
Abstract
Gaucher disease is a relatively rare metabolic disease caused by the inherited deficiency of the lysosomal enzyme glucocerebrosidase. Gaucher disease affects multiple organs, among which is the skeleton. Bone involvement occurs frequently in Gaucher disease, and is one of its most debilitating features, reducing the quality of life of patients. Bone status is an important consideration for treatment to ameliorate symptoms and reduce the risk of irreversible complications. We have conducted a systematic review of all the various aspects of Gaucher disease, focusing on different skeletal manifestations, pathophysiology of bone alterations, clinical symptoms, and current diagnostic and therapeutic approaches.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gaucher disease (GD) is an autosomal recessive disorder, characterized by the accumulation of glucosylceramide, mainly in cells of the reticuloendothelial system, due to mutations in the acid b-glucocerebrosidase (GBA) gene. GD is one of the most common of approximately 60 hereditary lysosomal storage disorders (LSD) [1]. Systemic accumulation of these glycolipid-lipid engorged cells results in variable combinations of splenomegaly, hepatomegaly, anemia, thrombocytopenia with bleeding tendency and a diverse pattern of bone disease [1]. The disease is heterogeneous even among patients with the same genotype, implicating that in addition to the different GBA mutations, there are other genetic and non-genetic factors, which play a role in the manifestations of the disease [2]. There is wide variability in the age of onset, clinical presentation, and disease severity, even within the three major clinical subtypes.
This article reviews various aspects of GD, including different skeletal manifestations, pathophysiology of bone alterations, clinical symptoms, and current diagnostic and therapeutic approaches. Finally, future goals will be discussed.
Historical Background
In 1882, Philippe Gaucher became the first to describe a disorder, which was later named after him, in a woman with an enlarged spleen that contained unusual engorged cells initially and incorrectly thought to be malignant, in his medical thesis. In 1904, Brill hypothesized that the disease was inherited, and he showed that the liver, lymph nodes and bones were involved. In the 1920s, patients with neurological involvement were first described. The biochemical basis was resolved in the 1960s, when Brady et al. demonstrated that the primary derivative pathway via glucocerebrosidase was markedly but variably defective [1]. Afterwards, glucocerebrosidase was identified as a lysosomal enzyme, and patients with GD were thereafter described as having a lysosomal storage disorder [3]. Subsequently, the GBA gene was localized to chromosome 1, q21, and more than 300 GBA1 mutations and polymorphisms have been reported [4]. Although the disorder was first described more than 130 years ago, an effective intravenous enzyme replacement therapy (ERT) has only been introduced in 1991. Our knowledge about this disorder has progressively increased, and today different disciplines of medicine are involved in the diagnosis, therapy and management of this multisystem inherited disorder.
GD Types and Epidemiology
GD has been traditionally classified into 3 clinical forms: the adult chronic non-neuronopathic type 1 GD (MIM no. 230800), the infantile, acute neuronopathic type 2 GD (MIM no. 230900), and the juvenile sub-acute neuronopathic type 3 GD (MIM no. 321000).
Type 1, the non-neuronopathic form, is the most common form in the Western hemisphere, and has an ethnic predilection among Ashkenazi Jews, especially the N370S and 84GG mutations (carrier ship 1:17; expected birth frequency of 1:850, definitely caused by founder effect and probably also by some selective advantage, that is yet to be determined) [5]. Age of onset of symptoms and the disease course are variable even among patients homozygous for the common N370S (1226G) mutation (typically viewed as “mild”) and even among siblings [6].
Type 2 is a fulminant neuronopathic disease, fatal during infancy [1].
Type 3 is predominant in Asian and Arab countries. It is further divided into type 3a, 3b and 3c. Type 3a is characterized by predominance of neurologic signs over visceral features, type 3b by predominance of visceral features over neurologic signs initially identified in northern Sweden, and type 3c by a very rare cardiac variant, described mainly among Palestinian Arabs [7].
