed to dissolve 1 g lignin, and the observed differences in delignification rates originate from different rates of achieving this level. Thus, the data suggest that delignification is essentially a process of lignin functionalization followed by its dissolution.

It was found that in kraft pulping the number of new phenolic hydroxyls is proportional to the degree of splitting of interunit alkylarylether bonds in lignin (Evstigneyev et al. 1991).
Based on these results, a method was developed to calculate the number of such bonds in dissolved and residual lignin at any given moment of pulping. It is based on the balance equation connecting the number of OHphen and the number of the said bonds in, on the one hand, dissolved and residual lignin and, on the other hand, in native lignin. The interunit linkage splitting releases the phenolic groups:

At any moment of the pulping,
where QOH,d is the number of free phenolic hydroxyls in dissolved lignin, QOH,r is the number of free phenolic hydroxyls in residual lignin, QOR,d is the number of bound phenolic hydroxyls in dissolved lignin, QOR,r is the number of bound phenolic hydroxyls in residual lignin and ∑Q is constant.
where QOH,i is the number of free phenolic hydroxyls in native (initial) lignin and QOR,i is the number of bound phenolic hydroxyls in native (initial) lignin.
In Eq. (4), all the values are numbers of the bonds and groups in lignin (mM) but not their contents. QOH,i, QOH,d and QOH,r values were calculated based on the analytical data on the quantities of initial, dissolved and residual lignins and content of phenolic hydroxyls in these lignins. All the data are given for 100 g o.d. spruce wood containing 27.2 g lignin. For pine wood, Gellerstedt’s experimental data (Gellerstedt and Lindfors 1984a, b; Robert et al. 1984) were utilized. Figure 6a gives a summary of data for OHphen (QOH). It should be noted that had demethylation contributed significantly to the mechanism of the release of free phenolic hydroxyl groups, the slope in the QOCH3–A graph would have the opposite direction while the same value as the one in the QOH–A graph at the same stage of pulping. Meanwhile, in all stages of pulping, the QOCH3–A graph for dissolved lignin is a straight line with the same slope.

Dependence of the quantities of free (a) and bound (b) phenolic hydroxyl groups on the degree of delignification in dissolved (1) and residual (2) lignins of kraft pulping of pine (Evstigneyev et al. 1991)
The numbers of bound phenolic hydroxyls in dissolved lignin are calculated based on the following equations:
where m is a slope of curve 1 (Fig. 6a) at the bulk stage (m = 0.98). The number of bound phenolic hydroxyls in residual lignin (QOR,r) is calculated based on Eq. (4). The constant ∑Q is measured by extrapolation of the part of curve 1 at A > 80% to A = 100%. Calculated QOR,d and QOR,r values are presented in Fig. 6b.
There is a linear correlation between the number of bonds split during pulping and the quantity of dissolved lignin (Fig. 6b, curve 2). When 1 mM (~ 1%) of the bonds is broken, 1% initial lignin is dissolved. To calculate the content of bonds from their number, one should divide all values in Eq. (4) by the quantity of lignin at the delignification stage under consideration and, using this correction, determine the number of such bonds per 100 PPU.
The described method was used to determine alkyl-O-aryl bonds content in both soluble and insoluble lignin samples of different origins (Evstigneyev et al. 2017). The content of alkyl-O-aryl bonds in native lignin of softwood (pine, spruce) is 79/100 PPU. In isolated lignin preparations, the content of these bonds decreases in the order: Freudenberg lignin (71/100 PPU) > Björkman lignin (61/100 PPU) > Pepper lignin (44/100 PPU). In dissolved alkaline lignins, a small amount of alkyl-O-aryl bonds: 36/100 PPU in soda lignin and an average of 23/100 PPU in soda–AQ lignin, kraft lignin and kraft–AQ lignin, is preserved. In residual lignin that represents the fraction of native lignin with interunit bonds, stable under the conditions of kraft cooking, 66/100 PPU of such bonds are contained. A relatively high content of alkyl-O-aryl bonds (61/100 PPU) is preserved in technical hydrolysis lignin. Currently, application of the solid-state 13C CP/MAS NMR method opens new avenues in the characterization of such chemical bonds (Evstigneyev et al. 2018).
Structure of residual lignin
The key unresolved issue in our understanding of the chemistry of alkaline delignification is the cause of the dramatic decrease in the rate of lignin dissolution at the end of pulping. As a possible cause, lignin–carbohydrate bonds (Fengel and Wegener 1989; Sarkanen and Ludwig 1971; Yamasaki et al. 1981), the effect of pores in a cell wall on lignin diffusion (Favis and Goring 1981) and lignin condensation (Sarkanen and Ludwig 1971) was suggested. The nature of residual lignin is still a subject of active discussions.
Gellerstedt (1996) noted that the conditions of kraft pulping favor lignin secondary condensation, and condensation reactions are observed in model studies; however, it is not proven if polymer components are affected by such reactions. It is important in this regard that residual lignin from kraft pulp does not contain diphenylmethane structures (Balakshin et al. 2000) and that chemicals known to condense with lignin model compounds have no effect on actual pulping of wood (Gellerstedt and Al-Adjani 1998).
Condensed structures in technical lignins (e.g., with 5-5, β-5 or 4-O-5 bonds) may originate from native lignin but not be a product of secondary reactions during pulping. Thus, based on 13C NMR, the content of condensed structures in both milled wood lignin and kraft lignin is around 55% (Kringstsad and Mörck 1983).
In residual lignin, the picture is more complicated. While generally it is believed that it contains more condensed structures, the accuracy of quantification is not quite certain (Dimmel and Gellerstedt 2010). There also are some uncertainties in the current understanding of lignin-carbohydrate bonds, e.g., if hemicelluloses or cellulose are bound to lignin, and if such bonds mostly form during pulping (Gellerstedt 1996; Helm 2000). Based on known mechanisms of lignification, it is believed that chemical bonds between hemicelluloses and lignin are intrinsicantly present in wood; however, the evidences of that are mostly indirect. Such bonds are relatively rare, by some estimates, just one per carbohydrate chain (Gellerstedt 1996).
It is not surprising then that no signal can be assigned to such bonds in the NMR spectra; see discussion in a review by Maunu (2002).
However, a review by Balakshin et al. (2008) claims, based mostly on the research by the authors, that in lignin–carbohydrate complex (LCC) samples isolated from both wood and kraft pulp, lignin–carbohydrate (LC) bonds could be identified when multi-dimensional NMR spectroscopic techniques were applied (phenyl glycoside, γ-ester and benzyl ether bonds).
Wood pulping slows down when lignin content in pulp reaches < 10%; therefore, to study the complexes, such methods as enzymatic hydrolysis (Yamasaki et al. 1981) are employed. A method was developed from separation of lignin–carbohydrate complexes (glucomannan, xylan, cellulose) based on endoglucanase treatment, dissolution of the residue in alkali and subsequent precipitation (Lawoko et al. 2004, 2005) that led to the conclusion that 90% residual lignin after kraft pulping of spruce is chemically bound to carbohydrate, with glucomannan–lignin structures predominating.
Physical association between lignin and carbohydrates through hydrogen and van der Waals interaction was also discussed (Dudkin and Gromov 1991, p. 178), but this type of interaction is likely secondary in an aqueous medium.
An alternative approach to residual lignin and other issues related to the chemistry of alkaline pulping was developed in recent publications (Evstigneyev et al. 1993, 1994, 1996). It is based on the studies of chemical transformations under the pulping conditions of isolated lignins structurally close to native lignin.
The dissolution curves of Freudenberg lignin and wood lignin under the conditions of kraft pulping are presented in Fig. 7. They both are S-shaped that reflect the three stages of delignification. At the initial ramping stage, Freudenberg lignin dissolves more slowly than native lignin, but the rate of dissolution increases significantly from 160 °C, and most of the sample moves into the solution in a short time.

