
Abstract
Vascular calcification significantly contributes to mortality in chronic kidney disease (CKD) patients. Sevelamer and pyrophosphate (PPi) have proven to be effective in preventing vascular calcification, the former by controlling intestinal phosphate absorption, the latter by directly interfering with the hydroxyapatite crystal formation. Since most patients present with established vascular calcification, it is important to evaluate whether these compounds may also halt or reverse the progression of preexisting vascular calcification. CKD and vascular calcification were induced in male Wistar rats by a 0.75 % adenine low protein diet for 4 weeks. Treatment with PPi (30 or 120 µmol/kg/day), sevelamer carbonate (1500 mg/kg/day) or vehicle was started at the time point at which vascular calcification was present and continued for 3 weeks. Hyperphosphatemia and vascular calcification developed prior to treatment. A significant progression of aortic calcification in vehicle-treated rats with CKD was observed over the final 3-week period. Sevelamer treatment significantly reduced further progression of aortic calcification as compared to the vehicle control. No such an effect was seen for either PPi dose. Sevelamer but not PPi treatment resulted in an increase in both osteoblast and osteoid perimeter. Our study shows that sevelamer was able to reduce the progression of moderate to severe preexisting aortic calcification in a CKD rat model. Higher doses of PPi may be required to induce a similar reduction of severe established arterial calcification in this CKD model.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Calcification in the arteries is the most common life-threatening complication in patients with chronic kidney disease (CKD). Current therapies to treat vascular calcification in CKD mainly consist of controlling the mineral disturbances such as hyperphosphatemia. Despite the fact that this treatment strategy has proven to reduce arterial calcification to some extent in new dialysis patients and in an experimental model of CKD [1, 2], research on alternative forms of treatment is required. Arterial calcification is a multifactorial complication induced by various promoters and a lack of inhibitors which makes it difficult to control the various provocative factors by current treatment strategies such as phosphate binding agents. Therefore, an alternative therapeutic approach for the treatment of vascular calcification consisting of the direct interference with the mineralization process in the vessel wall, independently of the causal factors, could create new opportunities for more efficient therapy.
Pyrophosphate (PPi) is an endogenous calcification inhibitor whose local concentration depends on three regulatory factors: (1) the rate-limiting enzyme ectonucleotide pyrophosphatase phosphodiesterase (Enpp1) which intracellularly hydrolyzes ATP to AMP and thereby generates PPi (2) the transmembrane protein ankyrin (ANK) responsible for shuttling PPi in the extracellular space and (3) tissue nonspecific alkaline phosphatase (TNAP) which degrades excess extracellular PPi into phosphate ions [3]. PPi exerts its function, in great part, by binding to nascent hydroxyapatite crystals and as such prevents further incorporation of inorganic phosphate into these crystals [4]. Both in vitro and in vivo experiments have shown PPi to be a potent inhibitor of vascular calcification. A study by Villa-Bellosta et al. [5] has reported that calcium phosphate deposition in the vessel wall can occur passively by the lack of calcification inhibitors which in a next step triggers osteogenic transdifferentiation of vascular smooth muscle cells thereby modulating mineralization of the extracellular matrix in a well-organized crystalline structure. The protective role of PPi was demonstrated in cultured rat aortas exposed to high phosphate levels [6]. In addition, exogenous PPi was shown to inhibit aortic calcification in vitamin D-treated rats [7], adenine-induced uremic rats [8] and a uremic apolipoprotein E gene knockout mouse model [9].
In hemodialysis patients, plasma PPi levels are reduced and decrease further after each dialysis session due to dialytic clearance [10]. The decrease in PPi has further been attributed to the upregulation of TNAP, as shown in arteries of uremic rats, which led to an increased hydrolysis of PPi [11]. Apart from this, a negative association of plasma PPi levels with vascular calcification has been demonstrated in hemodialysis and peritoneal dialysis patients as well as in CKD stage 4 patients not yet on dialysis [12]. A recent publication of O’Neill’s research group nicely showed that the systemic deficiency of PPi plays a direct role in aortic calcification [13]. These studies show that this small molecule is of substantial importance in CKD-related arterial calcifications.
For a number of years, focus had been on bisphosphonates, which are nonhydrolysable PPi analogs worldwide prescribed for the treatment of osteoporosis. As severe bone loss is a common complication of CKD, bisphosphonates were put forward as potential treatment to preserve bone density in this population. In fact, several experimental studies have shown the ability of bisphosphonates to reduce the development of experimentally induced medial calcification [14, 15]. However, the safety and efficacy of bisphosphonates in CKD patients, as compared to subjects with normal renal function, are called into question. In CKD, bisphosphonates pose potential risks for adynamic bone disease and defective mineralization which in turn may exacerbate vascular calcifications [14, 16].