The panethnic estimated frequency is 1:50.000 to 1:100.000, and the prevalence of GD in the general population is probably 1:40.000 [8].
Recently, the identification of new phenotypes and appreciation that even patients with type 1 may evince some (late-onset) neurologic manifestations, has supported the hypothesis of “a continuum of GD phenotypes” [9]. However, subcategorization into archetypical forms is useful for diagnostic and therapeutic procedures, and in particular as a basis for genetic counseling. In addition, it should be clarified that the neurologic features, peripheral neuropathy, Parkinsonism, and neurologic changes secondary to bone complications in type 1 GD are different from those reported in the neuronopathic forms (type 2 and 3) [10].
Enzymatic and Molecular Basis
GD is an autosomal recessive LSD caused by GBA gene mutations which is responsible for hydrolysis of glucocerebroside into glucose and ceramide. The result of these mutations is deficiency and dysfunction of this enzyme leading to multisystemic accumulation of glucocerebroside in lysosomes of macrophages in various organs, compromising spleen, liver, bone marrow, bone mineral, and, less often, lungs, skin, conjunctiva, kidneys, and heart. The large macrophages, also called “Gaucher cells”, store glucocerebroside, and histologically show a small eccentrically placed nuclei surrounded by a bright cytoplasm with striations or crinkles [11].
Glucocerebrosidase is synthesized on endoplasmic reticulum (ER)-bound polyribosomes and translocated into the ER. This lysosomal enzyme, following N-linked glycosylations, is transported to the Golgi apparatus, from which it is trafficked to the lysosomes [2]. Ron et al. tested glucocerebrosidase protein levels, N-glycans processing and intracellular localization in GD fibroblasts [2]. Their results strongly indicate that the misfolded mutant glucocerebrosidase variants present variable levels of ER retention and proteasomal degradation [2]. The degree of ER retention and proteasomal degradation is one of the factors that determine GD severity [2]. Finally, saposin C deficiency represents a rare variant of GD (only six cases have been reported so far); it is due to mutations in the prosaposin gene affecting saposin C expression and/or function [12].
Genetics
The GBA gene is located on chromosome 1q21, and is 7.6 kb in length with 11 exons, as well as a 5-kb pseudogene located 16 kb downstream [1]. It is transcriptionally active, although translationally nonfunctional, and should be recognized as separate from the GBA gene when molecular diagnoses are performed. More than 300 GBA mutations are listed to date in the Human Gene Mutation database. They include point mutations, splice site mutations, deletions and recombinant alleles, resulting from recombination between the glucocerebrosidase gene and a closely related pseudogene, occupying the same locus on chromosome 1q21. More than 80 % of these alleles are single nucleotide substitutions, whereas insertions, deletions, or other complex alleles account for the remainder [13]. Clinical heterogeneity is attributable to this elevated number of mutations; however, the importance of epigenetic and environmental influences is beginning to be appreciated.
The N370S allele, common among patients with Ashkenazi Jewish heritage, appears to have an ameliorative effect on bone marrow disease [6]. Moreover, homozygosity for the N370S allele is associated with a phenotype characterized by later onset and skeletal complications, while compound heterozygosity of N370S with another allele is associated with early onset and predominantly visceral/hematologic disease [14].
Clinical Presentation and Diagnosis
The signs and symptoms of GD vary from the lethal neonatal form to a completely asymptomatic form. In general, all types of GD have some degree of visceral involvement and bone disease, with a possible overlapping presentation among the various types (mainly between type 2 and more severe type 3a).
