
Abstract
Type 2 diabetes mellitus (T2DM) and osteoporosis are two major disorders which prevalence increases with aging and is predicted to worsen in the coming years. Preclinical investigations suggest common mechanisms implicated in the pathogenesis of both disorders. Recent evidence has established that there is a clear link between glucose and bone metabolism. The emergence of bone as an endocrine regulator through FGF23 and osteocalcin has led to the re-evaluation of the role of bone cells and bone-derived factors in the development of metabolic diseases such as T2DM. The development of bone morphogenetic proteins, fibroblast growth factor 23, and osteoprotegerin-deficient mice has allowed to elucidate their role in bone homeostasis, as well as revealed their potential important function in glucose homeostasis. This review proposes emerging perspectives for several bone-derived factors that may regulate glycemia through the activation or inhibition of bone remodeling or directly by regulating function of key organs such as pancreatic beta cell proliferation, insulin expression and secretion, storage and release of glucose from the liver, skeletal muscle contraction, and browning of the adipose tissue. Connections between organs including bone-derived factors should further be explored to understand the pathophysiology of glucose metabolism and diabetes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The skeleton has been demonstrated to be determinant to preserve the mechanical integrity of the organism and to regulate calcium and phosphorus homeostasis. The high bone remodeling activity and vascularization of the skeleton also suggest that the bone tissue by its surface can have additional contribution to physiology of the whole organism, such as in glucose homeostasis [1]. Type 2 diabetes mellitus (T2DM) and osteoporosis are two major chronic disorders which prevalence increases with aging and is predicted to worsen in the coming years [2]. As an example, the prevalence of diabetes is expected to increase by 55% in the next 20 years, rising from 382 million people worldwide in 2013 to 592 million by 2035. The estimated number of osteoporotic hip fractures worldwide will also rise from 1.66 million in 1990 to 6.26 million in 2050 [3]. Both osteoporosis and diabetes are associated with an increased risk of fragility fractures, resulting from alterations of bone quality as well as a propensity to falls, associated with a loss of muscle function [4], providing the clinical characteristics of diabetoporosis (or diabetes in osteoporosis). Recently, a clear link between glucose metabolism and bone has been highlighted [5, 6]. More specifically, in vitro investigation demonstrated that high bone resorption and low bone formation in type 2 diabetic patients can be explained by a direct up-regulation of sclerostin (a major Wnt inhibitor, specifically expressed in osteocyte) by high glucose levels [7]. However, other mechanisms have been mentioned to explain the decline of bone strength: a decrease in the neovascularization, an increase in the mesenchymal stem differentiation into adipocyte rather than into osteoblasts, and accumulation of glycation end products (AGEs) decreasing the mechanical properties of the matrix [8, 9]. Hence, the exact pathophysiology of bone fragility in T2DM patients remains to be determined.
More recently, based on the cardinal rule of physiology that a regulated organ takes back to a regulating one to limit its influence, bone has been shown to regulate the whole body glucose homeostasis. Bone is known as an insulin-regulated tissue [10], but importantly, existence of feedback has emerged, with osteocalcin, an osteoblast-derived osteokine, reported to increase insulin release from beta pancreatic cells and indirectly to increase insulin action through enhanced release of adiponectin from adipose tissue [11, 12]. Hence, bone might contribute to the regulation of glucose homeostasis and an impairment of this control loop can favor diabetes occurrence. The fact that bone remodeling occurs daily, in multiple locations and in an organ covering a very large surface area, suggests that bone has a high energy demand which needs to be tightly regulated. Therefore, bone may needs more than one osteokine to regulate glucose resources. Few analyses have already indicated that bone is the fourth organ after adipose tissue, liver, and skeletal muscle to store glucose [13, 14]. Altogether, these data indicate a need to study how the skeleton can regulate energy metabolism, i.e., glucose homeostasis. Glucose regulation by bone-derived factors will particularly be reviewed here.
Glucose Consumption by Bone Cells Contributes to Glucose Homeostasis
Bone remodeling activity performed by osteoblasts—the bone forming cells—and osteoclasts—the bone resorbing cells—and their coordination by osteocytes, the end-product of osteoblast differentiation requires an amount of energy which can be important by taking into account the number of cells in the skeleton. Osteoblast, osteocyte, and osteoclast exhibit well-developed Golgi apparatus, endoplasmic reticulum, and mitochondrial activity [15]. Interestingly, cellular glucose metabolism has been shown to regulate osteoblast biology [16], so osteoblast differentiation, collagen synthesis, and bone formation activity are tributary to the amount of energy which is available. More precisely, intracellular entry of glucose, regulated by Glut1 glucose transporter, promotes RUNX2 transcriptional factor and the accumulation of glucose into osteoblasts [14]. By using the euglycemic hyperinsulinemic clamp, we confirm data from Karsenty laboratory, showing that bone takes up one-fifth of the quantity of glucose taken by skeletal muscle (80 ng/mg tissue/min), the tissue taking up the majority of glucose in the mouse after the brown adipose tissue (290 ng/mg tissue/min) [14].
Intracellular machinery yield rate which determine the amount of glucose which will be consumed can change in response to different treatments. For example, rats receiving PTH roughly increased structure and enzymatic activity of endoplasmic reticulum mitochondria and Golgi apparatus of osteoblast and osteocyte without affecting those of osteoclast [17]. In accordance, a randomized clinical trial showed that PTH increased bone formation, i.e., osteocalcin, and decreased blood glucose without influencing insulin secretion or resistance [18]. Thus, the hypothesis that PTH contributes to glucose homeostasis through the bone tissue himself remains possible. A preclinical study recently highlighted that intermittent PTH reduces glycemia by increasing aerobic osteoblast glycolysis via IGF signaling [19].