In contrast to bisphosphonates, PPi is degraded in the bone by the high level of exposure to alkaline phosphatase, constantly expressed by osteoblasts, and keeping physiological bone mineralization intact. Therefore, it is of particular interest to investigate the effect of PPi on vascular calcification as well as on the bone status in rats with adenine-induced CKD. One model that has been extensively used to study vascular calcification in CKD is the adenine-induced CKD rat model. Rats fed a 0.75 % adenine low protein diet during 4 weeks show mineral deposits from week 3 on which aggregate to severe, macroscopic distinguishable calcifications in the tunica media of the aorta of all animals after 8 weeks of CKD [17, 18]. In previous studies, using this model, we evaluated the effect of phosphate-lowering agents on the development of vascular calcifications [19]. Also, O’Neill and Riser [8] used this model to show that peritoneal administration of PPi (simulating peritoneal dialysis treatment) was able to prevent vascular calcification. However, no experimental studies have been reported evaluating the effect of sevelamer or PPi on the progression or reversal of established medial calcification in this model.
Animals and Methods
Study Setup
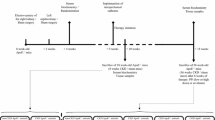
Male Wistar rats were purchased (250 g, Iffa Credo, Brussels, Belgium) and housed two per cage for the duration of the study in a temperature-controlled (22 °C) room with a strict 12-h light/dark cycle. Food and water were provided ad libitum. After a 2-week equilibration period on a high phosphate diet (1.03 % P, 1.06 % Ca) (SSNIFF Spezialdiäten, Soest, Germany), animals were maintained for 4 weeks on a 0.75 % adenine low protein diet (0.92 % P, 1.0 % Ca, 2.5 % protein) to induce CKD. Treatments were started for 3 weeks from stop of adenine treatment on. Following CKD induction, the animals were randomly assigned to the following treatment groups (Fig. 1): (1) CKD rats killed at the stop of adenine treatment (baseline uremic controls, n = 12), (2) CKD rats receiving peritoneal dialysis fluid containing no PPi during 3 weeks via intraperitoneal catheterization (Vehicle, n = 14), (3) CKD rats receiving 30 µmol/kg/day of PPi dissolved in peritoneal dialysis fluid during 3 weeks via intraperitoneal catheterization (PPi 30, n = 14), (4) CKD rats receiving 120 µmol/kg/day of PPi dissolved in peritoneal dialysis fluid during 3 weeks via intraperitoneal catheterization (PPi 120, n = 14) and (5) CKD rats receiving 1500 mg/kg/day of sevelamer carbonate (Renvela®) during 3 weeks via gavage (Sevelamer, n = 14). For the implantation of the intraperitoneal catheters, rats were anesthetized by intraperitoneal injection with 60 mg/kg sodium pentobarbital (Nembutal, Ceva Santé Animale, France).

Study design. CKD—chronic kidney disease
Blood and 24-h urine samples were collected at various time points for measurement of a series of biochemical parameters (Fig. 1). At killing, animals were exsanguinated through the retro-orbital plexus after anesthesia with 60 mg/kg sodium pentobarbital (Nembutal) via intraperitoneal injection.
Biochemical Analyses
Serum creatinine was determined according to the Jaffé method. Total calcium in serum and urine was measured with flame atomic absorption spectrometry (Perkin-Elmer, Wellesley, MA, USA) after appropriate dilution in 0.1 % La(NO3)3 to eliminate chemical interferences. Serum and urinary phosphorus were measured using the Ecoline®S Phosphate kit (DiaSys, Holzheim, Germany). Serum iPTH (Immutopics, San Clemente, CA, USA) levels were determined by use of an ELISA kit.
Assessment of Vascular Calcification
After isolation of the thoracic aorta, the tissue was fixed in neutral buffered formalin for 90 min and cut into sections of 2–3 mm. These sections were embedded upright in a paraffin block, and 4-µm sections were stained for calcification with von Kossa’s method and counterstained with hematoxylin and eosin. The percentage of calcified area was calculated using AxioVision image analysis software (Release 4.5, Carl Zeiss, Oberkochen, Germany) in which, based on two color separation thresholds, the total tissue area and the von Kossa-positive area are measured. After summing both absolute areas, the percentage of calcified area was calculated as the ratio of the von Kossa-positive area versus the total tissue area.