Type 1 GD affects children and adults at any age and typical clinical manifestations include: splenomegaly, hepatomegaly, anemia, thrombocytopenia, hemorrhagic manifestations (attributed to thrombocytopenia, although they may occur as a result of coagulation factors deficiency or abnormal platelet functions), asthenia (whether or not associated with anemia), early satiety, stunted growth and bone disease (acute bone crises, chronic bone pain, osteopenia, lytic lesions, fractures and osteonecrosis) [15]. According to the International Collaborative Gaucher Group (ICGG) Gaucher Registry, the clinical characteristics for all GD types occur with the following percentages: splenomegaly 85 %, hepatomegaly 63 %, anemia 34 %, thrombocytopenia (with or without bleeding manifestations) 68 %, osteopenia 55 %, fractures 7 %, bone crises 7 %, bone pain 33 %, and growth retardation 36 % [15].
Type 2 GD generally affects infants at 4–5 months of age, and in addition to the findings observed in type 1 GD, type 2 includes: developmental delay, strabismus, supranuclear gaze palsy, bulbar palsy, paresis, hypertonia, rigidity, opisthonus, dysphagia, and seizures [1]. The neurologic complications are serious, and the evolution is rapid, leading to death within the first 2 years of life, usually due to lung failure. A neonatal sub-type exists and is characterized by hydrops fetalis and congenital ichthyosis; these neonates die within the first 2 days of life, and resemble findings in the first knock-out mouse model reported by Tybulewicz VL in 1992 [16].
Type 3 GD is characterized by the presence of oculomotor apraxia (supra-nuclear gaze palsy) which either may be the predominant neurological abnormality or associated with developmental delay, myoclonus, convulsive crises and ataxia with various visceral manifestation, as in type 1.
In addition to the classic manifestations, patients with GD may develop atypical features such as: primary or secondary pulmonary hypertension [17], brownish pigmentation on face and legs [17], liver cirrhosis with portal hypertension (usually in splenectomized patients) [17], hepato-pulmonary syndrome, heart valve calcifications (type 3c), renal involvement (such as nephrotic syndrome) [17], recurrent bacterial infections, isolated vertebral compression, iliopsoas hematoma, gaucheromas, and mesenteric lymphadenopathy.
Patients with GD may also suffer from additional disorders that may complicate the diagnosis or impact the disease manifestations. We differentiate between “associated diseases” which are distinctly different disease entities but present at greater prevalence among patients with GD compared to the general population (such as Parkinson’s and multiple myeloma) [18, 19], and “co-morbidities” which are unrelated to GD and occur at a similar frequency as the general population, yet may have an impact (such as cataract, breast cancer, etc).
There have been reports suggesting that patients with GD have an increased risk of cancer compared with the general population, but this is considered controversial with many of the publications suffering from ascertainment bias.
Multiple myeloma, which is a classic example of an associated disorder, is of particular relevance to bone involvement (lytic lesions, osteoporosis and pathological fractures), as well as haematological features (such as anemia and bleeding tendency), as several of the key disease features may overlap.
An increased risk has also been reported for hepatocellular carcinoma and, to a lesser degree, renal cell carcinoma [20], but these are anecdotal observations that need further confirmation before one can consider them to be associated diseases and not just co-morbidities. Because of the high prevalence of monoclonal gammopathies and myeloma, the current recommendations for the follow-up of patients with GD suggest the performance of an immunoglobulin profile at diagnosis and every 2 years (patients <50 years) or every year (patients >50 years) [21].
Accurate and early diagnosis is critical, because most patients with significant visceral involvement (types 1 and 3) will benefit from ERT, and earlier ERT may prevent development of irreversible complications, such as avascular necrosis (AVN) of large joints and height retardation in children [22]. The “gold standard” for diagnosis of GD is still the detection of low enzymatic activity of b-glucocerebrosidase in peripheral blood cells compared with normal controls [22]. This is supplemented by mutation analysis at the DNA level, preferably via whole gene sequence to avoid pitfalls [23], which provides genotype definition. Bone marrow aspiration for diagnosis should not be performed as the diagnostic test, because it may be traumatic for patients, and because it is non specific, possibly resulting in a false-positive diagnosis [22].
