
Abstract
Our understanding of the control of skeletal metabolism has undergone a dynamic shift in the last two decades, primarily driven by our understanding of energy metabolism. Evidence demonstrating that leptin not only influences bone cells directly, but that it also plays a pivotal role in controlling bone mass centrally, opened up an investigative process that has changed the way in which skeletal metabolism is now perceived. Other central regulators of bone metabolism have since been identified including neuropeptide Y (NPY), serotonin, endocannabinoids, cocaine- and amphetamine-regulated transcript (CART), adiponectin, melatonin and neuromedin U, controlling osteoblast and osteoclast differentiation, proliferation and function. The sympathetic nervous system was originally identified as the predominant efferent pathway mediating central signalling to control skeleton metabolism, in part regulated through circadian genes. More recent evidence points to a role of the parasympathetic nervous system in the control of skeletal metabolism either through muscarinic influence of sympathetic nerves in the brain or directly via nicotinic receptors on osteoclasts, thus providing evidence for broader autonomic skeletal regulation. Sensory innervation of bone has also received focus again widening our understanding of the complex neuronal regulation of bone mass. Whilst scientific advance in this field of bone metabolism has been rapid, progress is still required to understand how these model systems work in relation to the multiple confounders influencing skeletal metabolism, and the relative balance in these neuronal systems required for skeletal growth and development in childhood and maintaining skeletal integrity in adulthood.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the past 20 years, a fascinating story has evolved relating what used to be considered two apparently unrelated organs–brain and bone. We now understand that pathways that connect brain and bone are fundamental in mediating bone metabolism and energy regulation. Even more fascinating to this story is the role of hormones and cytokines that were previously known to be involved in the regulation of energy metabolism and satiety are also in part responsible for the central control of bone mass. Central regulation of bone mass targets two fundamental cell types, osteoblasts and osteoclasts, and possibly the osteocyte. The relative actions of each cell type determines bone modelling changes and therefore longitudinal growth, as well as bone remodelling and bone repair. Over time, we have come to learn that a balance exists between centrally mediated neuronal pathways and neurotransmitters exerting a centrally mediated equilibrium between osteoblasts and osteoclasts, bone formation and bone resorption.
Linking the Brain and Bone Through Fat
The tenet that the brain could mediate bone turnover started with the discovery that leptin, a hormone exclusively produced by adipose tissue [1], had an impact on bone. Leptin is an adipocyte-produced hormone that inhibits appetite and favours energy expenditure primarily through its action on the arcuate nucleus of the hypothalamus [2, 3]. Given that adipocytes and osteoblasts differentiate from the same mesenchymal stem cell, paracrine crosstalk between these two cells was to be expected. Thus, following the discovery of leptin and its receptor, initial studies focused on its peripheral action on bone cells. Collectively, studies suggested that leptin functioned as an osteogenic hormone by directly acting on leptin receptors on the osteoblast [4, 5] and by diminishing osteoclast differentiation and proliferation either directly or indirectly by influencing the ratio of osteoprotegerin (OPG) and RANK ligand [6, 7]. However, this perspective, although classically one that bone researchers applied often, overlooked the physiologic importance of leptin in signalling other aspects of body composition and function, particularly appetite, energy expenditure and reproduction.
Original experiments from the Karsenty group in 2000 demonstrated that the leptin-deficient mouse (Ob/Ob) had a high bone mass phenotype that was primarily restricted to the trabecular compartment [8]. This was despite the absence of substantial gonadal steroid production and infertility [1, 9]. These observations implied that absolute leptin deficiency was skeletally protective despite hormonal changes that would normally result in profound osteopenia. Intracerebroventricular (ICV) infusion of leptin into the third ventricle corrected the high bone mass phenotype providing evidence for central leptinergic control of bone. The ventromedial hypothalamic (VMH) neurons that highly express the leptin receptor (ObRb) were identified through chemical lesioning studies as the central mediators of leptin’s central skeletal control and destruction of VMH neurones recapitulated the high bone mass phenotype. Paradoxically, however, targeted destruction of ObRb on the VMH neurons did not lead to the same high bone mass phenotype, implying that the VMH nucleus is required for central leptinergic control of bone mass, but direct activation was not required [10].
Brain and Bone–The Role of Serotonin
Serotonin (5-hydroxytryptamine–5-HT) is a monoamine neurotransmitter derived from tryptophan, primarily produced by enterochromaffin cells in the gastrointestinal tract by tryptophan hydroxylase 1 (Tph1) and in the central nervous system (CNS) by tryptophan hydorxylase 2 (Tph2) [11]. Centrally, serotonin is a neurotransmitter well known to support cognitive function and emotional well-being and happiness [12, 13]. As serotonin does not cross the blood–brain barrier, central serotonergic actions are not influenced by serum serotonin levels. Following the recent discovery that gut-derived serotonin regulated bone formation through LRP5-dependent pathways [14], attention was turned to the role of brain-derived serotonin in skeletal metabolism. Peripherally, synthesis of gut-derived serotonin by enterochromaffin cells is controlled by the negative regulation that Lrp5 exerts on the expression of tryptophan hydroxylase (Tph1), the rate-limiting enzyme in the serotonin biosynthetic pathway. Interestingly, inactivation of Tph1 is gut and not osteoblast Lrp5 derived. When gut serotonin is released in blood, its free circulating form negatively regulates the bone formation arm of bone remodelling via the osteoblast Htr1b receptor and CREB by inhibiting proliferation [15]. Clinically, this finding is supported by the increase in serum serotonin levels in Lrp5-deficient patients with osteoporosis pseudoglioma (OPPG) syndrome [16] and indirectly by the acceleration of postmenopausal bone loss and reduction in bone mass accrual following the use of selective serotonin uptake inhibitors (SSRIs) [17–19]. However, in contrast, Cui et al. [20] demonstrated that osteocyte specific mutations in LRP5 in mice result in high and low bone mass phenotypes similar to those seen in humans challenging the existence of bone mass regulation by gut-derived LRP5. Moreover, the same group demonstrated normal serotonin levels in three independently generated global Lrp5 knockout mouse strains and suggested that a difference in the measurement of serum serotonin between groups accounted for the difference in findings [21, 22].