The proximal part of the abdominal aorta and the left carotid and femoral artery were isolated and weighed on a precision balance. Subsequently, the samples were digested in 65 % HNO3 at 60 °C overnight. The calcium content of each artery was measured with flame atomic absorption spectrometry and expressed as mg calcium/g wet tissue.
Histomorphometric Analysis of Static Bone Parameters
Sections of the left tibia were Goldner stained for histomorphometric analysis of the proximal metaphysis with AxioVision Release 4.5 software. Out of primary measurements, the following static bone parameters were calculated: bone area, osteoid area, osteoid perimeter, osteoid width, eroded perimeter, osteoblasts on total perimeter and osteoclasts on total perimeter [20]. As a result of the severe secondary hyperparathyroidism, inherent to the CKD rat model used in the present study, and the chaotic incorporation of tetracycline labels going along herewith, quantitative measurement of dynamic bone parameters was not possible.
Statistical Analysis
Results were expressed as mean ± SD, unless indicated otherwise. Nonparametric statistical analyses were performed with SPSS 20.0 software. To compare results of one specific study group obtained at different time points, a Friedman test was followed by a Wilcoxon signed-rank test when significance was reached. Comparison among groups at one specific time point was performed by a Kruskall–Wallis test, followed by a Mann–Whitney U test when significant. Bonferroni correction was applied when more than two groups were compared. A p value <0.05 was considered statistically significant.
Results
Mortality was relatively low and similar in the different treatment groups: 9, 10, 11 and 10 animals out of 14 survived until the end of the study in the CKD group treated with vehicle, PPi 30, PPi 120 and sevelamer, respectively. No mortality was noted in the baseline uremic control group which was killed after 4 weeks of CKD. There were no differences in food, water consumption and body weight between the different study groups at the various time points.
Renal Function and Mineral Metabolism
As indicated by the serum creatinine concentrations (Fig. 2a), adenine treatment resulted in severe CKD. After stopping adenine feeding, partial recovery of renal function was seen due to discontinuation of this renal toxin. However, serum creatinine levels at the end of the study were still significantly elevated as compared to normal values at baseline and no significant differences between treatment groups were seen. Induction of CKD led to reduced phosphorus excretion in all groups (Fig. 3a), which in turn resulted in phosphorus retention. The high dose of PPi produced a partial blunting of this reduction at the end of the study as compared to the vehicle group. Serum phosphorus levels were in line with the respective serum creatinine values and show that all animals developed serious hyperphosphatemia after 4 weeks of CKD (Fig. 2b). Treatment with either PPi or sevelamer between 4 and 7 weeks after CKD induction did not affect serum phosphorus levels at the end of the study as compared to vehicle-treated rats. Inherent to the development of CKD, serum calcium levels were decreased after 4 weeks of adenine treatment, which was maintained at week 7 for all treatment groups with exception of the sevelamer group (Fig. 2c). Treatment with sevelamer significantly increased total calcium in the serum at the end of the study. Serum ionized calcium levels evolved in a more or less similar way, but no significant differences were seen between the study groups at any time point (Fig. 2e). Urinary calcium excretion was significantly increased in the groups treated with PPi 120 and sevelamer at the end of the study as compared to baseline levels, and in the sevelamer group as compared to the vehicle group at week 7 (Fig. 3b). Serum creatinine, phosphorus and calcium levels of the group that was killed after 4 weeks of CKD (i.e., the uremic baseline group) did not differ from the other study groups at baseline and after 4 weeks of CKD and are shown in a supplementary file (Online Resource 1).

Serum parameters of renal function and mineral metabolism: creatinine (a), phosphorus (b), calcium (c), PTH (d) and ionized calcium (e) in CKD rats treated with vehicle (n = 9), PPi 30 (n = 10), PPi 120 (n = 11) or sevelamer (n = 10). °p < 0.05 versus week 0 within the same group. *p < 0.05 versus vehicle at the same time point. # p < 0.05 versus normal values

Urinary parameters of mineral metabolism: phosphorus (a) and calcium (b) in CKD rats treated with vehicle (n = 9), PPi 30 (n = 10), PPi 120 (n = 11) or sevelamer (n = 10). °p < 0.05 versus week 0 within the same group. *p < 0.05 versus vehicle at the same time point
Consistent with the induction of renal failure and the development of hyperphosphatemia and hypocalcemia, a six-fold increase in serum iPTH levels was seen in all groups in comparison with historical values of rats with normal renal function [21] (Fig. 2d).