Bone Involvement in GD
Bone disease affects up to 90 % of GD patients, mainly present in type 1 and type 3 forms. Children with type 2 GD don’t show clinically relevant bone involvement because rapid neurological deterioration leads to death at 2–3 years of age, prior to the onset of bone pathology [24]. Bone involvement, instead, is a frequent manifestation in GD, but a correlation between bone and haematological or visceral involvement is not always demonstrated.
Pathogenesis of Bone Changes in GD
The process of bone remodeling, as already known, requires osteoclastic resorption of bone matrix and deposition of a new matrix by osteoblasts. The osteoclasts not only resorb bone, but also regulate the function of other cells such as osteoblasts, regulate hematopoietic cell egress from bone marrow, and function as immune cells during inflammation [25]. The pathogenesis of bone changes in GD has not been fully understood, but both mineralized bone and bone marrow seem to be involved, and the bone alterations may reflect the effects of nearby disease in marrow tissue.
The progressive accumulation of glucocerebrosides within the bone marrow cavity appears to be the first step of the pathological process, causing marrow expansion and progressive centrifugal expansion of the red bone marrow. The displacement of inactive yellow marrow by red marrow in the periphery alters vascularity; vascular occlusion and compression cause bone infarction, and increasing intraosseous pressure may lead to bone necrosis [24].
Moreover, glucocerebrosidase accumulation seems to induce macrophage activation, which may promote additional inflammatory processes due to the altered expression of different macrophage-derived factors and cytokines. Cytokine and inflammatory mediators, such as interleukin (IL)-1, IL-6, and tumour necrosis factor-alpha (TNF-a), influence osteoclast and osteoblast activity. In particular, IL-10 activity may inhibit the osteoblast activity [26], whereas IL-1b, IL-6, and M-CSF could enhance bone resorption due to increased osteoclast activation and formation [26]. Osteoporosis in GD may be caused by the changes of these cytokines. Moreover, macrophage inflammatory protein (MIP)-1a and MIP-1b, which increase the bone resorption by osteoclasts in multiple myeloma, are also elevated in GD patients with bone disease, probably contributing to pathological bone resorption in GD [27]. Changes in total T-lymphocyte numbers and alterations of CD4+/CD8+ T-lymphocyte ratios have also been reported in GD patients with bone involvement, as an overall decrease of T-lymphocytes with lower CD8+ T-lymphocyte numbers [28]. Therefore, the complex network of interacting factors between bone marrow, immune and bone cells, the altered vascularity, and various cytokines, may contribute to the bone manifestations described in GD [26]. Finally, another factor to consider is the role of hormones such as estrogen, testosterone, parathyroid, and thyroid hormone, which in patients with GD are usually within the normal range, but if altered they could influence the activity of osteoclasts and osteoblasts either directly by hormone receptors located on osteoblasts and osteoclasts or indirectly by various other cells of the immune system.
Histologically, bone tissue, analyzed after hip replacement surgery, showed a heterogeneous picture with areas of vital bone and bone marrow filled with Gaucher cells, islands with residual hematopoietic red bone marrow, zones with non-specific chronic inflammation and fibrosis, and areas of necrotic cells and bone material [29].
Clinical and Radiological Aspects of Bone Manifestations
Common bone findings include: decreased bone mineral density (BMD), Erlenmeyer flask deformity (EFD), bone crises, osteonecrosis, lytic bone lesions, osteosclerosis, increased fracture risk, cortical thinning, rarely acute osteomyelitis, and growth retardation. In the ICGG Gaucher Registry, 82 % of 1698 GD patients, 94 % of whom had type 1 GD, had radiological evidence of bone disease before treatment [15]. All patients with type 1 GD are at risk of bone complications regardless of age of disease onset, the presence and severity of visceral or haematological disease, or genotype [15]. In most GD patients, bone disease shows a progressive course over years, and is one of the most debilitating aspects of the disease, aggravating quality of life.