Lack of brain serotonin in Tph2 −/− mice results in a low bone mass phenotype resulting from a reduction in bone formation and increase in bone resorption, indicating that brain-derived serotonin exerts a paradoxical effect on bone compared to its peripheral action, in a similar manner to leptin. Furthermore, Tph2-expressing neurons express ObRb, and leptin reduces Tph2 expression and the action potential of brain serotonergic neurons [23]. The discovery that ObRb ablation on serotonergic neurons (Ob/ObSERT−/−) resulted in a high bone mass phenotype similar to that seen in the Ob/Ob mouse, suggested a link between brainstem neurons and the VMH in the central control of bone mass. A connection between the VMH and the brainstem was thus established using axon guidance studies. Finally, the manifestation of a high bone mass phenotype following the deletion of the serotonin receptor gene Htr2c in the VMH provided the receptor-mediated link between brainstem neurons and the VMH [23]. Serotonin initiates the phosphorylation of the transcription factor CREB (cAMP response element binding protein) using a calmodulin kinase (CaMK)-dependent signalling cascade involving CaMKKβ and CaMKIV to decrease the sympathetic tone and increase bone mass accrual [24]. Coincidentally, leptin’s regulation of appetite is also mediated by similar neuronal pathways extending from the brainstem to the arcuate nucleus (ARC) of the hypothalamus via Htr1a and 2b serotonin receptors [25]. Therefore, centrally leptin appears to exert an indirect osteogenic action through the suppression of serotonergic signalling to the VMH, but efferent pathways are required for this signal to be relayed to bone cells.
The Brain and Sympathetic Innervation of Bone
Several lines of evidence pointed towards an efferent sympathetic pathway from the brain controlling skeletal metabolism. Mice treated with isoproterenol, a beta-adrenergic receptor agonist, lose bone mass and conversely treatment with the beta-adrenergic receptor antagonist propranolol protects against ovariectomy-induced bone loss [10]. Surgical removal of the superior cervical ganglion increases bone resorption in rats [26] and mice deficient in dopamine beta-hydroxylase, an enzyme required for the synthesis of noradrenaline also develop high bone mass [10]. Additionally, patients with reflex sympathetic dystrophy, a disease characterised by high sympathetic tone, are prone to low bone mass, that at least in some cases can be mitigated by beta-blockers [27]. Sympathetic over activity has also been proposed as a contributing mechanism for microgravity-induced bone loss during space flight [28]. Recent evidence points to a role of the inner ear vestibular system in the regulation of sympathetic outflow, and thus bone remodelling. Vestibular lesioning in mice results in bone loss predominantly in the lower limbs [29]. An alteration in vestibular function related to space travel and the microgravity environment is therefore thought to contribute to an increase in sympathetic outflow leading to low bone mass independent of change in locomotor activity or change in vestibular function [30].
The link between leptin signalling and bone via a sympathetic efferent was established by Elefteriou et al. [31] through failure to rectify the high bone mass phenotype in beta-2 adrenergic receptor-deficient mice following ICV infusion of leptin. The expression of β2 adrenergic receptors (Adrβ2) in osteoblasts provided another clue to the link in the pathway between hypothalamus and osteoblast before signal transduction, and mice with selective knockout of Adrβ2 on osteoblasts have a high bone mass from increased bone formation and reduced bone resorption. Studies subsequent to this (see later) revealed that sympathetic signalling in osteoblasts is responsible for regulatory control of osteoblast function through inhibition of osteoblast proliferation via circadian clock genes and that the sympathetic nervous system also favours bone resorption by increasing the expression of RANKL [32]. The balance of bone formation and resorption from chronic stress-induced sympathetic activity may shift to favour bone resorption. This is seen with chronic stimulation of β-AR with low-dose agonist treatment in mice, which induces bone loss mainly via enhanced bone resorption [32], suggesting the control of each cell type by the SNS is temporal. Second generation anti-psychotics, such as risperidone, also up-regulate sympathetic tone and uncouple remodelling; these effects can also be blocked with propranolol [33].
Neuropeptide Y (NPY)
The hypothalamus has at least five Y receptors (Y1-5) that respond to Neuropeptide Y [34]. There is a high density of NPY neurons emanating from the arcuate nucleus of the hypothalamus and regulation of appetite is in part controlled by the suppression of NPY expression by leptin [35, 36]. NPY-deficient mice have significantly increased bone mass evident throughout the skeleton, including cortical and cancellous bone as well as axial and appendicular sites. This is associated with enhanced osteoblast activity and elevated expression of bone osteogenic transcription factors, Runx2 and Osterix [37, 38]. Y2 receptor (Y2−/−) deficient mice show an increase in trabecular bone mass, and cortical bone mass is elevated due to increased endosteal and periosteal bone formation secondary to a marked increased osteoblast activity. Conversely, intracerebroventricular infusion of NPY results in a reduction in bone formation [39]. Additionally, knockout of the hypothalamic Y4 receptor together with the Y2 receptor results in a greater increase in trabecular bone formation [40].
As NPY and leptin are associated in the process of energy regulation, and bone formation by hypothalamic neurons as observed in leptin-deficient ob/ob and Y2 receptor null mice, a common pathway relating NPY to leptin was sought by studying the interaction of concomitant leptin and Y2 receptor deficiency in controlling bone in Y2−/− ob/ob double mutant mice. The reduction in osteoblast activity by leptin following NPY overexpression occurred despite the constitutively high NPY, suggesting that the centrally mediated anti-osteogenic effects of leptin are independent of NPY. Conversely, the consistent stimulation of osteoblast activity in the Y2 knockout mouse model, despite suppression of osteoblast activity by increasing leptin levels, indicated that the pathway by which the Y2 receptor regulates bone formation is functionally distinct from the leptin hypothalamic pathway [37]. Baldock has also proposed that NPY may act locally in a paracrine fashion via osteoblastic Y1 receptor signalling to repress bone formation, and that NPY regulation of bone mass is linked with its role in energy metabolism such that in periods of starvation during which NPY levels are elevated, bone formation is down-regulated. Conversely, when energy intake increases, the suppression of NPY allows bone accretion to progress [41].