In contrast, additional sex combs-like (ASXL2), an enhancer of trithorax and polycomb family protein which interacts with PPARγ, promotes osteoclast mitochondrial biogenesis, i.e., glucose consumption, through PGC-1β independently of the c-Fos-NFATc1 pathway classically required for bone resorption activity. In accordance, authors demonstrated that ASXL2-deficient mice exhibit high glucose levels and are glucose intolerant [20]. In vitro, in basal condition, investigation of 2-[U-14C] deoxyglucose (2-DG) uptake illustrates that either osteoblast or osteoclast consumes around 20 μmol/g protein/min of glucose each compared to 60 μmol/g protein/min for myoblast, arguing that both formation and resorption cells are able to consume a substantial amount of glucose.
Since 10 years, a new bone endocrine function has emerged: the production of osteocalcin for the control of glucose homeostasis. In this model, osteocalcin modulates three other hormones: Insulin secreted by beta cells of the pancreas [21]; adiponectin secreted by adipocytes, known to reduce insulin resistance [12, 22]; and testosterone synthesized by Leydig cells and favoring fertility [23] (Fig. 1). Hence, we will review in the next paragraph whether, in addition to osteocalcin, other bone-derived factors could be able to modulate energy metabolism.

Bone-derived factor, control of glucose metabolism. Osteocalcin exhibits an important endocrine function by targeting multiple tissues: pancreas, adipose tissue, skeletal muscle, brain, and liver testis. However, osteocalcin might not be the only factor produced by bone cells (osteoclast, osteoblast, and/or osteocyte) involved in the regulation of glucose homeostasis
Glucose Regulation by Bone-Derived Factors
Osteocalcin
Osteocalcin also named bone gamma-carboxyglutamic acid-containing protein (BGLAP) is a non-collagenous protein secreted by osteoblast/osteocyte. Osteocalcin plays a role in the mineralization process by having a high affinity with calcium. In clinical practice, osteocalcin is used as a marker of bone formation and more broadly of bone remodeling. Osteocalcin can also be undercarboxylated (unOC), by the acidification of the matrix performed by osteoclast, exhibiting glutamic acid instead of gamma-carboxyglutamic acid. This form is released into the bloodstream and has been considered to be the bioactive form able to regulate energy metabolism. Historically, metabolic function of osteocalcin has been investigated by characterizing the phenotype of the enterococcal surface protein (Esp)-knockout mouse (Esp−/−). Esp gene encodes an osteotesticular protein tyrosine phosphatase (OST-PTP) in osteoblast, a tyrosine phosphatase which stimulates the carboxylation of osteocalcin. Hence, Esp−/− mice exhibit an increased level of unOC, as well as increased insulin and adiponectin expression, increased insulin secretion and sensitivity, increased lean, and decreased fat mass and triglyceride levels. In contrast, osteocalcin-deficient mice, which have low unOC circulating levels, are fat, insulin resistant, glucose intolerant, and hyperlipidemic [21]. The proof of concept that unOC is able to impact pancreatic beta cell, adipocyte, or myocyte has been demonstrated by co-culturing Esp −/− osteoblast with primary beta cell or adipocyte of WT [12, 21] and was confirmed later on subsequently with the treatment of WT mice with osteocalcin [24] and with the phenotypic characterization of the GPRC6A ‘osteocalcin receptors’- deficient mice [25, 26]. Several studies also show that both injection and oral administration of unOC can abrogate the deleterious effects of high-fat diet on glucose metabolism [11, 27].
In addition to the direct effect of unOC on insulin secretion, it has been shown that unOC increases insulin secretion indirectly through an increased secretion of glucagon-like peptide-1 (GLP1) from intestinal endocrine cells [27].
The effects of unOC on skeletal muscle have been suggested with the demonstration that unOC increases the fusion rate of the C2C12 lining cells in vitro [28, 29] and improves significantly grip strength in vivo by increasing muscle volume [30, 31]. In addition, unOC increases insulin signaling in muscle, contributing to glucose metabolism.
Transfer to humans has also been largely investigated. Several cross-sectional studies highlighted the association between osteocalcin serum level and blood glucose, and HbA1c and metabolic syndrome [32]. A proof of concept study using surgical resection of osteoma osteoid showed a direct effect of osteocalcin on glucose serum level [33]. In addition, the fact that patients with a mutation of GPRC6A receptor, the receptor of osteocalcin, do have an impaired glucose metabolism and a low sperm count [34] also sustains relevance of osteocalcin physiology in humans. Nevertheless, direct evidence of the role of osteocalcin in humans is scarce and large-scale prospective studies are needed. Several limitations need to be mentioned. Caution must be taken particularly because murine and human osteocalcins are not the same size and differ in some amino acids [35]. In humans, majorities of the studies show association between total osteocalcin and glucose metabolism (such as plasma glucose, fasting insulin, and resistance) rather than with unOC. Another reason is that it is not completely clarified whether the active form is the fully unOC or only the partially unOC form. In the same line, because research and commercial assays for measuring unOC are not standardized, it is difficult to compare results between studies, and this could influence the interpretation of the results. The specificity of osteocalcin to the bone tissue is now also questioned since the two forms of osteocalcin are also expressed in adipose tissue [36]. Osteocalcin is maximally expressed in preadipocytes and expression decreased during adipocyte differentiation [36]. Osteocalcin has also been suggested to be produced in the brain and to possibly function as a neuropeptide [37]. In this regard, further studies are needed to clarify the precise regulation of osteocalcin released from adipose and/or brain in different physiological contexts of obesity, metabolic syndrome, and T2DM. While osteocalcin replacement in osteocalcin-null mice can reverse the glucose intolerance and correct glucose levels, it could not restore insulin sensitivity, indicating that other bone-derived factors may also mediate insulin action [38]. More recent investigation on NPY signaling in early osteoblasts also argues for a control of glucose homeostasis by other osteokines than unOC [39].
Major factors controlling both glucose homeostasis and osteoblast/osteocyte differentiation are the bone morphogenetic proteins (BMPs) [40].