Kinetics of delignification curve of spruce chips (1) versus dissolution of spruce Freudenberg lignin (2) (Evstigneyev et al. 1996)
In both samples, the process slows down in the final stage. The reasons of this phenomenon were discussed above, and lignin–carbohydrate bonds are mentioned as one of the proposed causes (Yamasaki et al. 1981). Freudenberg lignin contains almost no carbohydrates and is not topologically hindered; therefore, this observation does not support the hypotheses that lignin–carbohydrate bonds or cell pores play any significant role in the kinetics of delignification. More likely, the primary factor affecting the kinetics is changing reactivity of lignin, i.e., easiness in intermolecular fission releasing free phenolic groups.
Close similarity of kinetic curves illustrated in Fig. 7 allows us to consider Freudenberg as an adequate lignin model in wood pulping studies.
FTIR data are consistent with this theory: The spectra of dissolved kraft pulping of wood and Freudenberg lignin are almost identical (Evstigneyev 2001). The difference in the slopes of kinetic curves at the bulk and final stages of pulping (Fig. 7) can be explained by the absence in Freudenberg lignin of carbohydrates, which decrease the pH of the liquor. Besides, Freudenberg lignin does not contain easily hydrolysable fraction of native lignin that is lost when the lignin is separated from wood. It also explains some differences in delignification curves at the initial stage of pulping.
FTIR studies of residual Freudenberg lignin after kraft pulping demonstrated the same bands as in dissolved lignins; however, relative band intensities varied dramatically. The bands at 2924–2934 (CH3, CH2) and 1369–1375 (CH3) cm−1 in residual lignin are twice as high as in dissolved one. The intensities were normalized to the aromatic ring; therefore, the difference indicates a higher content of aliphatic groups in residual lignin (Evstigneyev 2001).
Based on own experimental and literature data on the structure of residual lignin available to date, it might be suggested that the reason of a decreasing rate of delignification at the final stage of wood alkaline pulping is the absence of hydroxyl groups adjacent to β-O-4 bonds in some of the side chains of residual lignin:

The β-O-4 bond in such structures is stable toward liquor reagents because it is α- and γ-aliphatic hydroxyl groups that facilitate splitting of the bond in alkaline media (Gierer and Ljunggren 1983).
Indeed, structures with aliphatic side chains were found in native lignin (Table 1) and among products of kraft pulping of spruce wood (Gierer and Lindenberg 1980) and birch (Niemelä 1990). A dimer containing β-O-4 bond in its aliphatic side chain is mentioned as one of the products of kraft pulping of softwood (Rinaldi et al. 2016).

It was found that a β-O-4 dimer with γ-OH functionality is relatively stable under the conditions of kraft pulping; this suggests that it is the α-OH group that is most important for easy β-O-4 splitting. Residual lignin contains a noticeable quantity of β-O-4 structures (Maunu 2002).
Structure of native lignin
Currently, no method can tell us the MM of lignin in wood. Moreover, even the theoretical definition of the lignin MM in wood is not quite certain because, historically, there are two competing views on the native lignin topology. One concept treats lignin as an infinite three-dimensional network (Sarkanen and Ludwig 1971), while the other considers it as a branched molecule of a relatively small size (Shorygina et al. 1976). In early works of Freudenberg, lignin macromolecule was described as one of a relatively small size but branched and essentially three-dimensional (Freudenberg 1965). On the other hand, if lignin is an indefinite three-dimensional network, the very concept of a molecule does not apply as it is always the case with polymer networks (Irzhak et al. 1979, p. 7).
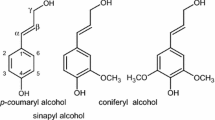
Another discrepancy in the current understanding of lignin is the degree of the order in native lignin as related to its biosynthesis. Specific traits of lignin biosynthesis are usually considered causing a multitude of interunit bonds and irregularities in lignin structure (Fengel and Wegener 1989; Ralph 2010; Vanholme et al. 2010; Shi et al. 2010; Weng and Chapple 2010; Bonawitz and Chapple 2010; Magalhaes et al. 2010; Zhao and Dixon 2011; Liu 2012; Mottiar et al. 2016; Rinaldi et al. 2016). Phenoxyl radicals are generated from lignin precursors (coniferyl and related unsaturated alcohols) by dehydrogenase and oxidase enzymes. Radical recombination is random, and the spin density in the radical is a primary factor in determining the type of a forming bond. Thus, enzymatic control is absent in building a lignin macromolecule that yields an irregular polymer. This is quite different from the process of polysaccharide biosynthesis.
However, some published data indicate a degree of regularity in isolated lignin samples. Thus, Karmanov (2004) suggests a nonlinear self-organization process in lignification of cell walls that results in the formation of an ordered structure of a fractal type. Computer modeling suggests a spiral shape of linear parts of a lignin macromolecule (Faulon 1994; Faulon and Hatcher 1994) that is quite typical of biopolymers.
MMD curves for hardwood and softwood lignins were obtained using a MALDI (matrix-assisted laser desorption ionization) technique that provides the most accurate absolute mass values to date (Metzger et al. 1992). Average MM values (Mn) were found to be 2600 ± 100 in hardwood and 2300 ± 100 in softwood lignins; own measurement for Björkman spruce lignin by MALDI was 2090 (Evstigneyev et al. 2016). The fine structure of the mass spectra suggests the presence of tetramer (in hardwood) and trimer (in softwood) oligomers (Srzic et al. 1995).
Some regularity in lignin was also observed at a supermolecular level. A scanning tunneling microscopy study of dehydrogenative polymerization of coniferyl alcohol (model lignification) suggested that the polymer has a modular structure (about 20 PPU in one module) associated in supermodules (larger particles consisting of about 500 PPU), which, in turn, form a web (Radotic et al. 1994). Separated lignins form monomolecular films on the surface of water that is sometimes considered as a (vague) indicator of possible self-organization (Luner and Roseman 1986).
Raman spectroscopy indicates orientation of lignin in the cell wall at a molecular level (Atalla and Agarwal 1985; Agarwal and Atalla 1986). It seems to be illogical that all wood constituents but lignin are genetically programmed and formed under enzymatic control; only lignin is considered as random (Ralph et al. 2004).
A modern theory of the structure of native lignin is presented by Gellerstedt and Henriksson (2008), where lignin is treated as a mixture of branched and linear macromolecules. Crestini et al. (2011) and Crestini (2014) consider lignin as just a linear polymer. Linear macromolecules in a low molecular mass fraction of eucalyptus lignin were proven by mass spectrometry (Evtuguin and Amado 2003).
The content of alkyl-O-aryl bonds in native lignin of softwood (pine, spruce) is 79/100 PPU (Evstigneyev et al. 2017). A model of alkylarylether bonds distribution in native lignin is proposed that differ in their chemical reactivities under the conditions of wood alkaline pulping (Fig. 8).

Model of alkylarylether bond distribution in native softwood lignin (Evstigneyev 2012)
According to this model, native lignin in wood consists of three fractions F1 (23%), F2 (70%) and F3 (7%) that differ in chemical reactivities of their alkylarylether bonds following the sequence: F1 > F2 ≫ F3. In the process of wood pulping, fractions F1 and F2 dissolve, thus yielding soluble lignin, while most of the F3 fraction remains yielding residual lignin. The idea of such three fractions in native lignin is consistent with known differences in the kinetics of the main stages of pulping: initial, bulk and final (Wilder and Daleski 1965; Kleinert 1966; Ljunggren 1980; Kondo and Sarkanen 1984; Chiang et al. 1990; Evstigneyev et al. 1996).
The authors’ studies support the idea that the F3 fraction of lignin is responsible for the known fact of virtual impossibility of complete lignin removal by cooking only; the subsequent bleaching stage is always needed to approach this goal. Chemical reactions of conventional cooking processes (kraft, sulfite) are predominantly nucleophilic, while traditional bleaching sequences involve electrophilic reactions (Fengel and Wegener 1989). Substrates for the nucleophilic attack resulting in breaking the polymer chain are mostly α-positions in lignin, and these positions are not chemically active in the F3 lignin fraction. Thus, under the cooking conditions, no active intermediates are formed in this fraction, and therefore, no bond splitting, lignin functionalization nor subsequent dissolution occurs. On the other hand, reaction centers for electrophilic reactions of residual lignin are, predominantly, aromatic rings that yield dicarboxylic acids upon reactions with oxidative bleaching chemicals.
Redox properties of lignin and delignification catalysts
Polarography can hardly be considered as a popular method of lignin analysis. To assess its usefulness and limitations, a systematic study of electrochemical reactions of lignin and its model compounds (more than 70 structures) was performed in Evstigneyev et al. (1992b, 1999, 2004) and Evstigneyev (2009). A detailed analysis of electrochemical reactions of lignin and lignin model compounds is provided in a review (Evstigneyev 2014).
A diagram in Fig. 9 based on the results obtained using monomer and dimer lignin model compounds shows structure–reactivity relationships in lignin structures in reductive electrochemical reactions.