CNS, Clock Genes and Remodelling
Nearly, all homeostatic functions work in a coordinated manner and are under circadian control. Bone remodelling is no different and is a dynamic process regulated by cycles of bone resorption, quiescence and subsequent bone formation. Given the diurnal variation in a number of bone turnover markers [42], it follows that bone remodelling may also fall under circadian control. There are both central and peripheral circadian rhythms, the former tied directly to light exposure as well nutrient status, the latter primarily mediated by energy availability. Central circadian clocks are located in the hypothalamic suprachiasmatic nucleus and subordinate clocks in peripheral tissues [43]. Circadian genes, such as, Clock, Bmal1, Period (Per1, 2, and 3), Cryptochrome (Cry1 and 2), and Rev-Erbα are central to circadian rhythms [44, 45]. Per1, Per2, Cry1, Bmal1, and Clock all demonstrate robust, rhythmic diurnal expression in bone and in particular osteoblasts [45]. Mice deficient in Per1 and Per2 genes (Per1 −/− ;Per2 −/−) or Cry1 and Cry 2 genes (Cry1 −/− ;Cry2 −/−) exhibit a high bone mass phenotype again with an increase primarily in trabecular bone as a result of increased osteoblast number and bone formation as seen in the Ob/Ob mouse [46]. Paradoxically though, Per1/2-deficient mice have an increase in serum leptin and an elevation in sympathetic tone, a finding previously associated with lower bone mass [10]. Moreover, ICV infusion of leptin in Per-deficient mice does not result in a reduction in bone mass and bone formation parameters as seen in wild-type mice [46]. Thus, clock genes in osteoblasts were found to mediate rather than regulate leptin central control of bone via sympathetic pathways. Clock genes such as Per1 and Per2 are upregulated by sympathetic signalling in osteoblasts and in turn down-regulate bone formation by down-regulating c-myc and in turn cyclin D1, important regulators of osteoblast proliferation. Paradoxically, in the absence of clock gene regulation, AP-1 (c-fos) is upregulated which promotes osteoblast proliferation and bone formation through upregulation of c-myc and cyclin-D1. More recently, a temporal relationship has been identified between the expression of Per1 and osteoblast mineralisation supporting the circadian control of bone formation [47], and the clock gene Bmal1 (brain and muscle Arnt-like protein 1) influences bone resorption by osteoclastic BMAL1 interactions with the SRC family and binding to the Nfatc1 promoter [48]. Clock genes also appear to exert control over stem cell fate as knockdown of Clock and Per2 results in the inhibition of adipocyte differentiation, whilst osteoblastic differentiation is unmodified [49]. Thus, a complex pathway is established whereby the efferent limb of centrally mediated leptin signalling is not only dependent on sympathetic nerves, but clock genes central to circadian regulation.
CART and Bone
Primarily, central regulation of bone mass appears be driven by control of osteoblast proliferation and bone formation. However, the ying and yang in nature should dictate that if there is central mediation of bone formation, then central pathways should exist that influence bone resorption. In fact, the HBM phenotype seen in the Ob/Ob mouse results from high bone turnover with a concomitant rise in both bone formation and bone resorption markers [31]. Indeed, the sympathetic nervous system plays a role in the control of bone resorption as well as bone formation. This occurs indirectly via an osteoblast Adrβ2 mediated increase in RANKL but not osteoprotegerin (OPG) resulting an increase in osteoclast differentiation and bone resorption. This is regulated by ATF4 phosphorylation, an osteoblast-specific CREB/ATF family member essential for osteoblast function [31].
Cocaine- and amphetamine-regulated transcript (Cart) has been identified as a candidate for the central control of bone resorption. CART is a neuropeptide that is expressed throughout the CNS [50] and other organs such as the adrenal glands and pancreas [51, 52]. The involvement of CART in leptinergic control of bone mass again derives from an understanding of the central control of appetite regulation and, in particular, the melanocortin-4 receptor (MC4R). Leptin promotes the expression of CART and mice deficient in Mc4r manifest a high bone mass phenotype with a concomitant rise in hypothalamic Cart expression that correlates with low bone resorption and high bone mass [31]. Conversely, Cart expression is regulated by leptin and is virtually absent in hypothalamic neurons of ob/ob mice [53]. Deletion of one allele of Cart rescues the high bone mass phenotype of the Mc4r −/− mouse by increasing bone resorption without any perturbation in the hormonal abnormalities seen in MC4R deficiency [54]. However, the mechanism by which Cart exerts its control of bone resorption is unclear. In Mc4r-deficient mice, Cart hypothalamic expression as well as CART serum levels are increased [54], and in Cart-deficient mice (Cart −/−), an increase in serum CART rescues the low bone mass phenotype, suggesting that its mode of action is peripheral and perhaps direct rather than central [55]. Thus, two counteracting pathways mediated by leptin appear to exist to control bone resorption. Leptin acts via the SNS and osteoblast Adrβ2 receptors to increase the expression of Rankl and thus promote osteoclast proliferation, whilst conversely, leptin increases the expression of CART which in turn reduces bone resorption by yet to be determined pathways [31]. Therefore, in effect leptin exerts a more balanced homeostatic control of bone resorption via independent central pathways.
The Ying and Yang of the Central Control of Bone Metabolism
The sympathetic (SNS) and parasympathetic nervous systems (PNS) form the two regulatory pathways of the autonomic nervous system. In general, the parasympathetic system is primarily responsible for control of the ‘rest-and-digest’ or ‘feed and breed’ activities that occur when the body is at rest, especially after eating, including sexual arousal, salivation, urination and digestion. Its action is described as being complementary to that of the sympathetic nervous system, which is responsible for activating activities associated with the ‘fight-or-flight’ response. In skeletal biology, this makes perfect sense. Bone formation is not required during the ‘fight-or-flight’ response, and energy expenditure is required in other organ systems thus, the sympathetic nervous system fundamentally controls the down-regulation of bone formation. As with other homeostatic functions, sympathetic activity is usually opposed by parasympathetic pathways acting directly or indirectly. Given that SNS activity results in the suppression bone formation, PNS activity should increase bone formation or reduced bone resorption. Concordantly, mice subjected to subdiaphragmatic sectioning of the vagus nerve, a cranial nerve that carries cholinergic fibres of the PNS, have low vertebral bone mass [56].