Evaluation of Vascular Calcification
Figure 4 shows the total calcium content in the abdominal aorta and the percentage calcified area, i.e., von Kossa-positive surface, in the thoracic aorta. Both von Kossa staining and measurement of the total calcium content in the aorta show the development of distinct vascular calcifications in all but one animal of the uremic control group; i.e., at the stop of adenine treatment. Although there was no significant progression in the area of calcification of the aorta, there was a significant increase in the calcium content of the aorta observed after 3 weeks of vehicle treatment (50 ± 25 mg calcium/g wet tissue) as compared to rats killed after 4 weeks of CKD (baseline) (20 ± 13 mg calcium/g wet tissue). Treatment with PPi either at 30 µmol/kg/day (55 ± 28 mg calcium/g wet tissue) or at 120 µmol/kg/day dose (49 ± 28 mg calcium/g wet tissue) did not halt or reverse this progression of calcification in the aorta. However, 3 weeks of sevelamer treatment at a 1500 mg/kg/day dose prevented the increase in aortic calcium content (33 ± 19 mg calcium/g wet tissue) between week 4 and 7. This beneficial effect was not confirmed in the femoral and carotid arteries where progression of calcification was not observed between weeks 4 and 7 (data not presented).

The calcium content of the aorta (a) and the percentage calcified aortic area (b) in untreated CKD rats killed at week 4 (n = 12) and CKD rats treated with vehicle (n = 9), PPi 30 (n = 10), PPi 120 (n = 11) or sevelamer (n = 10) for 3 weeks (from week 4 to week 7). Data are presented as individual values. *p < 0.05 versus CKD at week 4 (Mann–Whitney U test with Bonferroni correction for multiple comparisons)
Figure 5 presents representative photographs of aortic sections of untreated CKD rats killed at week 4 (baseline) and CKD rats treated with vehicle, PPi 30 or sevelamer at week 7.

Von Kossa-stained aortic sections of untreated CKD rats killed at week 4 and CKD rats treated with vehicle, PPi 30 or sevelamer at week 7
Histomorphometric Analysis of Static Bone Parameters
Static bone parameters are presented in Fig. 6. Vehicle-treated CKD animals showed no differences in static bone parameters after the 3-week treatment period versus the uremic control group at stop of adenine treatment. Treatment with PPi also did not induce significant changes in the various histological bone parameters during the treatment period at either dose tested. However, a significant increase in osteoid and osteoblast perimeter was seen in the sevelamer-treated animals (Fig. 6c, e).

Static bone parameters in the groups under study: bone area (a), osteoid area (b), osteoid perimeter (c), eroded perimeter (d), osteoblast perimeter (e) and osteoclast perimeter (f) in untreated CKD rats killed at week 4 (n = 12) and CKD rats treated with vehicle (n = 9), PPi 30 (n = 10), PPi 120 (n = 11) or sevelamer (n = 10) for 3 weeks (from week 4 to week 7). °p < 0.05 versus vehicle-treated CKD rats
Discussion
Vascular calcification is a prominent feature of vascular disease and is rapidly progressive in CKD patients [22]. Until now, experimental studies have focused on the effect of therapeutics in the prevention of vascular calcification [2, 19, 23], which means that compounds are administered before the onset of calcification in the arteries. However, at the time patients are being diagnosed with CKD, calcification in the vasculature is often present. In nondialysis CKD patients, the prevalence of vascular calcification has been reported to vary between 47 and 64 %, suggesting that the process of calcification starts at early stages of CKD [24–26]. As most patients present themselves in the clinic with a given degree of vascular calcification which progresses rapidly as renal function deteriorates, it is of utmost importance to evaluate the effects of (potential) therapeutic agents on the progression or even regression of preexisting calcification. Few clinical trials have reported the effect of phosphate binding agents or calcimimetics on the progression of established calcification. Sevelamer has been shown to attenuate the progression of vascular calcification in predialysis patients [27], as well as in prevalent [28, 29] and incident [1] hemodialysis patients in comparison with calcium containing phosphate binders, which caused a significant progression of coronary and aorta calcification. Lanthanum carbonate also inhibited the progression of mild aortic calcification in hemodialysis patients suffering from type II diabetes mellitus as compared to calcium carbonate treatment [30]. In addition, increases in calcification scores were consistently less in the aorta, aortic valve and mitral valve among hemodialysis patients treated with cinacalcet plus low-dose vitamin D sterols as compared to those treated with flexible doses of vitamin D sterols alone [31], suggesting that cinacalcet plus low-dose vitamin D sterols might attenuate vascular and cardiac valve calcification.