BMPs
BMPs are members of the transforming growth factor (TGF-β) superfamily, originally discovered for their ability to induce bone and cartilage formation [41]. BMPs are now known to regulate embryonic development of multiple tissues by the phosphorylation of the intracellular BMP effector proteins SMADs [42]. They have been shown to be crucial in metabolic pathologies such as T2DM and obesity by regulating inflammation, glycemia, and energy metabolism. We will particularly focus on BMP2, BMP4, and BMP7 already known to impact hepatic fibrosis, endocrine cell differentiation of the pancreas, and browning of the adipose tissue.
Interestingly, BMPs and their respective receptor are not systematically expressed in the same tissue suggesting an important endocrine function of BMPs. For example, BMP7 receptor is expressed in hepatocyte and its activation has an anti-apoptotic and anti-inflammatory effect improving liver regeneration; however, BMP7 is not expressed in liver [43], but is mainly expressed by the kidney, pulmonary artery, cartilage, and bone.
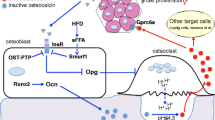
Hence, as for osteocalcin, the feedback loop could also exist for BMPs. Insulin signaling in osteoblast/osteocyte, decreased OPG/RANKL ratio, and during the initial phase of demineralization, BMPs initially embedded in the matrix can be released into the bloodstream (Fig. 2). This mechanism has already been suggested to explain the coupling between bone resorption and bone formation and is probably also involved in the coupling between bone, pancreas, and adipose tissue (Fig. 2).

Potential effects of BMPs released by the bone tissue on glucose homeostasis. BMP2, BMP4, BMP7, and BMP9 are all synthesized by the bone tissue and all regulate metabolic tissues such as the pancreas, liver, and adipose tissue. Insulin signaling in osteoblasts (Ob)/osteocytes (Ocy) decreases the expression of OPG, by decreasing OPG/RANKL ratio and increasing bone resorption. Osteoclastic (Oc) activity releases bone proteins embedded in the matrix such as bone morphogenetic proteins (BMPs) into the bloodstream. BMPs secondarily exert an action on key organs involved in glucose metabolism pancreas, liver, and adipose tissue
Effects of BMPs in Pancreatic Beta Cell
In the pancreas, ablation of BMP receptor (BMPr1a) in beta cells leads to glucose intolerance, decreased expression of genes involved in glucose sensing and metabolism, and decreased insulin production and secretion [44, 45]. On the contrary, overexpression of BMP4 improves glucose tolerance and insulin secretion as well as markers of islet function [44]. However, the interaction between BMP4 and insulin secretion during the development of diabetes is not so clear. It was reported that glucose-induced insulin secretion was significantly impaired in rodent and human islets pre-treated with BMP4, and inhibition of BMP activity resulted in enhanced insulin release, in part through an increased proliferation of beta cells [46]. On the contrary, recombinant human BMP3 in INS-1 potently increased insulin expression and protein [47]. The various effects among the BMPs on beta cell induce different phosphorylations of the SMAD signaling pathway, for example, BMP3 signals through phospho-SMAD2/3 similar to TGFβs, whereas BMP4 through phospho-SMAD1/5/8. Discordant data reported in the literature may also come from the differential effects of BMPs induced during developmental and adult lives. The specific contribution of BMPs secreted locally compared to circulating levels of BMPs arising from other tissues can also contribute to discrepancies observed between studies.
The complexity of BMPs effects on glucose homeostasis may come from their ability to target several tissues, as it is the case for BMP9 and BMP7. BMP9 targets both the liver and pancreas, respectively, by inhibiting hepatocyte glucose production, activating key enzyme of lipid metabolism (malic enzyme and fatty acid synthase), and stimulating insulin secretion by pancreatic beta cell in normal and diabetic mice [48]. In addition to its role in liver fibrosis (see above), BMP7 has been reported to improve glucose homeostasis by increasing insulin production through the conversion of pancreatic exocrine tissue (98% of the organ) to an endocrine one, expressing beta cell master genes [49].
Effects of BMPs on Browning of the Adipose Tissue and Consequences in Energy Metabolism
Among all BMPs, involvement of BMP-2, BMP4, BMP6, and BMP7 has been well described in the differentiation of the mesenchymal stem cell into the adipogenic lineage [50]. In recent findings, the evidence of a role for BMPs on adipose tissue comes from studies of the knockout models. BMP7-deficient mice exhibit a reduction in brown adipose tissue (BAT), without any changes in white adipose tissue (WAT) [51]. Morphologically, WAT and BAT differ by their droplet size and abundance. WAT stores triglycerides in a single large lipid droplet and contains few mitochondria, while BAT shows many small lipid droplets and a lot of mitochondria. BAT by expressing uncoupling protein 1 (UCP1) increases cellular respiration from ATP production and burns much more lipid and glucose than WAT. Hence, overexpression of UCP1 protects mice from diet induced obesity [52]. In particular, BMP7 stimulates the expression of PR domain containing 16 (PRDM16), a factor responsible for triggering commitment of mesenchymal stem cell into BAT rather than to WAT. BMP7 and BMP8B also act on mature BAT to promote thermogenesis by increasing UCP1 and lipolysis through phosphorylation of P38-MAPK. Unlike BMP-8B, BMP7 is not expressed in mature BAT, suggesting that it does not work in an autocrine or paracrine manner, but rather as an endocrine factor as seen for the liver [53]. Overexpression of BMP4 in WAT resulted in decreased fat mass with reduced adipocyte size coupled with an increased number of brown or beige adipocytes and an increase in insulin sensitivity [54].
On the contrary, BMP2 is expressed in adipose tissue and has been shown in vitro to promote adipogenesis into WAT [55]. Hence, BMP2 KO mice exhibit a reduction in white fat mass. However, conflicting data remain concerning BMP4 signaling depending on the cell lines investigated [56].