The key neurotransmitter of the PNS is acetylcholine. Acetylcholine is biosynthesised by choline acetyl transferase and is stored in small synaptic vesicles and once secreted, targets nicotinic or muscarinic receptors. Original evidence that parasympathetic control of bone may exist came from studies down-regulating muscarinic receptors [57]. Of the five known muscarinic receptors (M1R–M5R), M3R is the only muscarinic receptor subtype that has shown to be key to bone remodelling, and M3R fulfils this function through its neuronal expression and by decreasing sympathetic activity rather than by acting on osteoblasts or osteoclasts. M3R−/− mice are shorter and have a low bone mass phenotype affecting both the axial (vertebrae) and appendicular skeleton resulting from reduction in trabecular bone mass [57]. Although the reduction in bone mass in M3R−/− mice is due to a reduction in osteoblast number and bone formation rate, M3 receptors are not highly expressed in the osteoblast in the same manner as Adrβ2 and osteoblastic specific deletion of M3R (M3Rosb−/−) does not result in an osteogenic phenotype. Instead, parasympathetic pathways in the locus ceruleus located in the brain stem act indirectly via M3R to inhibit sympathetic nerve pathways. The finding that the low bone mass phenotype in M3R mice is rescued by the removal of one allele of Adrβ2 supports the relationship between these autonomic pathways. Further studies have demonstrated that central IL-1 (interleukin-1) signalling may be integral to parasympathetic regulation of bone [58] and that parasympathetic pathways may directly influence bone cells. Rather than an osteoblast-specific effect that has been identified with sympathetic nerve pathways, the PNS targets nicotinic receptors on osteoclasts through release of acetylcholine resulting in an increase in osteoclast apoptosis and favouring high bone mass. Nicotinic receptors are comprised of different subunits α, β, γ, σ, and ε that assemble to form ionic channels [59]. Of these, the nicotinic subtype-α2 receptor appears to be the key receptor involved in PNS osteoclast apoptosis [58]. Thus, the ying and yang of central control of bone mass appears to be closer to harmony. The balance is addressed by the central IL-1–parasympathetic–bone axis that antagonizes the skeletal sympathetic tone favours bone mass accrual either though osteoblast regulation by indirect action on central muscarinic receptors (M3R) or by direct control via nicotinic receptors.
However, our understanding of this pathway is by no means complete. The central neuronal pathways connecting IL1 and the PNS have yet to be established. The balance of autonomic regulation is also under the influence of circadian rhythmicity and interestingly, the autonomic nervous system follows a circadian pattern that matches the bone remodelling cycle. Alongside the circadian sympathetic control of bone metabolism, recent evidence indicates that osteoclast activity is also regulated by clock genes although a link with the PNS has not been established [48]. Sympathetic activity is dominant during day hours when bone resorption activity reaches its peak, whilst the parasympathetic activity dominates night hours when bone formation is more active. Overall, this implies that the remodelling cycle may be rhythmically controlled by the autonomic nervous system [42, 47].
Sensory Innervation of Bone
The strong similarity in the bone phenotype, resulting from sensory denervation and sympathetic hyperfunction, suggests that either sensory nerves functionally interact with the sympathetic nervous system in bone metabolism, or independently promote changes in bone cell activity in an opposing manner to the SNS. The effects of sensory denervation on bone metabolism have previously been examined in animals treated with capsaicin, which destroys unmyelinated and small-diameter myelinated sensory neurons. Previous animal studies examining the effect of sensory nerve denervation have demonstrated a reduction in bone mineral density largely due to an alteration in the trabecular microarchitecture in both the axial and appendicular skeleton related to an increase in osteoclast number, activity and surface area [60, 61]. In vitro evidence points towards a role of CGRP (Calcitonin Gene-Related Peptide) as an efferent neurotransmitter in the sensory innervation of bone. CGRP inhibits osteoclast differentiation and activity by inhibiting the response of osteoclast nuclear factor −κB (NFκB) to RANKL as well as down-regulating TRAP and cathepsin K but without influencing the osteoblast production of OPG and RANKL [62–64]. Recent evidence suggests that bone marrow stromal cells and osteoblasts also have CGRP receptors and CGRP promotes osteoblast proliferation and mineralisation [64]. Given the opposing actions of sympathetic and sensory nerves on skeletal metabolism, an interaction between these pathways has been postulated and evidence supports this. In hypertensive rats, treatment with beta-blockers rectifies the bone loss but also activates sensory neurones with a concomitant release of CGRP [65]; chemical sympathectomy is associated with an increase in the number of CGRP sensory nerve fibres [66]. Thus, in a similar manner to parasympathetic mediation of bone metabolism, sensory pathways provide another homeostatic balance to the reduction in bone formation and increased bone resorption directed by sympathetic pathways.
Other Central Mediators of Remodelling
The number of hormones and peptides centrally mediating the actions of bone cells continues to grow. Neuromedin U (NMU) is a 25-amino-acid neuropeptide located in the brain thought to play diverse roles in regulation of blood pressure, appetite, and gonadal function [67, 68]. NMU deficiency in mice results in a high bone mass phenotype caused by an increase in bone formation [69]. However, NMU receptors are not detectable in osteoblasts, and in vitro NMU appears to have no effect on osteoblast differentiation. However, ICV infusion of NMU in NMU-deficient mice and leptin-deficient mouse (Ob/Ob) rescues the HBM phenotype. NMU appears to act downstream of leptin though interaction with NMU2 receptors located in the paraventricular nucleus of the hypothalamus. The finding that the clock gene Per is down-regulated and that ICV infusion of leptin paradoxically increases bone mass in NMU-deficient mice suggests that NMU may be involved in the sympathetic regulation of clock genes.