Diagram of reduction potentials of polarographically active functional groups and bonds in lignin model compounds (DMSO/tetrabutylammonium perchlorate) (Evstigneyev et al. 2004)
A comparison between model compounds and chemically modified (NaBH4, LiAlH4, CH2N2, (CH3O)2SO2) lignin samples allowed ascribing peaks in differential pulse polarograms of lignin to certain bonds and functional groups (Fig. 10).

Diagram of reduction potentials of polarographically active functional groups and bonds in lignin (Evstigneyev et al. 2004)
Only carbonyl groups and conjugated double bonds in diphenyl structures are polarographically active. Polarography differentiates aldehyde and ketone groups and can, therefore, be used in studies on lignin reactions involving such structures.
Polarograms of soda and soda–AQ lignins are shown in Fig. 11. From the mechanistic point of view, the most interesting are two overlapping peaks at Ep − 1.98 B and − 2.07 V corresponding to reduction of carbonyl α-groups.

Polarograms of soda–AQ (1) and soda (2) lignins (DMSO/tetrabutylammonium perchlorate (3)) (Evstigneyev et al. 1992a)
The content of these groups in soda–AQ lignin is much higher than in soda lignin (Fig. 12). It reaches the maximum during a transition from the initial to bulk stage of pulping and decreases in the final stage. These results are discussed in more detail in connection to the mechanism of AQ lignin reactions (Evstigneyev 2014).

Dependence of α-carbonyl groups content on the degree of delignification in dissolved lignin (A, %) of soda (•) and soda–AQ (+) pulping of spruce wood (Maiyorova et al. 1995)
The data suggest that AQ may affect the pulping process in two ways. First, it involves lignin in reactions that do not take place without AQ present; this results in the formation of oxidized groups (compare polarograms in Fig. 11). Second, AQ accelerates reactions that take place without the catalyst present but at a slower rate (β-alkylarylether bond splitting).
Considering the redox mechanism of chemical transformations of AQ and others efficient during alkaline pulping (Shevchenko and Deineko 1983), one could expect that their activities depend on their reduction potentials, and there should be a correlation between redox properties of AQ and lignin. There is still no straightforward answer to the question about such dependencies. However, as a general trend it may be noticed increased activity with reduction of the potential, and the range Eo = 0.1 − 0.25 V seems to be optimal (Nomura and Nakamura 1978; Algar et al. 1979). Lindenfors (1980) reported a linear correlation between the efficiency and potential in small series of naphthoquinones, but it was not applicable to AQ derivatives. Besides, naphthacenequinone (Eo ~ 0.1 V) is less active than AQ (Eo ~ 0.15 V), while the opposite could be expected based on the potentials only.
The Eo (pH 0) values of a series of quinones corresponded to their catalytic activities in soda pulping (Nomura and Nakamura 1978; Lindenfors 1980). It could be expected that redox parameters of quinones in an alkaline medium better correlate with the efficiency. However, Eckert and Amos (1980, 1981) demonstrated in this case no correlation. They determined polarographically reduction potentials of fluorenone and structurally similar but inactive in pulping compounds. Based on these data, they suggested that any diarylketone can be considered as a potential catalyst if such a ketone reduces at E < − 1.2 V, is stable under the pulping conditions and bears some structural features. Later, Eckert and Amos (1982) and Amos and Eckert (1982) emphasized the effect of hydrophilicity on the catalytic activity. Werthemann et al. (1981) suggested the potential range from − 0.55 to − 1.05 V to be characteristic of active catalysts and emphasized the effect of xylophilicity/hydrophilicity on the activity (Werthemann 1981).
Thus, a linear correlation between the redox potential and catalytic activity hardly exists. On the other hand, there is clearly a range of potentials corresponding to the catalytic activity that is still not unambiguously explained. In addition, the role in delignification of ASQ, an alkaline-soluble product of one-electron reduction of AQ, is not quite clear; it may participate in lignin-splitting electron transfer mechanisms, but no direct evidence was provided. Solubility and stability under the pulping conditions are also critically important in the potential catalyst assessment.
Polarographic data from the authors’ work (Evstigneyev and Shalimova 1985a, b) are summarized in Table 8. Benzoquinone and naphthoquinone are unstable in an alkaline medium (50% DMFA, 0.55 N NaOH) at room temperature. Their polarograms show several reduction peaks that change their position and height upon re-recording, likely due to reactions of the quinones with hydroxide anions (Pedersen 1973). Phenanthrenequinone reduces at 0.59 V and decomposes under pulping conditions.
The substituent effect on the reduction mechanism and the value of reduction potential is clear in a series of AQ derivatives. The compounds presented in Table 8 can be divided into two groups based on one- or two-stage reduction. AQ, its amino-, chloro-, sulfo- and alkyl derivatives as well as compounds 27–30 (mixed substituents) reduce in two subsequent one-electron stages. Reduction of AQ in aqueous alkaline media containing organic solvents is proven to proceed via ASQ anion radical (Heifetz and Bezugly 1969):
Reductive potentials of AQ derivatives are consistent with known electron effects of the substituents. Electron–donor amino, ethyl and methyl groups move the potential to more negative values, while electron–acceptor chloro and sulfo groups move it in the opposite direction.
Most of AQ derivatives (4–12, 15, 16, 27–30; 1,5-dichloro-AQ is an exception) reduce reversibly that is confirmed by their cyclic voltamperograms (Fig. 13). Our interest in the electrochemical process is driven by the similarities between polarographic and redox potentials in reversibly reduced compounds so that polarographic values can be used in the assessment of redox properties in alkaline media (Heifetz and Bezugly 1969). Reversibility criterium was used in determining the character of the process (Bond 1980).

Cyclic voltamperogram of AQ in 0.55 N NaOH in 50% DMFA (Evstigneyev and Shalimova 1985a)
Sulfonate derivatives of AQ (15, 16, 27) were found to be unstable under the pulping conditions that is evidenced by the measured post-pulping potentials. 2-Sulfo-AQ and 1,8-disulfo-AQ are completely transformed, while 1-amino-2-sulfo-AQ only partially. AQ itself as well as its amino-, chloro- and alkyl derivatives (4–12, 29) is stable; the post-pulping polarograms are essentially the same.
All the studied chemicals to some extent accelerate delignification (Table 9). Figure 14 shows correlation between the redox potential and lignin content in pulp after pulping.