Children may have linear growth retardation and delayed puberty [1]. After initiation of ERT, most patients showed growth acceleration and regained normal weight [30].
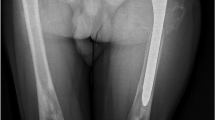
EFD describes a distinct abnormality of remodelling of the metaphyseal-diaphyseal areas with a progressive enlargement of the metaphyseal area, due to local bone marrow infiltration by Gaucher cells [31]. This leads to a lack of the typical concave di-metaphyseal curve resulting in an Erlenmeyer flask-like appearance. The sites affected are: the distal femora, occasionally also within the metaphyseal region of other tubular bones, and the proximal tibia or humerus. These changes occur before puberty, after which the bone alteration develops progressively. Modeling deformities, particularly the EFD in the distal femur, are often-described (30–45 % of adults) (Fig. 1), although they appear to be asymptomatic and not pathognomonic for GD. There is no relationship between this radiologic finding and other skeletal complications of GD [31].

X-ray examination showing bilateral EFD of distal femurs (white arrows)
Reduced BMD, expected for their age and sex, with an increased risk of fracture, is common in GD, and is progressive with age [15]. Patients may not achieve peak bone mass, resulting in subsequent osteopenia and osteoporosis. Localised or generalised osteopenia/osteoporosis affects both the trabecular and cortical bone. The generalised form can be determined by BMD decreasing measured by dual X-ray energy absorptiometry (DXA). In the first report of DXA studies in GD, Pastores GM et al. measured the BMD of the lumbar spine, femoral neck, trochanter, and distal radius in 61 adult patients (from 22 to 77 years of age) with type 1 GD [32]. The mean BMD at each site was significantly lower than expected for age and sex. The severity of the osteopenia correlated significantly with other clinical indicators of disease severity, including the N370S/84GG genotype, prior splenectomy, and hepatomegaly. The bone density measurements also correlated significantly with the overall severity of skeletal disease as assessed by skeletal radiography. This study suggested that BMD measurements allow a quantitative assessment of bone involvement, which may permit serial, readily available, non-invasive and inexpensive monitoring of bone changes in type 1 GD [32].
The finding of polymorphisms of genes (CYP1A1, BMP4, and VDR) that have been previously identified as associated with osteoporosis in non-GD populations and which are correlated with low BMD in GD, may implicate a constellation of non-Gaucher specific factors that are as predictive in patients with GD as in other osteoporotic persons [33]. Moreover, one haplotype of IL-1 genes was identified among patients with GD that correlates with osteopathy [33].
Focal osteolytic lesions, frequently described in GD, appear to be closely related to regions where Gaucher tissue is packed tightly with pathological macrophages in the intramedullary space [34]. Osteolytic lesions are small erosions with a “Worm-eaten” aspect, characterized by rarefied cortex with dentate endosteum, frosted glass or favus aspect. Osteolysis is associated with expanding lesions that typically cause cortical thinning and pathological fractures of the shaft of long bones [34]. Increased cathepsin K (CatK) excretion by activated osteoclasts may play a role in the development of osteolytic lesions [35].