Recent evidence suggests that melatonin (N-acetyl-5-methoxy tryptamine) is involved in the control of bone mass, albeit via peripheral action on bone cells. Melatonin is involved in the synchronisation of multiple circadian rhythms including sleep [70], blood pressure regulation [71] and temperature regulation [72]. Two membrane-bound melatonin receptors (MT1 and MT2) exist, and both are expressed on osteoblasts and osteoclasts [73]. As bone remodelling follows a circadian rhythm, the role of melatonin in bone turnover has been questioned. In vitro, melatonin down-regulates PPARγ, thus suppressing adipogenesis and promoting osteoblast lineage commitment [74, 75] and promotes osteoblast differentiation and mineralisation [76]. Osteoclast differentiation and bone resorption are reduced by melatonin indirectly through a reduction in RANKL and a marked rise in OPG. Despite in vitro evidence supporting osteogenic properties of melatonin, in vivo studies are conflicting. In normal physiological conditions, the daily oscillations of the osteogenic markers alkaline phosphatase and PICP are negatively correlated with melatonin in rats [77]. Melatonin supplementation in animals may have a positive [78–81] or negative impact on bone mass [82], or a positive effect when combined with oestrogen [83], but in humans, supplementation with melatonin in postmenopausal osteopenic women has shown positive effects on hip and vertebral BMD gains [84].
The endocannabinoid system also plays a role in the central control of bone mass. The endocannabinoid system mediates its actions via two cannabinoid receptors, CB1 and CB2 with CB1 receptors being predominantly located within the central nervous system, and CB2 receptors located peripherally [85–87]. In vitro studies suggest that CB1 receptor signalling on the presynaptic terminals in bone inhibits noradrenaline release by sympathetic nerves thus in turn, preventing the sympathetic inhibition of bone formation [88]. However, others have shown that in vivo, inactivating CB1 receptors, results in an increase in peak bone mineral density and protects against ovariectomy-induced bone loss, and in vitro promotes osteoclast apoptosis [89]. However, over time, CB1−/− mice develop age-related osteoporosis with reduced bone formation and accumulation of adipocytes in the bone marrow space as marrow stromal cells show an enhanced capacity for adipocyte differentiation [90]. Thus, CB1 regulation of bone mass appears to be maturity-dependent. Interestingly, the influence of CB1 activity on bone may be influenced by genetic background and in turn by gender [91]. In mouse studies, CB1 knockout in two different strains (C57 and CD1) resulted in opposing phenotypes with CD1CB1−/− mice demonstrating a high bone mass (HBM) phenotype and C57CB1−/− mice showing a low bone mass phenotype occurring in the absence of functional CB1 receptors and an increase in osteoclast number and reduction in osteoblast activity. Moreover, compartment specific differences were demonstrated in male and female CD1CB1−/− mice, with the HBM phenotype relating to an increase in trabecular density in male mice compared with cortical expansion in female mice [91]. The degree of maturity was suggested as a possible factor for the difference in skeletal phenotype between strains, but the gender difference in CB1 activity remains unexplained. In humans, traumatic brain injury (TBI) enhances peripheral osteogenesis and acutely stimulates bone formation at peripheral sites. Results from animal studies suggest that TBI induces an increase in diacylglycerol lipases, enzymes essential for the endocannabinoid 2-arachidonoylglyverol (2-AG), leading to an elevation in presynaptic 2-AG, which in turn activates prejunctional sympathetic CB1 receptors, inhibiting the release of norepipherine [92]. Peripherally, the CB2 receptor regulates osteoclast formation and contributes to ovariectomy-induced bone loss [93] and selective CB2 agonists stimulate osteoclastic bone resorption [93, 94]. Results from other studies, however, are contradictory to this demonstrating that CB2 receptor-deficient mice have accelerated age-related trabecular bone loss characterised by increased osteoclast and osteoblast number and activity but a reduced number of osteoblast precursors and that CB2 agonists in vitro promote direct and indirect RANKL-mediated inhibition of osteoclasts [95]. More recent evidence demonstrates that CB2 selective agonists exert a mitogenic effect on osteoblasts via activation of a Gi protein-cyclin D1 and ERK1/2 axis [96, 97]. A comprehensive review of the endocannabinoid regulation of bone mass is published elsewhere [98].
Adiponectin, like leptin, is an adipocyte-derived hormone well known for its role in energy metabolism and its insulin sensitising properties [99]. Paradoxically though, obesity results in a reduction in adiponectin production. Only two studies to date have examined the central effects of adiponectin on bone [100, 101]. Kajimura, Karsenty and colleagues initially demonstrated that adiponectin knockout (adiponectin −/−) mice have a high bone mass resulting from increased bone formation. However, over time, they develop severe low bone mass [101]. This was explained by the fact that early on adiponectin acts directly on osteoblasts to prevent their proliferation and increase osteoblast apoptosis; over time, this is obscured by adiponectin signalling centrally in neurons of the locus ceruleus through FoxO1 dependant pathway (and independent of the known adiponectin receptors-AdipoR1 and R2). This in turn decreases sympathetic tone, thereby increasing bone mass and decreasing energy expenditure thus partially opposing leptin’s influence on the sympathetic nervous system. In a subsequent study, ICV infusion of adiponectin in both wild-type and adiponectin knockout mice increased trabecular bone volume, density, and number and decreased trabecular spacing derived from an increase in osteoblast proliferation and mineralisation and a RANKL-mediated reduction in osteoclastogenesis. Given that the arcuate nucleus of the hypothalamus expresses AdipoR1 and R2 receptors, it was postulated that the hypothalamus may also be involved in adiponectin signalling. Adiponectin ICV infusion resulted in a down-regulation of hypothalamic CB1 receptors with a concurrent increase in the expression of Tph2, Htr2C and Cart [100]. Concordantly, down-regulation of hypothalamic APPL1 (adaptor protein, phosphotyrosine interacting with PH domain and leucine zipper 1) in vivo was associated with a concurrent reduction in Tph2 and a decrease in trabecular microacrhitectural parameters supporting the findings that centrally, adiponectin influences sympathetic tone and bone metabolism, and it does this partly by inducing the hypothalamic APPL1-TPH2 signalling pathway. Moreover, the increased expression of Tph2 and Htr2C and increase in trabecular bone mass with a reduction sympathetic tone agrees with previous evidence showing that serotonin decreases sympathetic tone and increases bone mass [20], and this is regulated by the opposing actions of leptin and adiponectin in the locus cerluleus and possibly the hypothalamus.