For the compounds that are reduced in two stages, the second peak potential is applied because it is widely accepted that the active species reacting with lignin during pulping is AHQ dianion (Shevchenko and Deineko 1983). In the chemicals characterized by reversible reduction, the polarographic potentials reflect both reductive and oxidative properties of the species.
As can be seen in Fig. 14, compounds 4, 6–9, 11 and 12 are the best catalysts, and in all of them, electrochemical reduction proceeds through two one-electron reductive steps, i.e., ASQ intermediate. These compounds are stable in both oxidized and reduced form and are to some extent soluble (1–10 × 10−5 mol/l). Lesser activity of compounds 5, 10, 15, 16, 27–30, which reduction is similar mechanistically, can be ascribed to their lesser stability.
Based on the described results, the following general requirements can be formulated toward potential quinone catalysts of soda pulping: Electrochemical reduction proceeds in two one-electron, fully reversible stages: the potential of the second stage being close to − 0.85 V; stability of the compound in its reduced and oxidized forms under the conditions of soda pulping; sufficient solubility (1–10 × 10−5 mol/l).
Applicability of the developed criteria to other classes of organic chemicals was tested on two additives described in the literature (Shevchenko and Deineko 1983), phenazine and fluorenone (31 and 32 in Table 9). They both are stable under pulping conditions and moderately soluble in aqueous alkali. The polarogram of phenazine contains only one peak, and the second reduction stage of fluorenone is irreversible and characterized by a very negative potential (− 1.29 V). Thus, they do not meet all the listed criteria. Not surprisingly, they are inferior catalysts to AQ.
The trends in activities of redox catalysts as dependent on their redox properties, stability and solubility as found in the studies of soda pulping are also fully applicable to kraft pulping that is confirmed by experimental data reported in Evstigneyev and Shalimova (1987).
Lignin solubility
All delignification technologies for plant biomass can be reduced to a chemical functionalization, i.e., adding hydrophilic groups to the macromolecule that leads to dissolution of the polymer. It is critically important to fully understand what factors determine solubility of lignin in process media.
The popular Schuerch method for evaluation of lignin solubility is based on the empirical correlation between, on the one hand, the Hildebrand parameter and power of hydrogen bonds in the solvent and, on the other hand, lignin solubility. This method is applicable to organic solvents only and gives just a qualitative estimate such as soluble, partially soluble or insoluble (Sarkanen and Ludwig 1971). Later, a Hildebrand parameter was supplemented with Hansen parameters that reflect nonpolar interactions, dipole–dipole interactions and hydrogen bonds (Hansen and Björkman 1998). However, these solubility parameters cannot reliably describe interaction of organic solvents with wood and its components.
A thermodynamic study on lignin solubility in dioxane and acetone demonstrated that the said solvents interact with acidic groups in the polymer and the quantity of available for solvation groups is more important for solubility than the size of the macromolecule (Pilyugina et al. 1974). In aqueous media, a thermal effect of interaction between lignin and alkali is determined by the content of phenolic groups (Andreyev et al. 1974). All commercial processes to date are based on aqueous media and the most popular of them (kraft, soda) on aqueous alkali. In recent years, there has been a lot of interest in the literature on organosolv pulping; however, the latter remains an academic exercise with no prospect of large-scale commercialization (Hergert and Pye 1993).
In studies by Evstigneyev (2010, 2011), solubility of lignin in aqueous alkali was quantified.
Solubility of kraft spruce lignin in aqueous sodium hydroxide is illustrated in Fig. 15. In the figure,
where S is the mass part of dissolved lignin; Ls is the mass of dissolved lignin, g; and Li is the initial quantity of lignin, g;
where f is the part of titrated phenolic hydroxyl groups in lignin; OH− is the quantity of hydroxide anions in the solution, mmol; and OHphen is the quantity of phenolic hydroxyls in lignin in the solution, mmol.

Dependence of solubility of kraft lignin of spruce in aqueous solution of sodium hydroxide on the quantity of titrated phenolic hydroxy groups (Evstigneyev 2010)
Figure 15 shows that full dissolution of lignin is achieved at f = 1, i.e., full ionization of all phenolic hydroxyl groups (1:1 phenolic hydroxyl–alkali stoichiometry). Dependence of lignin solubility on f value is an S-shaped curve with three distinct areas: At f = 0–0.2, the tangent of the slope is 1.25; at f = 0.2–0.6, it reaches 1.83; at f = 0.6–1.0, it is < 0.05. The cross-section points of the three linear parts of the curve correspond to S values 0.25 and 0.98 (shown). Thus, the analyzed sample consisted of three fractions of 25, 73 and 2% that are different in their solubility in alkali.
The first fraction dissolves at pH 7.65–9.04, the second one at pH 9.04–9.82 and the third one at pH 9.82–11.43. It should be noted that a glass electrode was not stable when measuring in lignin suspension (500 mg lignin in 20 ml solution of NaOH). Therefore, the pH values were calculated based on the equation for titration of a weak acid with strong base (Hulanicky and Masson 1987):
where a constant Ka = 2.25 × 10−10 is calculated based on the value pKa = 9.65 for kraft lignin (Kurzin et al. 1997).
Equation (9) can be written for the f parameter
at f = 0.5, i.e., pH = pKa, it can be presented as
Lignin solubility depends on f (Fig. 15); then,
Equation (12) shows that lignin solubility depends on the pH of the solution and pKa of acidic groups in lignin. It would be difficult to present this dependence through the entire range of f values for the curve in Fig. 15 as a simple equation because of the different slopes in the three areas. Therefore, it is more convenient to use S value at f = 0.5 as a parameter characterizing solubility of lignin samples in aqueous alkaline media; the physical sense of this parameter is the solubility of the sample, in which half of the phenolic hydroxyls is titrated. Then, there is a need for a method that would expediently and accurately determine the quantity of such groups.
Two methods are currently used to measure phenolic hydroxyls. Mänsson (1983) method is based on the pyrrolidine aminolysis reaction of acetylated lignin. An alternative method is based on the bathochromic shift of spectral bands of phenols upon ionization of their hydroxyl groups (Zakis 1987).
The method proposed in Evstigneyev (2011) combined elements of both techniques and included differential spectroscopy of a lignin sample with a known hydroxyl content determined by aminolysis. The band at 250 nm is symmetrical, has little overlap and, therefore, is suitable for making a calibration curve.
First, the calibration curve was made in coordinates optical density–lignin concentration. Then, based on the content of phenolic hydroxyls in the sample, it was re-done in coordinates optical density—content of phenolic hydroxyls. Standard deviation was found to be ± 0.13 mmol/g.
Dependence of lignin solubility on the molecular mass is shown in Fig. 16. Solubility of all polymers decreases with mass; lignin though has some specifics. It was found thermodynamically that the quantity of groups available for solvation is more important than the molecular mass in determining lignin solubility (Pilyugina et al. 1974).

Solubility of fractions of dioxane lignin of spruce versus molecular mass (Evstigneyev 2011)
In several fractions of dioxane lignin, the content of phenolic hydroxyls was measured using the method described above. It was found to be decreasing with the increase in MM: 3.40; 3.15; 2.95; 2.05 mmol/g (62, 58, 54, 38 per 100 PPU). Lignin solubility was found to be proportional to the number of hydrophilic groups per PPU in the macromolecule.
A minimum ratio required for solubilization of lignin can be determined from the solubility–phenolic hydroxyl content plot. Linear extrapolation gives the ratio ~ 1.7 mmol/g or 31/100 PPU. For comparison, as an independent test, changes in phenolic hydroxyls in dissolved and residual lignin after kraft pulping of softwood and Freudenberg lignin of spruce were analyzed (Evstigneyev et al. 1996). Through the full length of the pulping process, the content of OHphen in residual (solid phase) lignin did not exceed 30/100 PPU. In dissolved lignins, the minimum OHphen content was 40/100 PPU. Thus, the extrapolated value of 31/100 PPU is consistent with the actual solubility data. It is applicable to both lignin in wood and separated samples such as dioxane and Freudenberg lignins.
Temperature-solubility dependence Solubility of lignin increases linearly proportional to temperature, with a rather low temperature coefficient: Sf=0.5/1 °C = 0.004; it dissolves completely at 100 °C and is rather soluble even at room temperature.
Dependence of lignin solubility on the hydromodule is illustrated in Fig. 17. It is changing stepwise, the solubility decreasing with hydromodule increasing. Maximum solubility is reached at H = 5. This can be explained considering that in all experiments the solubility was measured at f = 0.5, i.e., the same ratio between hydroxide anions in the liquid phase and phenolic hydroxyls. There is more water in the system with a higher hydromodule, and sodium hydroxide becomes more dilute. In the range of values used in this study H = 5, 10, 20, 30, 40, 50, 60, concentration of the alkali is 0.4; 0.2; 0.1; 0.07; 0.05; 0.04; 0.03 mol/l, respectively. Thus, solubility of lignin depends on absolute concentration of the base. The discussed results are consistent with the understanding of lignin dissolution as fractionation based on the acid–base properties of dissolving fractions.