In summary, in addition to their major contribution to skeletal remodeling, BMPs also play a key role in glucose homeostasis. However, several discordances remain and additional experiments with tissue-specific strategies will be required to better characterize the tissue specificity of BMPs as well as the contribution of bone to circulating levels of BMPs.
OPG
Osteoprotegerin (OPG) is a soluble glycoprotein that belongs to the tumor necrosis factor (TNF) receptor superfamily. In the bone tissue, it is secreted by osteoblast and acts as a decoy soluble receptor for the receptor activator of nuclear factor kB (NF-kB) ligand (RANKL), thus preventing RANKL from binding to its receptor on osteoclasts, thereby inhibiting osteoclastogenesis. Denosumab, a human monoclonal antibody against RANKL, has been used for nearly 5 years to treat osteoporosis and appears to be the most powerful inhibitor of bone remodeling [57]. It is now known that RANK and RANKL are both expressed in other tissues such as liver, pancreatic beta cell, skeletal muscle and in several tumors. Hence, activation of RANK has been reported to be involved in several diseases impairing glucose homeostasis. In a mouse model of T2DM, inhibition of RANKL signaling improves hepatic insulin sensitivity and normalizes plasma glucose concentration [58]. In skeletal muscle, inhibition of RANKL by OPG-immunoglobulin fragment complex (OPG-Fc) treatment increases skeletal muscle force from dystrophic mice [59]. OPG-Fc specifically reduces muscle inflammation, i.e., neutrophil and macrophages cells numbers and muscle damage, evaluated by hematoxylin and eosin staining. Such investigations need to be confirmed since these effects are observed only in extensor digitorum longus of dystrophic mdx mice and not in wild-type mice. However, such data argue for an improvement of glucose homeostasis by OPG-Fc treatment [59].
In a model of inflammation by microbial invasion, OPG production by beta cell is increased, which inhibits insulin secretion. In accordance, in vitro, OPG treatment of MIN6 pancreatic beta cell lines decreased insulin release following glucose stimulation. Hence, OPG would locally act as a negative feedback, preventing exhaustion of beta cell endocrine function [60].
In women with diabetes who were not receiving any anti-diabetic medication, RANKL neutralizing antibody not only reduced fractures but also significantly decreased fasting serum glucose [61]. However, associations between the levels of OPG and/or RANKL and parameters of glucose homeostasis such as HOMA, insulin sensitivity, and fasting glucose are producing conflicting results [62–64].
Hence, further study is needed to elucidate several points: (1) the direct effect of OPG/RANKL on each tissue involved in the regulation of glucose homeostasis; (2) the skeletal contribution versus non-skeletal of OPG and/or RANKL to circulating levels; (3) the role of RANKL/OPG on decarboxylation of osteocalcin, i.e., metabolic active form of osteocalcin, an indirect mechanism for RANKL/OPG to control glycemia.
To date, a real clinical trial with denosumab is needed with glucose homeostasis as a primary endpoint.
FGF23
In the bone, the most abundant cell is osteocyte also described as the chief of orchestra of bone modeling and remodeling. The first discovered hormone expressed by bone and mostly by osteocytes was FGF23. FGF23 is well characterized for its important role in regulating serum phosphate levels. Surprisingly, in addition to their expected altered phospho-calcic metabolism, FGF23-deficient mice exhibit hypoglycemia as well as increased insulin sensitivity [65]. Interestingly, clinical studies also confirm an association between FGF23 and energy metabolism. In two independent cohorts, ‘Osteoporotic Fractures in Men Study’ and ‘Vasculature Uppsala Seniors study,’ an association between FGF23, dyslipidemia, and fat mass has been reported [66]. Hence, FGF23 levels were higher in subjects with metabolic syndrome compared to healthy patients [66]. However, the precise role of FGF23 in energy metabolism is not well understood, and precision must be given on its direct and/or indirect action. Two reviews reported that FGF23 pathway could be involved in energy metabolism, and this could be mediated by bone [67, 68]. FGF23 would inhibit enterococcal surface protein (see above) and decreased bone resorption, decreasing the release of unOC.
SOST
As described above, low bone turnover in T2DM patients can be explained by an up-regulation of sclerostin accompanied by an uncoupling remodeling (decrease in bone formation and increase in bone resorption) [7]. Type 2 diabetic patients have increased plasma sclerostin associated with BMI, abdominal fat, higher fasting plasma glucose, and blood insulin sensitivity, suggesting that sclerostin may be implicated in diabetes pathogenesis [69, 70]. These associations led to the question whether circulating levels of sclerostin have a biological effect? In that regard, serum sclerostin is associated with vascular calcification in postmenopausal women [71], T2DM [72], and chronic kidney diseases and hemodialysis patients [73]. More recently, preliminary data from MINOS cohort indicated in men that the highest quartile of sclerostin was associated with the highest odds ratio (OR 2.40 [95% CI 1.33–4.41] p < 0.005) of metabolic syndrome (defined by a glycemia >5.6 mmol/l, arterial pressure >130/85 mmHg, triglycerides >1.7 mmol/l; HDL-cholesterol <1.03 mmol/l) compared to the lowest quartile [74]. The Wnt signaling pathway is active in key organs of glucose homeostasis such as pancreas, adipose tissue, liver, and skeletal muscle [75–78]. Hence, all the regulators of this signaling pathway can exert an effect on glucose homeostasis. For example, in a preclinical study, Bmp/Wnt signaling has been shown to be important in islet development, function, and insulin production and secretion [79]. More recently, the loss of expression of Wnt inhibitor (Sostdc-1) in mice has been shown to enhance insulin secretion and glucose homeostasis. These studies provide insight into modulators of BMP/Wnt pathway in the endocrine pancreas and reveal potential avenues for Wnt inhibitors in beta cell function such as sclerostin [80].