Future Directions
Central neuronal pathways controlling skeletal metabolism are clearly numerous and complex. As with many physiological systems in nature, a neuronal pathway controlling bone formation or resorption will be balanced by a pathway mediating an opposing action. The greatest challenge in the field currently is to develop a model system that addresses those complexities more fully and is able to control for the multiple confounding factors that influence skeletal metabolism. And to address that challenge, it will require not just more sophisticated technology, but also a more complete understanding of the physiologic aspects of bone remodelling. We have taken some tentative first steps linking bone turnover to metabolic homeostasis, particularly in relation to bone specific proteins that modulate insulin sensitivity and production. But we have yet to fully appreciate how bone cells use fuel to power formation and resorption, nor do we understand where that energy comes from, and how signals from the bone multicellular unit are transmitted back to the brain to control substrate utilisation. The best news is that the brain has become front and centre in the regulation of bone modelling (Fig. 1).

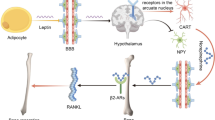
Central neuronal pathways controlling osteoblast and osteoclast proliferation and action. 1 Leptin down-regulates serotonergic neurons via its receptor (ObRb) by reducing Tph2 expression. Serotonin activates sympathetic neurons in the ventromedial hypothalamus, 2 the sympathetic nervous system (SNS) regulates osteoblast function through activation of β2 adrenergic receptors (Adrβ2) to control osteoblast clock genes, and activates AT4 phosphorylation to promote RANK-L production and osteoclast proliferation, 3 leptin promotes the expression of CART (cocaine- and amphetamine-regulated transcript) in the arcuate nucleus of the hypothalamus and reduces bone resorption via undetermined pathways, 4 endocannabinoids act via CB1 receptors at the presynaptic terminals of the sympathetic neurons to inhibit the release of noradrenaline, 5 neuromedin U (NMU) acts via NMU receptors (NMU2R) in the hypothalamic paraventricular nucleus to regulate clock genes by a sympathetic independent pathway, 6 neuropeptide Y acts via Y2 and possibly Y4 receptors in the arcuate nucleus to suppress osteoblast proliferation and function, 7 central interleukin-1 (IL-1) regulates parasympathetic neurons in the locus ceruleus of the brainstem. Parasympathetic nerves indirectly suppress sympathetic control of bone mass via M3R receptors located in the ventromedial hypothalamus, 8 the parasympathetic nervous system (PNS) promotes osteoclast apoptosis via the nicotinic receptor subtype 2 (NRα2), 9 adiponectin acts via hypothalamic AdipoR1 and R2 receptors in the arcuate nucleus to upregulate CART and down-regulate CB1. Adiponectin also acts in the Locus Ceruleus (receptor not yet known) to promote the serotonergic regulation of bone via FoxO1, 10 calcitonin gene-related peptide (CGRP) from sensory nerves inhibits the response of osteoclasts to RANKL
References
Zhang Y et al (1994) Positional cloning of the mouse obese gene and its human homologue. Nature 372(6505):425–432
Halaas JL et al (1995) Weight-reducing effects of the plasma protein encoded by the obese gene. Science 269(5223):543–546
Friedman J (2014) 20 years of leptin: leptin at 20: an overview. J Endocrinol 223(1):T1–T8
Thomas T et al (1999) Leptin acts on human marrow stromal cells to enhance differentiation to osteoblasts and to inhibit differentiation to adipocytes. Endocrinology 140(4):1630–1638
Cornish J et al (2002) Leptin directly regulates bone cell function in vitro and reduces bone fragility in vivo. J Endocrinol 175(2):405–415
Gordeladze JO et al (2002) Leptin stimulates human osteoblastic cell proliferation, de novo collagen synthesis, and mineralization: impact on differentiation markers, apoptosis, and osteoclastic signaling. J Cell Biochem 85(4):825–836
Holloway WR et al (2002) Leptin inhibits osteoclast generation. J Bone Miner Res 17(2):200–209
Ducy P et al (2000) Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell 100(2):197–207
Tartaglia LA et al (1995) Identification and expression cloning of a leptin receptor. OB-R. Cell 83(7):1263–1271
Takeda S et al (2002) Leptin regulates bone formation via the sympathetic nervous system. Cell 111(3):305–317
Walther DJ et al (2003) Synthesis of serotonin by a second tryptophan hydroxylase isoform. Science 299(5603):76
Schmitt JA et al (2006) Serotonin and human cognitive performance. Curr Pharm Des 12(20):2473–2486
Alenina N, Klempin F (2015) The role of serotonin in adult hippocampal neurogenesis. Behav Brain Res 277:49–57
Kode A et al (2014) Lrp5 regulation of bone mass and serotonin synthesis in the gut. Nat Med 20(11):1228–1229
Yadav VK et al (2008) Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell 135(5):825–837
Gong Y et al (2001) LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 107(4):513–523
Feuer AJ et al (2015) Use of selective serotonin reuptake inhibitors and bone mass in adolescents: an NHANES study. Bone 78:28–33
Sheu YH et al (2015) SSRI use and risk of fractures among perimenopausal women without mental disorders. Inj Prev 21(6):397–403
Rauma PH et al (2016) Effects of antidepressants on postmenopausal bone loss—A 5 year longitudinal study from the OSTPRE cohort. Bone 89:25–31
Cui Y et al (2011) Lrp5 functions in bone to regulate bone mass. Nat Med 17(6):684–691
Cui Y et al (2014) Reply to Lrp5 regulation of bone mass and gut serotonin synthesis. Nat Med 20(11):1229–1230
Kode A et al (2014) Lrp5 regulation of bone mass and serotonin synthesis in the gut. Nat Med 20(11):1228–1229
Yadav VK et al (2009) A serotonin-dependent mechanism explains the leptin regulation of bone mass, appetite, and energy expenditure. Cell 138(5):976–989