Solubility (S) of kraft lignin versus hydromodule (H) (Evstigneyev 2011)
Dependence of lignin solubility on the ionic strength was studied in the range of sodium chloride concentrations 0.01–0.4 mol/L. The effect is minor, and up to 0.1 mol/L, lignin solubility does not change (Sf = 0.5 = 0.7), and at higher concentrations, it decreases linearly to Sf = 0.5 = 0.6.
Lignin solubility in the studied samples is presented in Table 10, which clearly shows that in all lignin samples solubility increases with the quantity of phenolic hydroxyl groups.
Considering the conditions of sample preparation, it is obvious that it also corresponds to decreasing molecular mass of studied lignins. Thus, in a series Björkman—soda–soda/AQ–kraft–kraft–AQ lignin, an average MM is 2088, 1593, 1556, 1587 and 1560, respectively (vapor osmometry, Table 7). As already mentioned, it means that lignin solubility is determined by the ratio of hydrophilic groups to PPU in the macromolecule. Freudenberg lignin (22 OHphen/100 PPU) is insoluble under the conditions because the minimum value is 31 OHphen/100 PPU.
The data presented in Table 10 can be used for calculating solubility of a lignin sample under certain conditions. Sf = 0.5 values can easily be translated into solubility (g/L) based on the following equation:
where H is a hydromodule, L/g. For example, solubility of kraft lignin at H = 40 is 22.5 g/L. Maximum solubility for lignin in aqueous alkali solutions can be estimated based on the values Sf = 0.5 = 1 and H = 5 as 200 g/L.
Generally, solubility for lignin in aqueous alkali solutions in g/L is determined by the following equation:
where Sf is the lignin solubility at a certain f value and Hf is the hydromodule at that f value, L/g.
The solubility for lignin in aqueous alkali solutions is determined by the quantity of phenolic hydroxyls per PPU, and the minimum value at which lignin starts dissolving is 31 OHphen/100 PPU.
Lignin valorization
Conversion of lignin into value-added products is covered in several fundamental reviews published in recent years (Ragauskas et al. 2014; Doherty et al. 2011; Laurichesse and Averous 2014; Thakur et al. 2014; Sen et al. 2015; Ten and Vermerris 2015); here, major current directions in the area are listed, with brief explanations.
Chemical transformation of lignin into value-added products is considered a priority in developing a cost-efficient biorefinery; without efficient lignin utilization, the biorefinery cannot be economical. Selective cleavage of alkylaryl ether interunit bonds, mostly of the β-O-4 type, that yields monomeric aromatic products is considered as one of the most promising avenues in this development (Rinaldi et al. 2016; Galkin and Samec 2016; Kärkäs et al. 2016). The yield of monomeric products of catalytic hydrogenolysis of native (wood) lignin is close to theoretically possible: 23% in softwood and 51% in hardwood lignin (Evstigneyev 2018).
Valorization of hydrolysis lignin (HL) is a part of development of cost-efficient bioethanol and biobutanol production. Nowadays, common raw materials in this process are feedstock such as corn, sugar beet, sorghum, cassava and sugar cane, which could alternatively be used in food production. It seems logical to expand using wood waste instead. One of the main problems in this field, when wood is used, is the formation of a large amount of HL waste. One ton of coniferous wood yields 160–175 kg (45–49 gallons) of ethanol and 350–400 kg of the lignin by-product. Significant research efforts to find applications for HL so far did not yield results that would change the market situation; currently, there is little demand for HL and its derivatives (Rabinovich 2010).
Analytical characterization of HL and other technical lignins is presented in Table 11.
HL is not soluble (in any known solvents); therefore, its MM is not measured. Comparing with other known lignins, HL carries much less reactive functionalities such as carboxyl or phenolic hydroxyl groups, and it also is more condensed (cross-linked) and, therefore, less chemically reactive (Evstigneyev 2013). A method of oxidation of HL with hydrogen peroxide under acidic conditions was developed (Evstigneyev 2013; Evstigneyev et al. 2015, 2016) that yields oxidated hydrolysis lignin (OHL) containing 8.9% carboxylic groups (Table 11) in muconic acid-type structure. The solubility of lignin in dilute alkali increases from 16.2 to 91.8% upon such transformation (Evstigneyev et al. 2016), the values of \(\bar{M}_{n}\) being comparable to \(\bar{M}_{n}\) of kraft and organosolv lignins.
Physicochemical properties of OHL suggest some new avenues of waste lignin utilization. Thus, for this product, methylene blue sorption capacity is 97.8 mg/g that is twice as much as the one of a known enterosorbent Polyphepan™ (Rabinovich 2010; Evstigneyev et al. 2015).
OHL also is a more efficient surfactant than lignosulfonates (Evstigneyev et al. 2015). It could be important because the market demand for lignosulfonate-based surfactants currently exceeds the manufacturing capacity. Production of lignosulfonates that are side products of sulfite pulping is in decline for many years (Lora 2008). Meanwhile, their applications as surfactants are quite broad—they are used as dispersants, emulsifiers, floatation agents, additives in mining solutions, concrete, washing compositions, etc. Considering higher activity and similar structural features, using OHL as a lignosulfonate substituent in surfactant compositions seems to be a natural suggestion. In Russia, the stock of HL is estimated to be around 95 million tons (Rabinovich 2009).
Conclusion
Basic principles of a functionalization theory of delignification that summarizes the data discussed in this review and explains how structure, chemical reactivity and solubility of lignin are interconnected, are as follows.
Native lignin in wood consists of three fractions F1 (23%), F2 (70%) and F3 (7%), which are different in chemical reactivity of their alkylarylether bonds: F1 > F2 ≫ F3. During the pulping, fractions, F1 and F2 move into the solution giving rise to dissolved lignin, while most of F3 fraction stays with the pulp and becomes residual lignin.
Solubility of lignin in the pulping liquor is determined by the content and pKa of acidic functional groups, and the pH of the solution.
At the bulk stage of pulping, the rate of delignification is proportional to the rate of lignin functionalization.
Cooking additives for accelerated wood delignification in the alkaline process should satisfy certain requirements based on our knowledge of chemistry of the process: some similarity in redox properties between the catalyst and lignin, stability under cooking conditions, in both oxidized and reduced forms, and sufficient solubility.
Altogether, the data on wood delignification by different chemical methods unequivocally indicate that in all the processes, delignification is related to lignin functionalization, i.e., formation of functional groups providing solubility in the process liquors.
References
Agarwal UP, Atalla RH (1986) In situ Raman microprobe studies of plant cell walls: macromolecular organization and compositional variability in the secondary wall of Picea mariana. Planta 169(3):325–332
Algar WH, Farrington A, Jessup B, Vanderhock N (1979) The mechanism of soda-quinone pulping. Appita 33(1):33–37