Nevertheless, it would be of substantial interest to integrate this interesting and relevant approach in experimental studies which have the advantage to evaluate the capacity of different therapeutics in blocking the progression of calcified lesions in the vessel wall without the interference of standard medication often prescribed to CKD patients, and to have the opportunity to quantify vascular calcification by reliable and precise methods. The effectiveness of potential therapeutics on the progression of established vascular calcification has only been reported very recently by the group of Massy and Riser in the uremic apolipoprotein E knockout (apoE KO) mouse model of CKD [32]. Progression of calcification was halted by PPi and sevelamer hydrochloride. In comparison with the model used in the current study, the apoE KO in general, is primarily considered a model for atherosclerosis-related intimal calcifications. In the present study, the rat model with adenine-induced CKD and vascular media calcification was used. With this model, mortality is limited and did not differ between the control and treatment groups indicating that outcomes were not biased by differences in mortality. In our study, we showed that sevelamer carbonate appeared to prevent the further progression of vascular medial calcification, at least in the aorta. The calcium content of the aorta did not significantly increase over a 3-week treatment period with this phosphate binding agent, whereas a significant progression of aortic calcification was observed in the vehicle-treated CKD group. This beneficial effect of sevelamer was not seen on the percent calcified surface as measured on von Kossa-stained aortic sections. However, there was no progression of the percent calcified area between baseline values of CKD rats at week 4 and values of vehicle-treated CKD rats at the end of the study. Histomorphometric analysis of von Kossa-stained sections as a measure for aortic calcification is a less sensitive and less quantitative method than calcium bulk analysis of the aorta which may explain the absence of progression in calcified aortic surface found in vehicle-treated CKD animals. Histomorphometric measurement only takes into account the surface taken by the calcium phosphate crystals and does not give any information about the calcium density. Furthermore, in contrast to the aortic calcium content, the calcium content of the carotid and femoral arteries did not significantly progress over the 3-week treatment period in the vehicle-treated rats; therefore, no conclusions can be drawn with regard to the effect of the compounds on the progression of preexisting calcification in these smaller arteries.
Despite comparable doses of PPi as used in the study of Massy and Riser [32], we could not confirm the beneficial effect of PPi on the progression of established vascular medial calcification in the adenine-induced CKD rat model. In addition, both PPi and sevelamer treatment had no effect on calcium and phosphorus metabolism as compared to vehicle-treated CKD rats after 3 weeks of administration, except for a higher total serum calcium concentration in the sevelamer group. The discrepancy between both experimental studies is most likely due to the difference in the time course and severity of vascular calcification of the two different CKD models used. Uremic apoE knockout mice develop mild aortic intimal calcification, whereas adenine-induced CKD rats develop moderate to severe vascular calcification in the tunica media with arterial calcium concentrations up to 100 times higher as compared to the uremic apoE knockout mouse model. The adenine-induced CKD rat model may therefore reflect the clinical situation of moderate to severe vascular calcifications to a better extent [33]. It also remains that the concentrations of PPi required to induce an effect in this more “aggressive” adenine model could have been greater than those tested in the current study. Therefore, both findings suggest that PPi and sevelamer are capable of blocking at least mild aortic calcification. However, when calcifications are well established (i.e., in an advanced stage), it appears more difficult to halt its progression.
The efficiency of phosphate binders on vascular calcification is rather limited because these agents only target one of the causal factors of vascular calcification, i.e., hyperphosphatemia, and suffer from a poor compliance due to the high pill burden. As such, in a complex disease such as CKD–mineral and bone disorder (MBD) in which multiple organs are involved and various factors are known to play a role in the onset and progression of vascular calcification, calcifications should be detected in an early stage and the further expansion of the lesions should be stopped, preferably by directly and locally acting on the formation of calcium phosphate crystals in the vessel wall. For this purpose, PPi should reasonably be considered an ideal compound as it exerts its effect by binding to hydroxyapatite crystals to prevent further crystal growth [4]. An additional advantage of this promising molecule is the route of administration. In contrast to the orally prescribed current therapies, PPi can be added to the dialysate in patients on peritoneal dialysis or hemodialysis thereby avoiding gastro-intestinal side effects whilst dramatically increasing therapeutic compliance. Apart from its preventive effect on the development of vascular calcification and its beneficial effect on the progression of limited calcified lesions, further studies using higher PPi doses are needed to evaluate whether this compound can also halt the progression of more aggressive calcification in the tunica media of the aorta and peripheral vessels.