Oury F et al (2010) CREB mediates brain serotonin regulation of bone mass through its expression in ventromedial hypothalamic neurons. Genes Dev 24(20):2330–2342
Yadav VK et al (2011) Leptin-dependent serotonin control of appetite: temporal specificity, transcriptional regulation, and therapeutic implications. J Exp Med 208(1):41–52
Sandhu HS, Herskovits MS, Singh IJ (1987) Effect of surgical sympathectomy on bone remodeling at rat incisor and molar root sockets. Anat Rec 219(1):32–38
Schwartzman RJ (2000) New treatments for reflex sympathetic dystrophy. N Engl J Med 343(9):654–656
Mano T, Nishimura N, Iwase S (2010) Sympathetic neural influence on bone metabolism in microgravity. Acta Physiol Hung 97(4):354–361
Vignaux G et al (2015) Inner ear vestibular signals regulate bone remodeling via the sympathetic nervous system. J Bone Miner Res 30(6):1103–1111
Vignaux G et al (2013) Bone remodeling is regulated by inner ear vestibular signals. J Bone Miner Res 28(10):2136–2144
Elefteriou F et al (2005) Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature 434(7032):514–520
Kondo H, Togari A (2011) Continuous treatment with a low-dose beta-agonist reduces bone mass by increasing bone resorption without suppressing bone formation. Calcif Tissue Int 88(1):23–32
Motyl KJ et al (2015) Propranolol attenuates risperidone-induced trabecular bone loss in female mice. Endocrinology 156(7):2374–2383
Blomqvist AG, Herzog H (1997) Y-receptor subtypes–how many more? Trends Neurosci 20(7):294–298
Stanley BG et al (1986) Neuropeptide Y chronically injected into the hypothalamus: a powerful neurochemical inducer of hyperphagia and obesity. Peptides 7(6):1189–1192
Erickson JC, Hollopeter G, Palmiter RD (1996) Attenuation of the obesity syndrome of ob/ob mice by the loss of neuropeptide Y. Science 274(5293):1704–1707
Baldock PA et al (2005) Hypothalamic control of bone formation: distinct actions of leptin and Y2 receptor pathways. J Bone Miner Res 20(10):1851–1857
Baldock PA et al (2009) Neuropeptide Y knockout mice reveal a central role of NPY in the coordination of bone mass to body weight. PLoS One 4(12):e8415
Baldock PA et al (2006) Hypothalamic regulation of cortical bone mass: opposing activity of Y2 receptor and leptin pathways. J Bone Miner Res 21(10):1600–1607
Sainsbury A et al (2003) Synergistic effects of Y2 and Y4 receptors on adiposity and bone mass revealed in double knockout mice. Mol Cell Biol 23(15):5225–5233
Shi YC, Baldock PA (2012) Central and peripheral mechanisms of the NPY system in the regulation of bone and adipose tissue. Bone 50(2):430–436
Shao P, Ohtsuka-Isoya M, Shinoda H (2003) Circadian rhythms in serum bone markers and their relation to the effect of etidronate in rats. Chronobiol Int 20(2):325–336
Buijs FN et al (2016) The circadian system: a regulatory feedback network of periphery and brain. Physiology (Bethesda) 31(3):170–181
Reppert SM, Weaver DR (2002) Coordination of circadian timing in mammals. Nature 418(6901):935–941
Schibler U, Sassone-Corsi P (2002) A web of circadian pacemakers. Cell 111(7):919–922
Fu L et al (2005) The molecular clock mediates leptin-regulated bone formation. Cell 122(5):803–815
McElderry JD et al (2013) Tracking circadian rhythms of bone mineral deposition in murine calvarial organ cultures. J Bone Miner Res 28(8):1846–1854
Xu C et al (2016) Circadian clock regulates bone resorption in mice. J Bone Miner Res 31(7):1344–1355
Boucher H et al (2016) Circadian clock genes modulate human bone marrow mesenchymal stem cell differentiation, migration and cell cycle. PLoS One 11(1):e0146674
Kuhar MJ et al (2002) CART peptides. Neuropeptides 36(1):1–8
Vicentic A (2006) CART peptide diurnal variations in blood and brain. Peptides 27(8):1942–1948
Wierup N, Sundler F (2006) CART is a novel islet regulatory peptide. Peptides 27(8):2031–2036
Kristensen P et al (1998) Hypothalamic CART is a new anorectic peptide regulated by leptin. Nature 393(6680):72–76
Ahn JD et al (2006) Cart overexpression is the only identifiable cause of high bone mass in melanocortin 4 receptor deficiency. Endocrinology 147(7):3196–3202
Singh MK, Elefteriou F, Karsenty G (2008) Cocaine and amphetamine-regulated transcript may regulate bone remodeling as a circulating molecule. Endocrinology 149(8):3933–3941
Tien D et al (2003) Vagal afferents are necessary for the establishment but not the maintenance of kainic acid-induced hyperalgesia in mice. Pain 102(1–2):39–49
Shi Y et al (2010) Signaling through the M(3) muscarinic receptor favors bone mass accrual by decreasing sympathetic activity. Cell Metab 11(3):231–238
Bajayo A et al (2012) Skeletal parasympathetic innervation communicates central IL-1 signals regulating bone mass accrual. Proc Natl Acad Sci USA 109(38):15455–15460
Eimar H et al (2013) Cholinergic regulation of bone. J Musculoskelet Neuronal Interact 13(2):124–132
Offley SC et al (2005) Capsaicin-sensitive sensory neurons contribute to the maintenance of trabecular bone integrity. J Bone Miner Res 20(2):257–267
Ding Y et al (2010) Effects of capsaicin-induced sensory denervation on bone metabolism in adult rats. Bone 46(6):1591–1596
Cornish J et al (2001) Effects of calcitonin, amylin, and calcitonin gene-related peptide on osteoclast development. Bone 29(2):162–168
Ishizuka K et al (2005) Inhibitory effect of CGRP on osteoclast formation by mouse bone marrow cells treated with isoproterenol. Neurosci Lett 379(1):47–51
Wang L et al (2010) Calcitonin-gene-related peptide stimulates stromal cell osteogenic differentiation and inhibits RANKL induced NF-kappaB activation, osteoclastogenesis and bone resorption. Bone 46(5):1369–1379
Okajima K et al (2004) Activation of capsaicin-sensitive sensory neurons by carvedilol, a nonselective beta-blocker, in spontaneous hypertensive rats. J Pharmacol Exp Ther 309(2):684–691