In a preclinical study, we clearly demonstrated that sclerostin production is highly regulated by periostin signaling pathway in osteoblast/osteocyte in response to anabolic stimuli [81, 82]. Moreover, we demonstrated that periostin also directly regulates beta catenin signaling pathway independently of sclerostin. We found that after 5 weeks of intermittent PTH, circulating periostin levels were significantly increased compared to placebo [83]. Recent investigations highlighted periostin as a new molecule talented at pancreatic beta cell regeneration. Periostin injection for 8 weeks enhances glucose tolerance, increases insulin staining in pancreatic tissue, and increases the number of islets [84]. Hence, circulating levels could affect glucose metabolism. It has, however, to be remembered that periostin is not specific to bone and that serum levels represent an additional contribution, and the contribution of each one remains to be determined.
Lastly, because type I collagen is widely distributed in a variety of organs, particularly during pathophysiological conditions such as inflammation and fibrosis, bone matrix protein initially localized in the skeleton can be expressed significantly in soft tissue (Fig. 3). Hence, osteocalcin, periostin, and all others bone matrix proteins can regulate key organs involved in glucose homeostasis (liver, pancreas, adipocyte, smooth, and skeletal muscle) in an endocrine but also in paracrine way.

Soft tissue calcification and association with human diseases impacting glucose homeostasis. Bone matrix proteins are expressed in different organs, particularly during inflammation or fibrosis occurring in several diseases [83, 85–91]. These tissues expressing bone matrix proteins are involved directly or indirectly in the regulation of glucose homeostasis, leading to the idea that ‘extra skeletal tissues’ contribute to the regulation of energy metabolism. Type 2 diabetes mellitus (T2DM), non-alcoholic fatty liver disease (NAFLD). As an example, pancreatic calcification is seen on radiographs in about 30–50% of patients with chronic pancreatitis in adults and increases the risk of secondary diabetes [92]. However, the specific role of bone matrix protein in such observation needs to be further investigated
Conclusion
Developing evidence from preclinical to clinical studies argues for a control of glucose homeostasis through the bone tissue by targeting key organs of the energy metabolism such as pancreas, adipose tissue, liver, and muscle. Bone endocrine function on glucose metabolism cannot be summarized only by osteocalcin secretion and other osteokines—incl. BMPs need to be further investigated. Moreover, osteoclasts, osteoblasts, and osteocytes have a high glucose demand; therefore, the bone tissue should be taken into account in the control loop of glycemia. With a key corollary question: what is the contribution of the bone tissue in the development of insulin resistance and diabetes?
From an evolutionary perspective, organs adaptation has always been driven by one goal: efficient energy conservation and storage that enables survival through periods of food shortage. Accordingly, a tempting hypothesis would be that high bone remodeling and its subsequent bone loss with aging could be a mechanism developed throughout evolution in order to spare energy (and thereby improve longevity). However, these adaptations in the context of our current affluent lifestyles full of food are inappropriate and lead to an excess of energy which leads to many disorders rallied under the so-called metabolic syndrome. Whether maintenance of a normal bone mass could therefore increase glucose consumption and insulin sensitivity to prevent diabetes remains to be investigated.
To conclude, connections or ‘networks’ between organs including the bone tissue should further be explored to understand the pathophysiology of glucose metabolism and diabetes.
References
Karsenty G, Ferron M (2012) The contribution of bone to whole-organism physiology. Nature 481(7381):314–320
Ferrari S (2013) Diabetes and osteoporosis. Rev Med Suisse 9(390):1258–1259
Gullberg B, Johnell O, Kanis JA (1997) Worldwide projections for hip fracture. Osteoporos Int 7:407–413
Hita-Contreras F, Martínez-Amat A, Cruz-Díaz D, Pérez-López FR (2015) Osteosarcopenic obesity and fall prevention strategies. Maturitas 80(2):126–132
Ducy P, Amling M, Takeda S, Priemel M, Schilling AF, Beil FT et al (2000) Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell 100(2):197–207
Yadav VK, Oury F, Suda N, Liu ZW, Gao XB, Confavreux C et al (2009) A serotonin-dependent mechanism explains the leptin regulation of bone mass, appetite, and energy expenditure. Cell 138(5):976–989
Tanaka K, Yamaguchi T, Kanazawa I, Sugimoto T (2015) Effects of high glucose and advanced glycation end products on the expressions of sclerostin and RANKL as well as apoptosis in osteocyte-like MLO-Y4-A2 cells. Biochem Biophys Res Commun 461(2):193–199
Wongdee K, Charoenphandhu N (2011) Osteoporosis in diabetes mellitus: possible cellular and molecular mechanisms. World J Diabetes 2(3):41–48
Furst JR, Bandeira LC, Fan WW, Agarwal S, Nishiyama KK, McMahon DJ et al (2016) Advanced glycation endproducts and bone material strength in type 2 diabetes. J Clin Endocrinol Metab 101(6):2502–2510