Amos LW, Eckert RC (1982) Influence of methylation on the solubility and efficiency of anthraquinone in soda pulping. In: Proceedings of canadian wood chem symposium Niagara Falls, NY, pp 7–10
Andreyev VI, Vasilieva TM, Grigoriev GP, Vlasova KI (1974) Study on interaction of dioxane lignin with aqueous solutions of sodium hydroxide by calorimetry. In: Chemistry and use of lignin, Zinatne, Riga, pp 129–133
Asikkala J, Tamminen T, Argyropoulos DS (2012) Accurate and reproducible determination of lignin molar mass by acetobromination. J Agric Food Chem 60:8968–8973
Atalla RH, Agarwal UP (1985) Raman microprobe evidence for lignin orientation in cell walls of native woody tissue. Science 227(4687):636–638
Balakshin MYu, Capanema EA, Chen C-L, Gratzl JS, Gracz H (2000) The use of 2D NMR spectroscopy on structural analysis of residual and technical lignins. In: Proceedings of 6th European workshop on lignocellulosics and pulp, Bordeaux, France, pp 11–14
Balakshin MYu, Capanema EA, Chang H (2008) Recent advances in the isolation and analysis of lignins and lignin-carbohydrate complexes. In: Hu TQ (ed) Characterization of lignocellulosic materials. Blackwell Publishing Ltd, Oxford, pp 148–170
Bonawitz ND, Chapple C (2010) The genetics of lignin biosynthesis: connecting genotype to phenotype. In: Campbell A, Lichten M, Schupbach G (eds) Annual review of genetics. Annual reviews 44: 337–363
Bond AM (1980) Modern polarographic methods in analytical chemistry. M Dekker, New York
Brunow G, Miksche G (1976) Some reactions of lignin in kraft and polysulfide pulping. Appl Polym Symp 28:1155–1168
Brunow G, Poppius KA (1981) Kinetic study on the mechanism of β-O-4 ether cleavage in soda-anthraquinone pulping. Paperi ja Puu 63(12):783–785
Chiang VL, Kolppo K, Stokke DD (1989) Structure changes of lignin in kraft and acid sulphite delignification of western hemlock. In: Proceedings of international symposium on wood and pulping chemistry, Raleigh, NC, pp 593–597
Chiang VL, Yu J, Eckert RC (1990) Isothermal reaction kinetics of kraft delignification of Douglas-fir. J Wood Chem Technol 10(3):293–310
Crestini C (2014) Lignin structure: a revisitation of current paradigms through NMR analysis. In: Proceedings of 13th European workshop on lignocellulosics and pulp, Seville, Spain, pp 59–62
Crestini C, Melone F, Sette M, Saladino R (2011) Milled wood lignin: a linear oligomer. Biomacromolecules 12(11):3928–3935
Dimmel D, Gellerstedt G (2010) Chemistry of alkaline pulping. In: Heitner C, Dimmel DR, Schmidt JA (eds) Lignin and lignans. Advances in chemistry series. CRC Press, Boca Raton, pp 349–391
Dimmel DR, Schuller LF (1986a) Structural/reactivity studies (I): soda reactions of lignin model compounds. J Wood Chem Technol 6(4):535–564
Dimmel DR, Schuller LF (1986b) Structural/reactivity studies (II): reactions of lignin model compounds with pulping additives. J Wood Chem Technol 6(4):565–590
Doherty WOS, Mousavioun P, Fellows CM (2011) Value-adding to cellulosic ethanol: lignin polymers. Ind Crops Prod 33(2):259–276
Dudkin MS, Gromov VS (eds) (1991) Hemicelluloses. Zinatne, Riga
Eckert RC, Amos LW (1980) Catalysis of alkaline pulping by fluorenone. Tappi J 63(11):89–93
Eckert RC, Amos LW (1981) Prediction of chemical structures leading to catalysis of alkaline pulping. Tappi J 64(6):123–124
Eckert RC, Amos LW (1982) Influence of hydrophilicity on the delignification efficiency of anthraquinone derivatives. J Wood Chem Technol 2(1):57–71
Evstigneyev EI (2001) Structural changes in lignin during alkaline wood pulping and their effect on the rate of delignification and on properties of pulp. D.Sc. thesis, St-Petersburg Forest Technical Academy, St-Petersburg, Russia
Evstigneyev EI (2003) Basic theory of alkaline pulping. In: Pulp and paper technology. Politechnica, St-Petersburg, Russia 2(2): 7–16
Evstigneyev EI (2009) Chemical reactivity of lignin in reactions of electrochemical reduction. In: IV national conference on progress in chemistry and chemical technology of plant biomass, Barnaul, Russia, vol 1, pp 80–83
Evstigneyev EI (2010) Specific features of lignin dissolution in aqueous and aqueous-organic media. Russ J Appl Chem 83(3):509–513
Evstigneyev EI (2011) Factors affecting lignin solubility. Russ J Appl Chem 84(6):1040–1045
Evstigneyev EI (2012) On the structure of native lignin. Bull St-Petersburg Forest Technical Academy (Izvestiya SPBLTA), vol 198, pp 164–172
Evstigneyev EI (2013) Oxidation of hydrolysis lignin with hydrogen peroxide in acid solutions. Russ J Appl Chem 86(2):258–265
Evstigneyev EI (2014) Electrochemical reactions of lignin: a review. Khimija Rastitel nogo Syr’ja. Chem Plant Resour 3:5–42
Evstigneyev E (2018) Selective depolymerization of lignin: assessment of the yield of monomeric products. J Wood Chem Technol 1:2–3. https://doi.org/10.1080/02773813.2018.1500607
Evstigneyev EI, Rusakov AE (1990) The use of high performance liquid chromatography for studying lignin. In: Proceedings of 1st European workshop on lignocellulosics and pulp. Hamburg, Germany, pp 327–332
Evstigneyev EI, Shalimova TV (1985a) Redox properties, catalytic activity and stabilizing effect of some quinones in soda pulping. 1. Reduction potentials and solubility. Wood Chem (Riga) 1:50–54
Evstigneyev EI, Shalimova TV (1985b) Redox properties, catalytic activity and stabilizing effect of some quinones in soda pulping. 1. Effect on pulping. Wood Chem (Riga) 1:55–60
Evstigneyev EI, Shalimova TV (1987) Correlation of redox properties and catalytic activity of anthraquinone derivatives in sulfate pulping. Coll Works VNIIB VNPOBumprom, pp 27–32
Evstigneyev EI, Rusakov AE, Shalimova TV, Zakharov VI (1987a) Molecular mass distribution in lignin at various stages of soda and soda-AQ pulping. Wood Chem (Riga) 2:51–55
Evstigneyev EI, Rusakov AE, Shalimova TV, Zakharov VI (1987b) Study on changes in molecular mass distribution in spruce Björkman lignin under the conditions of soda, soda-AQ and kraft pulping. In: Abstract of 7 conference chemistry and use of lignin. Riga, Latvia (U.S.S.R.), pp 88–89
Evstigneyev EI, Maiyorova ED, Platonov AYu (1990) Alkaline delignification of wood and lignin functionalization. Wood Chem (Riga) 6:41–46
Evstigneyev E, Maiyorova H, Platonov A (1991) Lignin functionalization and reactivity in alkaline pulping. In: Proceedings of 6th international symposium on wood and pulping chemistry. Melbourne, Australia, vol 2, pp 131–138
Evstigneyev E, Maiyorova H, Platonov A (1992a) Lignin functionalization and the alkaline delignification rate. Tappi J 75(5):177–182
Evstigneyev E, Shevchenko SM, Apushkinsky AG, Semenov SG (1992b) Electrochemistry of lignin model p-quinone methides. Ligno-cellulosics: science, technology. development and use. Ellis Horwood, New York, pp 657–670
Evstigneyev E, Maiyorova H, Kurzin A, Platonov A (1993) About native lignin model. In: Proceedings of 7th International symposium on wood and pulping chemistry. Beijing, China, vol 3, pp 25–31
Evstigneyev E, Kurzin A, Platonov A, Maiyorova H (1994) About residual lignin nature. In: Ext Abstract 3rd European workshop on lignocellulosics and pulp. Stockholm, pp 232–233
Evstigneyev EI, Kurzin AV, Platonov AYu, Maiyorova ED (1996) Freudenberg lignin as a model for studying alkaline delignification of wood. Zhurn Prikl Khim 69(1):148–153
Evstigneyev E, Maiyorova H, Platonov A (1999) Polarographically active structural fragments of lignin. I. Monomeric model compounds. J Wood Chem Technol 19(4):379–407