Targeting the calcification process itself in the vessel wall entails the possible interference of the compound with bone mineralization. For the therapeutic evaluation of compounds on CKD-related pathological vascular calcification, it is a prerequisite to examine and exclude whether or not these agents negatively affect physiological bone mineralization which is already compromised in CKD. Vascular calcification and bone pathology are both hallmarks of CKD and highly interconnected [34], as disorders in bone mineralization are key determinants of the severity of vascular osteogenesis. Although the adenine-induced CKD rat model has been proven a valuable model of CKD-induced vascular (medial) calcifications, it does not allow quantitative histomorphometric measurement of dynamic bone parameters because of the chaotic incorporation of tetracyclines into the woven bone inherent to the high bone turnover and the development of severe osteitis fibrosa. Hence, in the present study, only static bone parameters were measured. Whereas PPi treatment did not affect any of the bone parameters under study, a significant increase in the amount of osteoid was seen in the CKD animals treated with sevelamer. This is likely due to the phosphate binding capacity of sevelamer and in line with previous findings of our group showing that strong intestinal phosphate binding can induce disturbances in bone mineralization either because of an insufficient supply of phosphate to be incorporated in the bone and/or an increased efflux of phosphate out of bone to allow systemic restoration of phosphate homeostasis [35, 36]. Whether the increased urinary calcium excretion was due to a concomitant efflux out of bone, or results from an increased calcium absorption inherent to the use of phosphate binding agents cannot be deduced from the present data. However, this in line with previous observations [37] and requires further investigation to determine the effect of overall bone health. The concomitant increased osteoblast perimeter in sevelamer-treated uremic rats also supports the fact that the increased osteoid was not caused by a direct toxic effect on the bone forming cells. Like our study here, an absence of overt harmful consequences on bone by PPi treatment was also reported in apoE KO mice [38] and adenine-induced CKD rats exposed to calcitriol [8].
In conclusion, we found that sevelamer carbonate was able to reduce the further progression of moderate to severe calcification in the aorta. However, in the present study, PPi, at the concentrations tested, was unable to induce significant attenuation and neither drug was able to reverse the established calcification. Based on these observations, previously reported effects of drug treatment on the prevention of vascular calcification are a starting point to our understanding, but are difficult to extrapolate in humans when considering established arterial calcifications. It remains, however, that the preexisting vascular calcifications in the present study might have been too severe to allow the observation of subtle effects, which could be enhanced with longer treatment time or alternative dosing. In view of the latter, a model with both a milder degree of CKD and vascular calcifications which also allow evaluation of dynamic bone parameters has recently been developed and further characterized [39, 40]. Collectively, these data indicate that future studies to determine whether compounds are capable of directly intervening with the calcification process to halt or reverse the progression of established vascular calcification are likely to be fruitful, but may require examination in multiple animal models.
References
Block GA, Spiegel DM, Ehrlich J, Mehta R, Lindbergh J, Dreisbach A et al (2005) Effects of sevelamer and calcium on coronary artery calcification in patients new to hemodialysis. Kidney Int 68:1815–1824
Neven E, Dams G, Postnov A, Chen B, De Clerck N, De Broe ME et al (2009) Adequate phosphate binding with lanthanum carbonate attenuates arterial calcification in chronic renal failure rats. Nephrol Dial Transplant 24:1790–1799
Terkeltaub RA (2001) Inorganic pyrophosphate generation and disposition in pathophysiology. Am J Physiol Cell Physiol 281:C1–C11
Fleisch H, Russell RG, Straumann F (1966) Effect of pyrophosphate on hydroxyapatite and its implications in calcium homeostasis. Nature 212:901–903