Fulzele K, Riddle RC, DiGirolamo DJ, Cao X, Wan C, Chen D et al (2010) Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell 142(2):309–319
Ferron M, McKee MD, Levine RL, Ducy P, Karsenty G (2012) Intermittent injections of osteocalcin improve glucose metabolism and prevent type 2 diabetes in mice. Bone 50(2):568–575
Wei J, Ferron M, Clarke CJ, Hannun YA, Jiang H, Blaner WS et al (2014) Bone-specific insulin resistance disrupts whole-body glucose homeostasis via decreased osteocalcin activation. J Clin Invest 124(4):1–13
Ducy P, Schinke T, Karsenty G (2000) The osteoblast: a sophisticated fibroblast under central surveillance. Science 289(5484):1501–1504
Wei J, Shimazu J, Makinistoglu MP, Maurizi A, Kajimura D, Zong H et al (2015) Glucose uptake and Runx2 synergize to orchestrate osteoblast differentiation and bone formation. Cell 161(7):1576–1591
Pritchard JJ (1972) General histology of bone. In: Bourne GH (ed) The biochemistry and physiology of bone structure, vol 1, 2nd edn. Academic press, New York, pp 1–20
Esen E, Long F (2014) Aerobic glycolysis in osteoblasts. Curr Osteoporos Rep 12(4):433–438
Weisbrode SE, Capen CC, Nagode LA (1974) Effects of parathyroid hormone on bone of thyroparathyroidectomized rats, an ultrastructural and enzymatic study. Am J Pathol 75(3):529–542
D’Amelio P, Sassi F, Buondonno I, Spertino E, Tamone C, Piano S et al (2015) Effect of intermittent PTH treatment on plasma glucose in osteoporosis: a randomized trial. Bone 76:177–184
Esen E, Lee SY, Wice BM, Long F (2015) PTH promotes bone anabolism by stimulating aerobic glycolysis via IGF signaling. J Bone Miner Res 30:2137
Izawa T, Rohatgi N, Fukunaga T, Wang QT, Silva MJ, Gardner MJ et al (2015) ASXL2 regulates glucose, lipid, and skeletal homeostasis. Cell Rep 11(10):1625–1637
Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C et al (2007) Endocrine regulation of energy metabolism by the skeleton. Cell 130(3):456–469
Otani T, Mizokami A, Hayashi Y, Gao J, Mori Y, Nakamura S et al (2015) Signaling pathway for adiponectin expression in adipocytes by osteocalcin. Cell Signal 27(3):532–544
Oury F, Sumara G, Sumara O, Ferron M, Chang H, Smith CE et al (2011) Endocrine regulation of male fertility by the skeleton. Cell 144(5):796–809
Ferron M, Hinoi E, Karsenty G, Ducy P (2008) Osteocalcin differentially regulates beta cell and adipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proc Natl Acad Sci U S A 105(13):5266–5270
Pi M, Wu Y, Quarles LD (2011) GPRC6A mediates responses to osteocalcin in β-cells in vitro and pancreas in vivo. J Bone Miner Res 26(7):1680–1683
Wei J, Hanna T, Suda N, Karsenty G, Ducy P (2014) Osteocalcin promotes β-cell proliferation during development and adulthood through Gprc6a. Diabetes 63(3):1021–1031
Mizokami A, Yasutake Y, Higashi S, Kawakubo-Yasukochi T, Chishaki S, Takahashi I et al (2014) Oral administration of osteocalcin improves glucose utilization by stimulating glucagon-like peptide-1 secretion. Bone 69:68–79
Levinger I, Lin X, Zhang X, Brennan-Speranza TC, Volpato B, Hayes A et al (2016) The effects of muscle contraction and recombinant osteocalcin on insulin sensitivity ex vivo. Osteoporos Int 27(2):653–663
Tsuka S, Aonuma F, Higashi S, Ohsumi T, Nagano K, Mizokami A et al (2015) Promotion of insulin-induced glucose uptake in C2C12 myotubes by osteocalcin. Biochem Biophys Res Commun 459(3):437–442
Fernández-Real JM, Izquierdo M, Ortega F, Gorostiaga E, Gómez-Ambrosi J, Moreno-Navarrete JM et al (2009) The relationship of serum osteocalcin concentration to insulin secretion, sensitivity, and disposal with hypocaloric diet and resistance training. J Clin Endocrinol Metab 94(1):237–245
Shen H, Grimston S, Civitelli R, Thomopoulos S (2015) Deletion of connexin43 in osteoblasts/osteocytes leads to impaired muscle formation in mice. J Bone Miner Res 30(4):596–605
Kunutsor SK, Apekey TA, Laukkanen JA (2015) Association of serum total osteocalcin with type 2 diabetes and intermediate metabolic phenotypes: systematic review and meta-analysis of observational evidence. Eur J Epidemiol 30(8):599–614
Confavreux CB, Borel O, Lee F, Vaz G, Guyard M, Fadat C et al (2012) Osteoid osteoma is an osteocalcinoma affecting glucose metabolism. Osteoporos Int 23(5):1645–1650
Oury F, Ferron M, Huizhen W, Confavreux C, Xu L, Lacombe J et al (2013) Osteocalcin regulates murine and human fertility through a pancreas-bone-testis axis. J Clin Invest 123(6):2421–2433
Desbois C, Hogue DA, Karsenty G (1994) The mouse osteocalcin gene cluster contains three genes with two separate spatial and temporal patterns of expression. J Biol Chem 269(2):1183–1190
Foresta C, Strapazzon G, De Toni L, Gianesello L, Calcagno A, Pilon C et al (2010) Evidence for osteocalcin production by adipose tissue and its role in human metabolism. J Clin Endocrinol Metab 95(7):3502–3506
Patterson-Buckendahl P, Sowinska A, Yee S, Patel D, Pagkalinawan S, Shahid M et al (2012) Decreased sensory responses in osteocalcin null mutant mice imply neuropeptide function. Cell Mol Neurobiol 32(5):879–889
Yoshikawa Y, Kode A, Xu L, Mosialou I, Silva BC, Ferron M et al (2011) Genetic evidence points to an osteocalcin-independent influence of osteoblasts on energy metabolism. J Bone Miner Res 26(9):2012–2025
Lee NJ, Nguyen AD, Enriquez RF, Luzuriaga J, Bensellam M, Laybutt R et al (2015) NPY signalling in early osteoblasts controls glucose homeostasis. Mol Metab 4(3):164–174
Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M et al (2013) PPARγ signaling and metabolism: the good, the bad and the future. Nat Med 19(5):557–566
Wozney JM (1992) The bone morphogenetic protein family and osteogenesis. Mol Reprod Dev 32:160–167
Wagner DO, Sieber C, Bhushan R, Börgermann JH, Graf D, Knaus P (2010) BMPs: from bone to body morphogenetic proteins. Sci Signal 3:107
Sugimoto H, Yang C, LeBleu VS, Soubasakos MA, Giraldo M, Zeisberg M et al (2007) BMP-7 functions as a novel hormone to facilitate liver regeneration. FASEB J 21(1):256–264
Goulley J, Dahl U, Baeza N, Mishina Y, Edlund H (2007) BMP4-BMPR1A signaling in beta cells is required for and augments glucose-stimulated insulin secretion. Cell Metab 5(3):207–219
Scott GJ, Ray MK, Ward T, McCann K, Peddada S, Jiang FX et al (2009) Abnormal glucose metabolism in heterozygous mutant mice for a type I receptor required for BMP signaling. Genesis 47(6):385–391
Bruun C, Christensen GL, Jacobsen ML, Kanstrup MB, Jensen PR, Fjordvang H et al (2014) Inhibition of beta cell growth and function by bone morphogenetic proteins. Diabetologia 57(12):2546–2554
Bonner C, Farrelly AM, Concannon CG, Dussmann H, Baquié M, Virard I et al (2011) Bone morphogenetic protein 3 controls insulin gene expression and is down-regulated in INS-1 cells inducibly expressing a hepatocyte nuclear factor 1A-maturity-onset diabetes of the young mutation. J Biol Chem 286(29):25719–25728
Chen C, Grzegorzewski KJ, Barash S, Zhao Q, Schneider H, Wang Q et al (2003) An integrated functional genomics screening program reveals a role for BMP-9 in glucose homeostasis. Nat Biotechnol 21(3):294–301
Klein D, Álvarez-Cubela S, Lanzoni G, Vargas N, Prabakar KR, Boulina M et al (2015) BMP-7 induces adult human pancreatic exocrine-to-endocrine conversion. Diabetes 64(12):4123–4134
Kang Q, Song WX, Luo Q, Tang N, Luo J, Luo X et al (2009) A comprehensive analysis of the dual roles of BMPs in regulating adipogenic and osteogenic differentiation of mesenchymal progenitor cells. Stem Cells Dev 18(4):545–559
Tseng YH, Kokkotou E, Schulz TJ, Huang TL, Winnay JN, Taniguchi CM et al (2008) New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 454(7207):1000–1004
Poher AL, Altirriba J, Veyrat-Durebex C, Rohner-Jeanrenaud F (2015) Brown adipose tissue activity as a target for the treatment of obesity/insulin resistance. Front Physiol. doi:10.3389/fphys.2015.00004
Whittle AJ, Carobbio S, Martins L, Slawik M, Hondares E, Vázquez MJ et al (2012) BMP8B increases brown adipose tissue thermogenesis through both central and peripheral actions. Cell 149(4):871–885
Qian SW, Tang Y, Li X, Liu Y, Zhang YY, Huang HY et al (2013) BMP4-mediated brown fat-like changes in white adipose tissue alter glucose and energy homeostasis. Proc Natl Acad Sci U S A 110(9):E798–E807
Taha MF, Valojerdi MR, Mowla SJ (2006) Effect of bone morphogenetic protein-4 (BMP-4) on adipocyte differentiation from mouse embryonic stem cells. Anat Histol Embryol 35(4):271–278
Xue R, Wan Y, Zhang S, Zhang Q, Ye H, Li Y (2014) Role of bone morphogenetic protein 4 in the differentiation of brown fat-like adipocytes. Am J Physiol Endocrinol Metab 306(4):E363–E372
Rinotas V, Niti A, Dacquin R, Bonnet N, Stolina M, Han CY et al (2014) Novel genetic models of osteoporosis by overexpression of human RANKL in transgenic mice. J Bone Miner Res 29(5):1158–1169
Kiechl S, Wittmann J, Giaccari A, Knoflach M, Willeit P, Bozec A et al (2013) Blockade of receptor activator of nuclear factor-κB (RANKL) signaling improves hepatic insulin resistance and prevents development of diabetes mellitus. Nat Med 19(3):358–363
Dufresne SS, Dumont NA, Bouchard P, Lavergne É, Penninger JM, Frenette J (2015) Osteoprotegerin protects against muscular dystrophy. Am J Pathol 185(4):920–926
Kuroda Y, Maruyama K, Fujii H, Sugawara I, Ko SB, Yasuda H et al (2016) Osteoprotegerin regulates pancreatic β-cell homeostasis upon microbial invasion. PLoS ONE 11(1):e0146544
Napoli N, Vittinghoff E, Pannacciulli N, Crittenden D, Yun J, Wang A et al (2014) Effect of denosumab on fasting glucose concentrations in postmenopausal women with osteoporosis: results from subjects with diabetes or pre-diabetes from the freedom trial. JBMR. Abstract supple 59
Gannagé-Yared MH, Fares F, Semaan M, Khalife S, Jambart S (2006) Circulating osteoprotegerin is correlated with lipid profile, insulin sensitivity, adiponectin and sex steroids in an ageing male population. Clin Endocrinol (Oxf) 64(6):652–658
Gannagé-Yared MH, Yaghi C, Habre B, Khalife S, Noun R, Germanos-Haddad M et al (2008) Osteoprotegerin in relation to body weight, lipid parameters insulin sensitivity, adipocytokines, and C-reactive protein in obese and non-obese young individuals: results from both cross-sectional and interventional study. Eur J Endocrinol 158(3):353–359
Lasco A, Morabito N, Basile G, Atteritano M, Gaudio A, Giorgianni GM et al (2015) Denosumab inhibition of RANKL and insulin resistance in postmenopausal women with osteoporosis. Calcif Tissue Int 98(2):123–128
Hesse M, Fröhlich LF, Zeitz U, Lanske B, Erben RG (2007) Ablation of vitamin D signaling rescues bone, mineral, and glucose homeostasis in Fgf-23 deficient mice. Matrix Biol 26(2):75–84
Mirza MA, Alsiö J, Hammarstedt A, Erben RG, Michaëlsson K, Tivesten A et al (2011) Circulating fibroblast growth factor-23 is associated with fat mass and dyslipidemia in two independent cohorts of elderly individuals. Arterioscler Thromb Vasc Biol 31(1):219–227