Evstigneyev E, Shevchenko S, Maiyorova H, Platonov A (2004) Polarographically active structural fragments of lignin II Dimeric model compounds and lignins. J Wood Chem Technol 24(3):263–278
Evstigneyev EI, Yuzikhin OS, Gurinov AA, Ivanov AYu, Artamonova TO, Khodorkovskiy MA, Bessonova EA, Vasilyev AV (2015) Chemical structure and physicochemical properties of oxidized hydrolysis lignin. Russ J Appl Chem 88(8):1295–1303
Evstigneyev EI, Yuzikhin OS, Gurinov AA, Ivanov AYu, Artamonova TO, Khodorkovskiy MA, Bessonova EA, Vasilyev AV (2016) Study of structure of industrial acid hydrolysis lignin, oxidized in the H2O2–H2SO4 system. J Wood Chem Technol 36(4):259–269
Evstigneyev E, Kalugina AV, Ivanov AYu, Vasilyev AV (2017) Contents of α-O-4 and β-O-4 bonds in native lignin and isolated lignin preparations. J Wood Chem Technol 37(4):294–306
Evstigneyev EI, Mazur AS, Kalugina AV, Pranovich AV, Vasilyev AV (2018) Solid-state 13C CP/MAS NMR for alkyl-O-aryl bonds determination in lignin preparations. J Wood Chem Technol 38(4):137–148
Evtuguin DV, Amado FML (2003) Application of electrospray ionization mass spectrometry to the elucidation of the primary structure of lignin. Macromol Biosci 3:339–343
Faulon JL (1994) Stochastic generator of chemical structure. 1. Application to the structure elucidation of large molecules. J Chem Inf Comput Sci 34(5):1204–1218
Faulon JL, Hatcher PG (1994) Is there any order in the structure of lignin? Energy&Fuels 8(2):402–407
Favis BD, Goring DAI (1984) A model for the leaching of lignin macromolecules from pulp fibers. J Pulp Pap Sci 10(5):139–143
Fengel D, Wegener G (1989) Wood: Chemistry, ultrastructure, reactions. Berlin-NY, Walter de Gruyter [Quoted pages refer to Russian translation: Фeнгeл Д, Beгeнep Г Дpeвecинa Xимия, yльтpacтpyктypa, peaкции Пep c aнгл M: Лecн пpoм-cть, 1988]
Freudenberg K (1965) Lignin: its constitution and formation from p-hydroxycinnamyl alcohols. Science 148(3670):595–600
Galkin MV, Samec JSM (2016) Lignin valorization through catalytic lignocellulose fractionation: a fundamental platform for the future biorefinery. Chemsuschem 9:1544–1558
Gellerstedt G (1996) Chemical structure of pulp components. In: Dence CW, Reeve DW (eds) Pulp bleaching principles and practice. Tappi Press, Atlanta, pp 93–111
Gellerstedt G, Al-Adjani WW (1998) The influence on bleachability of changes in pulping chemistry. In: Proceedings of 5th European workshop on lignocellulosics and pulp Aveiro, Portugal, pp 547–550
Gellerstedt G, Henriksson G (2008) Lignins: major sources, structure and properties. In: Belgacem MN, Gandini A (eds) Monomers, polymers and composites from renewable resources. Elsevier, Amsterdam, pp 201–224
Gellerstedt G, Lindfors E (1984a) Structural changes in lignin during kraft cooking. Part 4. Phenolic hydroxyl groups in wood and kraft pulps. Svensk Papperstidn 87(15):R115–R118
Gellerstedt G, Lindfors EL (1984b) Structural changes in lignin during kraft pulping. Holzforschung 38(3):151–158
Gierer J (1980) Chemical aspects of kraft pulping. Wood Sci Technol 14(4):241–266
Gierer J (1982a) The chemistry of delignification. A general concept. Part I. Holzforschung 36(1):43–51
Gierer J (1982b) The chemistry of delignification. A general concept. Part II. Holzforschung 36(2):55–64
Gierer J, Lindenberg O (1980) Reaction of lignin during sulfate pulping. Part XIX. Isolation and identification of new dimmers from spent sulfate liquor. Acta Chem Scand 34(3):161–170
Gierer J, Ljunggren S (1979) The reaction of lignin during sulfate pulping. 17. Kinetic treatment of the formation and competing reactions of quinone methide intermediates. Svensk Papperstidn 82(17):503–512
Gierer J, Ljunggren S (1983) Comparative studies of the participation of different neighboring groups in the alkaline cleavage of β-aryl ether bonds in lignin. Svensk Papperstidn 86(9):R100–R106
Gierer J, Ljunggren S, Ljungquist P, Noren I (1980) The reaction of lignin during sulfate pulping. 18. The significance of α-carbonyl groups for the cleavage of β-aryl ether structure. Svensk Papperstidn 83(3):75–82
Guizani C, Lachenal D (2017) Controlling the molecular weight of lignosulfonates by an alkaline oxidative treatment at moderate temperatures and atmospheric pressure: a size-exclusion and reverse-phase chromatography study. Int J Mol Sci 18(12):2520–2538
Gullichsen J (1999) Fiber line operations. In: Gullichsen J, Paulapuro H (eds) Papermaking science and technology series, Yväskylä: Finnish Paper Engineers’ Association and TAPPI, Fapet Oy, Book 6, Chapter 2
Hansen CM, Björkman A (1998) The ultrastructure of wood from a solubility parameter point of view. Holzforschung 52(4):335–344
Heifetz LYa, Bezugly VD (1969) Polarography of anthraquinone and its derivatives. In: Organic intermediates and dyes. Chemistry of anthraquinone. Khimiya, Moscow, Russia, vol 4, pp 164–193
Helm RF (2000) Lignin-polysaccharide interactions in woody plants. In: Glasser WG, Northey RA, Schultz TP (eds) Lignin: historical, biological, and materials perspectives, vol 742. ACS Symp Series, Washington, pp 161–171
Hergert HL, Pye EK (1993) Recent history of organosolv pulping. In: Proceedings of 2nd international technical conference on PapFor-93. St Petersburg, Russia, pp 79–100
Hulanicky A, Masson MR (1987) Reactions of acids and bases in analytical chemistry. E Horwood, Chichester
Irzhak VI, Rosenberg BA, Enikolopyan NS (1979) Web polymers: synthesis, structure, properties. Nauka, Moscow
Kärkäs MD, Matsuura BS, Monos TM, Magallanes G, Stephenson CRJ (2016) Transition-metal catalyzed valorization of lignin: the key to a sustainable carbon-neutral future. Org Biomol Chem 14:1853–1914
Karmanov AP (2004) Self-organization and structure of lignin. Yekaterinburg
Kleinert TN (1966) Mechanisms of alkaline delignification. I. The overal reaction pattern. Tappi J 49(2):53–57
Kondo R, Sarkanen KV (1984) Kinetics of lignin and hemicellulose dissolution during the initial stage alkaline pulping. Holzforschung 38(1):31–36
Kondo R, Tsutsumi Y, Imamura H (1987) Kinetics of β-aryl ether cleavage of phenolic syringyl type lignin model compounds in soda and kraft systems. Holzforschung 41(2):83–88
Kringstad KP, Mörck R (1983) 13C-NMR spectra of kraft lignins. Holzforschung 37(5):237–244
Kurzin AV, Platonov AYu, Evstigneyev EI Mayorova ED (1997) Nucleophilicity and basicity of phenols in aminolysis of their acetates with piperidine. Zhurn Obsch Khim 67(9):1568–1571
Laurichesse S, Averous L (2014) Chemical modification of lignins: towards biobased polymers. Progr Polymer Sci 39:1266–1290
Lawoko M, Berggren R, Berthold F, Henriksson G, Gellerstedt G (2004) Changes in the lignin-carbohydrate complex in softwood kraft pulp during kraft and oxygen delignification: lignin-polysaccharide networks. II. Holzforschung 58:603–610
Lawoko M, Henriksson G, Gellerstedt G (2005) Structural differences between the lignin-carbohydrate complexes in wood and chemical pulps. Biomacromolecules 6:3467–3473
Lindberg O (1979) Studies on the chemistry of delignification in alkaline media. Chem Commun Univ Stockholm 6:1–51
Lindenfors S (1980) Additives in alkaline pulping—What reduces what? Svensk Papperstidn 83(6):165–173
Lindner A, Wegener G (1900) Characterization of lignins from organosolv pulping according to the Organocell process. Part 3. Molecular weight determination and investigation of fractions isolated by GPC. J Wood Chem Technol 10(3):331–350
Lindner A, Wegener G (1988) Characterization of lignins from organosolv pulping according to the Organocell process. Part 1. Elemental analysis, nonlignin portions and functional groups. J Wood Chem Technol 8(3):323–340