Villa-Bellosta R, Millan A, Sorribas V (2011) Role of calcium-phosphate deposition in vascular smooth muscle cell calcification. Am J Physiol Cell Physiol 300:C210–C220
Lomashvili KA, Cobbs S, Hennigar RA, Hardcastle KI, O’Neill WC (2004) Phosphate-induced vascular calcification: role of pyrophosphate and osteopontin. J Am Soc Nephrol 15:1392–1401
Schibler D, Russell RG, Fleisch H (1968) Inhibition by pyrophosphate and polyphosphate of aortic calcification induced by vitamin D3 in rats. Clin Sci 35:363–372
O’Neill WC, Lomashvili KA, Malluche HH, Faugere MC, Riser BL (2011) Treatment with pyrophosphate inhibits uremic vascular calcification. Kidney Int 79:512–517
Riser BL, Barreto FC, Rezg R, Valaitis PW, Cook CS, White JA et al (2011) Daily peritoneal administration of sodium pyrophosphate in a dialysis solution prevents the development of vascular calcification in a mouse model of uraemia. Nephrol Dial Transplant 26:3349–3357
Lomashvili KA, Khawandi W, O’Neill WC (2005) Reduced plasma pyrophosphate levels in hemodialysis patients. J Am Soc Nephrol 16:2495–2500
Lomashvili KA, Garg P, Narisawa S, Millan JL, O’Neill WC (2008) Upregulation of alkaline phosphatase and pyrophosphate hydrolysis: potential mechanism for uremic vascular calcification. Kidney Int 73:1024–1030
O’Neill WC, Sigrist MK, McIntyre CW (2010) Plasma pyrophosphate and vascular calcification in chronic kidney disease. Nephrol Dial Transplant 25:187–191
Lomashvili KA, Narisawa S, Millan JL, O’Neill WC (2014) Vascular calcification is dependent on plasma levels of pyrophosphate. Kidney Int 85:1351–1356
Lomashvili KA, Monier-Faugere MC, Wang X, Malluche HH, O’Neill WC (2009) Effect of bisphosphonates on vascular calcification and bone metabolism in experimental renal failure. Kidney Int 75:617–625
Tamura K, Suzuki Y, Matsushita M, Fujii H, Miyaura C, Aizawa S et al (2007) Prevention of aortic calcification by etidronate in the renal failure rat model. Eur J Pharmacol 558:159–166
Ott SM (2012) Bisphosphonate safety and efficacy in chronic kidney disease. Kidney Int 82:833–835
Neven E, Persy V, Dauwe S, De ST, De Broe ME, D’Haese PC (2010) Chondrocyte rather than osteoblast conversion of vascular cells underlies medial calcification in uremic rats. Arterioscler Thromb Vasc Biol 30:1741–1750
Price PA, Roublick AM, Williamson MK (2006) Artery calcification in uremic rats is increased by a low protein diet and prevented by treatment with ibandronate. Kidney Int 70:1577–1583
De Schutter TM, Behets GJ, Geryl H, Peter ME, Steppan S, Gundlach K et al (2013) Effect of a magnesium-based phosphate binder on medial calcification in a rat model of uremia. Kidney Int 83:1109–1117
Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H et al (2013) Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone MinerRes 28:2–17
Neven E, De Schutter TM, Dams G, Gundlach K, Steppan S, Buchel J et al (2014) A magnesium based phosphate binder reduces vascular calcification without affecting bone in chronic renal failure rats. PLoS One 9:e107067
Goodman WG, Goldin J, Kuizon BD, Yoon C, Gales B, Sider D et al (2000) Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N Engl J Med 342:1478–1483
Phan O, Maillard M, Peregaux C, Mordasini D, Stehle JC, Funk F et al (2013) PA21, a new iron-based noncalcium phosphate binder, prevents vascular calcification in chronic renal failure rats. J Pharmacol Exp Ther 346:281–289
Porter CJ, Stavroulopoulos A, Roe SD, Pointon K, Cassidy MJ (2007) Detection of coronary and peripheral artery calcification in patients with chronic kidney disease stages 3 and 4, with and without diabetes. Nephrol Dial Transplant 22:3208–3213
Sigrist M, Bungay P, Taal MW, McIntyre CW (2006) Vascular calcification and cardiovascular function in chronic kidney disease. Nephrol Dial Transplant 21:707–714
Spiegel DM, Raggi P, Mehta R, Lindberg JS, Chonchol M, Ehrlich J et al (2004) Coronary and aortic calcifications in patients new to dialysis. Hemodial Int 8:265–272
Russo D, Miranda I, Ruocco C, Battaglia Y, Buonanno E, Manzi S et al (2007) The progression of coronary artery calcification in predialysis patients on calcium carbonate or sevelamer. Kidney Int 72:1255–1261
Chertow GM, Burke SK, Raggi P (2002) Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int 62:245–252
Asmus HG, Braun J, Krause R, Brunkhorst R, Holzer H, Schulz W et al (2005) Two year comparison of sevelamer and calcium carbonate effects on cardiovascular calcification and bone density. Nephrol Dial Transplant 20:1653–1661