Rowe PS (2012) Regulation of bone-renal mineral and energy metabolism: the PHEX, FGF23, DMP1, MEPE ASARM pathway. Crit Rev Eukaryot Gene Expr 22(1):61–86
Martin A, David V, Quarles LD (2012) Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev 92(1):131–155
García-Martín A, Rozas-Moreno P, Reyes-García R, Morales-Santana S, García-Fontana B, García-Salcedo JA et al (2012) Circulating levels of sclerostin are increased in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab 97(1):234–241
Daniele G, Winnier D, Mari A, Bruder J, Fourcaudot M, Pengou Z et al (2015) Sclerostin and insulin resistance in prediabetes: evidence of a cross talk between bone and glucose metabolism. Diabetes Care 38(8):1509–1517
Hampson G, Edwards S, Conroy S, Blake GM, Fogelman I, Frost ML (2013) The relationship between inhibitors of the Wnt signalling pathway (Dickkopf-1(DKK1) and sclerostin), bone mineral density, vascular calcification and arterial stiffness in post-menopausal women. Bone 56(1):42–47
Morales-Santana S, García-Fontana B, García-Martín A, Rozas-Moreno P, García-Salcedo JA, Reyes-García R et al (2013) Atherosclerotic disease in type 2 diabetes is associated with an increase in sclerostin levels. Diabetes Care 36(6):1667–1674
Pelletier S, Confavreux CB, Haesebaert J, Guebre-Egziabher F, Bacchetta J, Carlier MC et al (2015) Serum sclerostin: the missing link in the bone-vessel cross-talk in hemodialysis patients? Osteoporos Int 26(8):2165–2174
Confavreux CB, Casey R, Varennes A, Goudable J, Chapurlat RD, Szulc P (2016) Has sclerostin a true endocrine metabolic action complementary to osteocalcin in older men? Osteoporos Int 27(7):2301–2309
Elghazi L, Gould AP, Weiss AJ, Barker DJ, Callaghan J, Opland D et al (2012) Importance of β-Catenin in glucose and energy homeostasis. Sci Rep 2:693
Wang RN, Green J, Wang Z, Deng Y, Qiao M, Peabody M et al (2014) Bone morphogenetic protein (BMP) signaling in development and human diseases. Genes Dis 1(1):87–105
Liu H, Fergusson MM, Wu JJ, Rovira II, Liu J, Gavrilova O et al (2011) Wnt signaling regulates hepatic metabolism. Sci Signal 4(158):ra6. doi:10.1126/scisignal.2001249
Krützfeldt J, Stoffel M (2010) Regulation of wingless-type MMTV integration site family (WNT) signalling in pancreatic islets from wild-type and obese mice. Diabetologia 53(1):123–127
Liu Z, Habener JF (2010) Wnt signaling in pancreatic islets. Adv Exp Med Biol 654:391–419
Henley KD, Gooding KA, Economides AN, Gannon M (2012) Inactivation of the dual Bmp/Wnt inhibitor Sostdc1 enhances pancreatic islet function. Am J Physiol Endocrinol Metab 303(6):E752–E761
Bonnet N, Standley KN, Bianchi EN, Stadelmann V, Foti M, Conway SJ et al (2009) The matricellular protein periostin is required for sclerostin inhibition and the anabolic response to mechanical loading and physical activity. J Biol Chem 284(51):35939–35950
Bonnet N, Conway SJ, Ferrari SL (2012) Regulation of beta catenin signaling and parathyroid hormone anabolic effects in bone by the matricellular protein periostin. Proc Natl Acad Sci U S A 109(37):15048–15053
Bonnet N, Garnero P, Ferrari S (2015) Periostin action in bone. Mol Cell Endocrinol 7207(15):30170–30172
Smid JK, Faulkes S, Rudnicki MA (2015) Periostin induces pancreatic regeneration. Endocrinology 156(3):824–836
Vladimirova V, Waha A, Lückerath K, Pesheva P, Probstmeier R (2008) Runx2 is expressed in human glioma cells and mediates the expression of galectin-3. J Neurosci Res 86(11):2450–2461
Zamani N, Brown CW (2011) Emerging roles for the transforming growth factor-{beta} superfamily in regulating adiposity and energy expenditure. Endocr Rev 32(3):387–403
Perros F, Bonnet S (2015) Bone morphogenetic protein receptor type II and inflammation are bringing old concepts into the new pulmonary arterial hypertension world. Am J Respir Crit Care Med 192(7):777–779
Derwall M, Malhotra R, Lai CS, Beppu Y, Aikawa E, Seehra JS et al (2012) Inhibition of bone morphogenetic protein signaling reduces vascular calcification and atherosclerosis. Arterioscler Thromb Vasc Biol 32(3):613–622
Sartori R, Gregorevic P, Sandri M (2014) TGFβ and BMP signaling in skeletal muscle: potential significance for muscle-related disease. Trends Endocrinol Metab 25(9):464–471
Li RX, Yiu WH, Tang SC (2015) Role of bone morphogenetic protein-7 in renal fibrosis. Front Physiol 6:114
Kayed H, Bekasi S, Keleg S, Michalski CW, Giese T, Friess H et al (2007) BGLAP is expressed in pancreatic cancer cells and increases their growth and invasion. Mol Cancer 6:83
Pan J, Xin L, Wang D, Liao Z, Lin JH, Li BR et al (2016) Risk factors for diabetes mellitus in chronic pancreatitis: a cohort of 2011 patients. Medicine (Baltimore) 95(14):e3251
Acknowledgements
I thank Dr. Dominique Pierroz for providing constructive feedback throughout the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was not supported by any funding.
Conflict of interest
Nicolas Bonnet declare that he has no conflict of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
Rights and permissions
About this article

Cite this article
Bonnet, N. Bone-Derived Factors: A New Gateway to Regulate Glycemia. Calcif Tissue Int 100, 174–183 (2017). https://doi.org/10.1007/s00223-016-0210-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-016-0210-y