Liu C-J (2012) Deciphering the enigma of lignification: precursor transport, oxidation, and the topochemistry of lignin assembly. Mol Plant 5:304–317
Ljunggren S (1980) The significance of aryl ether cleavage in kraft delignification of softwood. Svensk Papperstidn 83(13):363–369
Lora J (2008) Industrial commercial lignins: sources, properties and applications. In: Belgacem MN, Gandini A (eds) Monomers, polymers and composites from renewable resources. Elsevier, Amsterdam, pp 225–241
Luner P, Roseman G (1986) Monomolecular film properties of isolated lignins. Holzforschung 40(Suppl.):61–66
Magalhaes Silva Moura JC, Valencise Bonine CA, de Oliveira Fernandes Viana J, Dornelas M C, Mazzafera P (2010) Abiotic and biotic stresses and changes in the lignin content and composition in plants. J Integr Plant Biol 52(4):360–376
Maiyorova H, Platonov A, Evstigneyev E (1995) About significance of nonphenolic lignin structure in alkaline pulping with anthraquinone. In: Proceedings of 8th international symposium on wood and pulping chemistry Helsinki, Finland, vol 2, pp 291–296
Mänsson P (1983) Quantitative determination of phenolic and total hydroxyl groups in lignins. Holzforschung 37(3):143–146
Maunu SL (2002) NMR studies of wood and wood products. Progr Magn Reson Spectrosc 40:151–174
Metzger JO, Bicke O, Faix O, Tuszynski W, Angermann R, Karas M, Strupat K (1992) Matrix-assisted laser desorption mass spectrometry of lignins. Angew Chem Int Ed Eng 31(6):762–764
Mörck R, Yoshida H, Kringstad KP, Hatakeyama H (1986) Fractionation of kraft lignin by successive extaction with organic solvents. Holzforschung 40(suppl):51–60
Mottiar Y, Vanholme R, Boerjan W, Ralph J, Mansfield SD (2016) Designer lignins: harnessing the plasticity of lignification. Curr Opin Biotechnol 37:190–200
Niemelä K (1990) Low-molecular-weight organic compounds in birch kraft black liquor. Ann Acad Sci Fennica Series A II Chemica 229:1–142
Nimz HH (1995) Analytical methods in wood, pulping and bleaching chemistry. In: Proceedings of 8th international symposium on wood and pulp chemistry Helsinki, Finland, vol 1, pp 1–32
Nomura Y, Nakamura M (1978) Studies on quinone additive cooking. 1. Effect of quinone addition on alkaline cooking. Jpn Tappi 32(12):713–721
Obst J (1983) Kinetics of alkaline cleavage of β-aryl ether bonds in lignin models: significance to delignification. Holzforschung 37(1):23–28
Pedersen JA (1973) Electron spin resonance studies of oxidative processes of quinones and hydroquinones in alkaline solution. Formation of primary and secondary semiquinone radicals. J Chem Soc Perkin Trans Part 2(4):424–431
Pilyugina LG, Haponen IL, Vasilieva TM, Mischenko KP (1974) Comparison of thermodynamic characteristics of separated lignins. Chemistry and use of lignin. Zinatne, Riga, pp 113–122
Rabinovich ML (2009) Wood hydrolysis industry in the Soviet Union and Russia: What can be learned from history? In: Rautakivi A (ed) Oral presentations, Proceedings of NWBC 2009, Helsinki, Finland, 2–4 September, pp 111–120
Rabinovich ML (2010) Wood hydrolysis industry in the Soviet Union and Russia: a mini-review. Cellulose Chem Technol 44(4–6):173–186
Radotić K, Simić-Krstić J, Jeremić M, Trifunović MA (1994) Study of lignin formation at the molecular level by scanning tunneling microscopy. Biophys J 66(6):1763–1767
Ragauskas AJ, Beckham GT, Biddy MJ, Chandra R, Chen F, Davis M, Davison BH, Dixon RA, Gilna P, Keller M, Langan P, Naskar AK, Saddler JN, Tschaplinski TJ, Tuskan GA, Wyman CE (2014) Lignin valorization: improving lignin processing in the biorefinery. Science 344:1246843-1–1246843-10
Ralph J (2010) Hydroxycinnamates in lignification. Phytochem Rev 9:65–83
Ralph J, Lundquist K, Brunow G, Lu F, Kim H, Schatz PF, Marita JM, Hatfield RD, Christensen JH, Boerjan W (2004) Lignins: natural polymer from oxidative coupling of 4-hydroxyphenylpropanoids. Phytochem Rev 3(1):29–60
Rinaldi R, Jastrzebski R, Clough MT, Ralph J, Kennema M, Bruijnincx PCA, Weckhuysen BM (2016) Paving the way for lignin valorisation: recent advances in bioengineering, biorefining and catalysis. Angew Chem Int Ed 55:8164–8215
Robert DR, Bardet M, Gellerstedt G, Lindfors E-L (1984) Structural changes in lignin during kraft cooking. Part 3. On the structure of dissolved lignins. J Wood Chem Technol 4(3):239–263
Sakakibara A (1991) Chemistry of lignin. In: Hon DN-S, Shirashi N (eds) Wood and cellulosic chemistry. Marcel Dekker, New York, pp 111–175
Santos RB, Jameel H, H-m Chang, Hart PW (2013) Impact of lignin and carbohydrate chemical structures on degradation reactions during hardwood kraft pulping processes. BioResources 8(1):158–171
Sarkanen KV, Ludwig CH (eds) (1971) Lignins: Occurrence, Formation, and Reactions, Wiley, NY [Quoted pages refer to Russian translation: Лигнины Cтpyктypa, cвoйcтвa и peaкции/Пoд peд КB Capкaнeнa, КX Людвигa Пep c aнгл M: Лecн пpoм-cть, 1975]
Sen S, Patil S, Argyropoulos DS (2015) Thermal properties of lignin in copolymers, blends, and composites: a review. Green Chem 17:4862–4887
Shevchenko SM, Deineko IP (1983) Chemistry of anthraquinone pulping. Wood Chem (Riga) 6:3–32
Shi R, Sun Y-H, Li Q, Heber S, Sederoff R, Chiang VL (2010) Towards a systems approach for lignin biosynthesis in Populus trichocarpa: transcript abundance and specificity of the monolignol biosynthetic genes. Plant Cell Physiol 51:144–163
Shorygina NN, Reznikov VM, Yelkin VV (1976) Chemical reactivity of lignin. Nauka, Moscow
Sjöström E (1981) Wood chemistry (Riga): fundamentals and applications. Academic Press, New York
Srzić D, Martinović S, Lj Tolić Paša, Kezele N, Shevchenko SM, Klasinc L (1995) Laser desorption Fourier-transform mass spectrometry of lignins. Rapid Commun Mass Spectr 9:245–249
Ten E, Vermerris W (2015) Recent developments in polymers derived from industrial lignin. J Appl Polymer Sci 132:42069–42082
Thakur VK, Manju Kumari Thakur M, Raghavan P, Michael R, Kessler M (2014) Progress in green polymer composites from lignin for multifunctional applications: a review. ACS Sustain Chem Eng 2(5):1072–1092
Vanholme R, Demedts B, Morreel K, Ralph J, Boerjan W (2010) Lignin biosynthesis and structure. Plant Physiol 153:895–905
Weng J-K, Chapple C (2010) The origin and evolution of lignin biosynthesis. New Phytol 187:273–285
Werthemann DP (1981) The xylophilicity/hydrophilicity balance of quinoid pulping additives. Tappi 64(3):140–142
Werthemann DP, Huber-Emden H, Bersier PM, Kelemen J (1981) High catalytic activity of rosindone and related compounds in alkaline pulping. J Wood Chem Technol 1(2):185–197
Wilder HD, Daleski EJ (1965) Part II of a series on kraft pulping kinetics. Tappi J 48(5):293–297
Yamasaki T, Hosoya S, Chen C-L, Gratzl JS, Chang H-M (1981) Characterization of residual lignin in kraft pulp. In: Proceedings of international symposium on wood pulp chemistry Stockholm, Sweden, vol 2, pp 34–42
Zakis GF (1987) Functional analysis of lignins and their derivatives. Zinatne, Riga
Zhao Q, Dixon RA (2011) Transcriptional networks for lignin biosynthesis: more complex than we thought? Trends Plant Sci 16(4):227–233
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Evstigneyev, E.I., Shevchenko, S.M. Structure, chemical reactivity and solubility of lignin: a fresh look. Wood Sci Technol 53, 7–47 (2019). https://doi.org/10.1007/s00226-018-1059-1
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00226-018-1059-1