Cherruau M et al (2003) Chemical sympathectomy-induced changes in TH-, VIP-, and CGRP-immunoreactive fibers in the rat mandible periosteum: influence on bone resorption. J Cell Physiol 194(3):341–348
Brighton PJ, Szekeres PG, Willars GB (2004) Neuromedin U and its receptors: structure, function, and physiological roles. Pharmacol Rev 56(2):231–248
Vigo E et al (2007) Novel role of the anorexigenic peptide neuromedin U in the control of LH secretion and its regulation by gonadal hormones and photoperiod. Am J Physiol Endocrinol Metab 293(5):E1265–E1273
Sato S et al (2007) Central control of bone remodeling by neuromedin U. Nat Med 13(10):1234–1240
Redman J, Armstrong S, Ng KT (1983) Free-running activity rhythms in the rat: entrainment by melatonin. Science 219(4588):1089–1091
Grossman E, Laudon M, Zisapel N (2011) Effect of melatonin on nocturnal blood pressure: meta-analysis of randomized controlled trials. Vasc Health Risk Manag 7:577–584
Cagnacci A, Elliott JA, Yen SS (1992) Melatonin: a major regulator of the circadian rhythm of core temperature in humans. J Clin Endocrinol Metab 75(2):447–452
Slominski RM et al (2012) Melatonin membrane receptors in peripheral tissues: distribution and functions. Mol Cell Endocrinol 351(2):152–166
Radio NM, Doctor JS, Witt-Enderby PA (2006) Melatonin enhances alkaline phosphatase activity in differentiating human adult mesenchymal stem cells grown in osteogenic medium via MT2 melatonin receptors and the MEK/ERK (1/2) signaling cascade. J Pineal Res 40(4):332–342
Zhang L et al (2010) Melatonin inhibits adipogenesis and enhances osteogenesis of human mesenchymal stem cells by suppressing PPARgamma expression and enhancing Runx2 expression. J Pineal Res 49(4):364–372
Park KH et al (2011) Melatonin promotes osteoblastic differentiation through the BMP/ERK/Wnt signaling pathways. J Pineal Res 51(2):187–194
Ostrowska Z et al (2002) The relationship between the daily profile of chosen biochemical markers of bone metabolism and melatonin and other hormone secretion in rats under physiological conditions. Neuro Endocrinol Lett 23(5–6):417–425
Koyama H et al (2002) Melatonin at pharmacologic doses increases bone mass by suppressing resorption through down-regulation of the RANKL-mediated osteoclast formation and activation. J Bone Miner Res 17(7):1219–1229
Uslu S et al (2007) Constructive effect of exogenous melatonin against osteoporosis after ovariectomy in rats. Anal Quant Cytol Histol 29(5):317–325
Egermann M et al (2011) Pinealectomy affects bone mineral density and structure–an experimental study in sheep. BMC Musculoskelet Disord 12:271
Witt-Enderby PA et al (2012) Effects on bone by the light/dark cycle and chronic treatment with melatonin and/or hormone replacement therapy in intact female mice. J Pineal Res 53(4):374–384
Ladizesky MG et al (2001) Effect of melatonin on bone metabolism in ovariectomized rats. Life Sci 70(5):557–565
Cardinali DP et al (2003) Melatonin effects on bone: experimental facts and clinical perspectives. J Pineal Res 34(2):81–87
Amstrup AK et al (2015) Melatonin improves bone mineral density at the femoral neck in postmenopausal women with osteopenia: a randomized controlled trial. J Pineal Res 59(2):221–229
Howlett AC (2002) The cannabinoid receptors. Prostaglandins Other Lipid Mediat 68–69:619–631
Pertwee RG, Ross RA (2002) Cannabinoid receptors and their ligands. Prostaglandins Leukot Essent Fatty Acids 66(2–3):101–121
Mackie K (2008) Signaling via CNS cannabinoid receptors. Mol Cell Endocrinol 286(1–2 Suppl 1):S60–S65
Ishac EJ et al (1996) Inhibition of exocytotic noradrenaline release by presynaptic cannabinoid CB1 receptors on peripheral sympathetic nerves. Br J Pharmacol 118:2023–2028
Idris AI et al (2005) Regulation of bone mass, bone loss and osteoclast activity by cannabinoid receptors. Nat Med 11(7):774–779
Idris AI et al (2009) Cannabinoid receptor type 1 protects against age-related osteoporosis by regulating osteoblast and adipocyte differentiation in marrow stromal cells. Cell Metab 10(2):139–147
Tam J et al (2006) Involvement of neuronal cannabinoid receptor CB1 in regulation of bone mass and bone remodeling. Mol Pharmacol 70(3):786–792
Tam J et al (2008) The cannabinoid CB1 receptor regulates bone formation by modulating adrenergic signaling. FASEB J. 22(1):285–294
Idris AI et al (2008) Regulation of bone mass, osteoclast function, and ovariectomy-induced bone loss by the type 2 cannabinoid receptor. Endocrinology 149(11):5619–5626
Whyte LS et al (2012) Cannabinoids and bone: endocannabinoids modulate human osteoclast function in vitro. Br J Pharmacol 165(8):2584–2597
Ofek O et al (2006) Peripheral cannabinoid receptor, CB2, regulates bone mass. Proc Natl Acad Sci USA 103(3):696–701
Ofek O et al (2011) CB2 cannabinoid receptor targets mitogenic Gi protein-cyclin D1 axis in osteoblasts. J Bone Miner Res 26(2):308–316
Sophocleous A et al (2011) The type 2 cannabinoid receptor regulates bone mass and ovariectomy-induced bone loss by affecting osteoblast differentiation and bone formation. Endocrinology 152(6):2141–2149
Idris AI, Ralston SH (2012) Role of cannabinoids in the regulation of bone remodeling. Front Endocrinol (Lausanne) 3:136
Stefan N, Stumvoll M (2002) Adiponectin–its role in metabolism and beyond. Horm Metab Res 34(9):469–474
Wu Y et al (2014) Central adiponectin administration reveals new regulatory mechanisms of bone metabolism in mice. Am J Physiol Endocrinol Metab 306(12):E1418–E1430
Kajimura D et al (2013) Adiponectin regulates bone mass via opposite central and peripheral mechanisms through FoxO1. Cell 17(6):901–915
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Dimitri, P., Rosen, C. The Central Nervous System and Bone Metabolism: An Evolving Story. Calcif Tissue Int 100, 476–485 (2017). https://doi.org/10.1007/s00223-016-0179-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-016-0179-6