Wada K, Wada Y (2014) Evaluation of aortic calcification with lanthanum carbonate vs. calcium-based phosphate binders in maintenance hemodialysis patients with type 2 diabetes mellitus: an open-label randomized controlled trial. Ther Apher Dial 18:353–360
Raggi P, Chertow GM, Torres PU, Csiky B, Naso A, Nossuli K et al (2011) The ADVANCE study: a randomized study to evaluate the effects of cinacalcet plus low-dose vitamin D on vascular calcification in patients on hemodialysis. Nephrol Dial Transplant 26:1327–1339
de Oliveira RB, Louvet L, Riser BL, Barreto FC, Benchitrit J, Rezg R et al (2015) Peritoneal delivery of sodium pyrophosphate blocks the progression of pre-existing vascular calcification in uremic Apolipoprotein-E knockout mice. Calcif Tissue Int 97:179–192
Ballanti P, Silvestrini G, Pisano S, De PP, Di GS, Mantella D et al (2011) Medial artery calcification of uremic patients: a histological, histochemical and ultrastructural study. Histol Histopathol 26:191–200
Persy V, D’Haese P (2009) Vascular calcification and bone disease: the calcification paradox. Trends Mol Med 15:405–416
Behets GJ, Dams G, Vercauteren SR, Damment SJ, Bouillon R, De Broe ME et al (2004) Does the phosphate binder lanthanum carbonate affect bone in rats with chronic renal failure? J Am Soc Nephrol 15:2219–2228
Behets GJ, Gritters M, Dams G, De Broe ME, D’Haese PC (2005) Effects of efficient phosphate binding on bone in chronic renal failure rats. Ren Fail 27:475–484
Behets GJ, Dams G, Damment SJ, Martin P, De Broe ME, D’Haese PC (2014) Differences in gastrointestinal calcium absorption after the ingestion of calcium-free phosphate binders. Am J Physiol Renal Physiol 306:F61–F67
Barreto FC, de Oliveira RB, Benchitrit J, Louvet L, Rezg R, Poirot S et al (2014) Effects of pyrophosphate delivery in a peritoneal dialysis solution on bone tissue of apolipoprotein-E knockout mice with chronic kidney disease. J Bone Miner Metab 32:636–644
Neven E, Bashir-Dar R, Dams G, Behets GJ, Verhulst A, Elseviers M et al (2015) Disturbances in bone largely predict aortic calcification in an alternative rat model developed to study both vascular and bone pathology in chronic kidney disease. J Bone Miner Res 30:2313–2324
Shobeiri N, Pang J, Adams MA, Holden RM (2013) Cardiovascular disease in an adenine-induced model of chronic kidney disease: the temporal link between vascular calcification and haemodynamic consequences. J Hypertens 31:160–168
Acknowledgments
We especially thank Hilde Geryl, Ludwig Lamberts and Simonne Dauwe for their excellent technical assistance and Dirk De Weerdt for his help with the graphics. Ellen Neven and Anja Verhulst are postdoctoral fellows of the Fund for Scientific Research-Flanders (FWO).
Funding
This work was supported by a research Grant from Baxter Healthcare, USA (A10/0650).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
At the time of the study, Bruce L. Riser was an employee of Baxter Healthcare, USA, a company with potential commercial interest in this research. Presently, Dr. Riser is the President and CEO of BLR Bio, a biotechnology company focused on the treatment of fibrosis and cancer. This focus does not presently include the use of the technology described in this publication. Ellen Neven, Britt Opdebeeck, Annelies De Maré, Rida Bashir-Dar, Geert Dams, Rita Marynissen, Geert J. Behets, Anja Verhulst, Bruce L. Riser and Patrick C. D’Haese have no conflict of interest to declare.
Ethical Approval
All applicable international, national and institutional guidelines for the care and use of animals were followed. All experimental procedures were performed according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals (2011) and approved by the University of Antwerp Ethical Committee for Animal Experiments (Permit Number: 2015-53).
Additional information
Dr. Patrick C. D’Haese and Dr. Bruce L. Riser equally contributed as organizers and supervisors to the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Neven, E., Opdebeeck, B., De Maré, A. et al. Can Intestinal Phosphate Binding or Inhibition of Hydroxyapatite Growth in the Vascular Wall Halt the Progression of Established Aortic Calcification in Chronic Kidney Disease?. Calcif Tissue Int 99, 525–534 (2016). https://doi.org/10.1007/s00223-016-0178-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-016-0178-7