Bone infarcts may be caused by infiltration of Gaucher cells leading to alterations of vascularity and additional inflammatory processes [24]. Bone pain may be related to the pathologic processes evident by radiography, magnetic resonance imaging (MRI), and computed tomography, or have the character of a “crisis” which is a self-limiting, albeit exquisitely painful event, associated with signs of acute local and/or systemic inflammation. Bone crisis is an independent predictor for future osteonecrosis; during pregnancy the risk is increased [36]. Bone infarcts may occur with no clinical symptoms, or slight pain, or sudden onset of severe localized pain which may be the first feature of osteonecrosis. Bone crisis is typically but not necessarily associated with local tenderness, erythema, and swelling, and it may be accompanied by fever, elevated white blood cell count, and an accelerated erythrocyte sedimentation rate [33]. The debilitation may last a few days, usually requiring immobilization of the affected area and narcotics for pain relief and hydration. Spontaneous bacterial osteomyelitis is rare in GD, and the differentiation between aseptic (“bone crisis”) and pyogenic osteomyelitis is difficult or even impossible at the time of onset if this is the presenting manifestation of GD. The only way to avoid misdiagnosis is when the physician is aware of GD and looks for other disease features such as splenomegaly or thrombocytopenia. Otherwise, negative blood cultures and sterile aspirates are used to exclude pyogenic osteomyelitis [24]. Bone infarcts are often followed by necrosis and fractures [36]. Osteonecrosis, also known as AVN, and episodes of bone infarction are the most relevant and invalidating skeletal manifestation. Osteonecrosis is irreversible and predominantly affects the femoral heads (Figs. 2, 3), knees or proximal humerus, and less commonly the vertebral bodies. Osteonecrosis will typically lead to joint collapse with consequent pain, disability, and a poor quality of life [12, 24]. This devastating feature was more common in the pre-ERT era, particularly after splenectomy, and usually occurred in teenagers and young adults. Mistry et al. [37] have explored the link between osteonecrosis and the interval between diagnosis and initiation of specific treatment. The authors have demonstrated that the risk of AVN was increased if the initiating of treatment occurred 2 years or more after diagnosis had been made. This study also confirmed that the higher risk of AVN was among splenectomized patients [37].

MRI of the pelvis showing bilateral osteonecrosis of femoral heads in a 33 year old splenectomized female patient who was poorly compliant with enzyme therapy (white arrows)

X-ray examination showing osteonecrosis of the right femoral head (white arrows)
Extraosseous extension of Gaucher cells is a very rare skeletal complication of GD, and has occured after cortical destruction (during surgical procedures, for example) and leakage of infiltrates into tissue adjacent to the bone [38]. Cortical thinning promoted by increased local pressure, altered vascularity, and increased cortical porosity due to cytokines activating osteoclasts may precede cortical destruction [38].
Bone involvement in GD requires an assessment with bone imaging. Imaging of bone and bone marrow in GD aims to evaluate the disease burden, evidence the presence of skeletal complications, and monitor disease progression and response to therapy.
Monitoring of bone disease in children and adults for initial assessment is carried out with X-ray examination of the femora, spine, and any other symptomatic sites. X-ray is commonly used for the detection of fractures, dislocation of joint replacements, the evaluation of joint arthroplasty, and can detect local deformities of bone including EFD, cystic or tumorous lesions, and localized cortical thinning [39]. However, X-ray shows a low sensitivity in detecting morphological bone manifestations in GD [39]. X-ray survey is useful at first assessment, and it is repeated only if there is a medical reason, while BMD or MRI can be used for follow up [22, 40].
MRI has high sensitivity for all kinds of skeletal pathologies in GD [39]. T1-weighted MRI is recommended to detect and quantify the extent of marrow infiltration, and T2-weighted MRI is recommended to identify focal lesions, active bone infarcts, osteonecrosis, and osteomyelitis. T1-weighted and T2-weighted spin echo sequences, short tau inversion recovery sequences, and turbo spin echo are different MRI modalities used in the evaluation of bone involvement in GD [39]. MRI spin echo sequences can show the fat content of bone marrow in adults [39]. Normal yellow marrow creates a hyperintense T1 and an intermediate to hyperintense T2-weighted signal; instead, the infiltration of bone marrow by Gaucher cells creates hypointense signals. ERT, determining the reduction of Gaucher cell deposition, increases fatty yellow marrow and therefore a normalization of signal intensity. MRI is the method of choice to evaluate the extent of bone disease prior to therapy and during follow-up in patients on therapy, and is also the most sensitive method to detect femoral head necrosis [39]. Quantitative chemical shift imaging (QCSI) is a quantitative MRI technique that measures fat content in the axial bone marrow and the extent of its displacement by Gaucher cells. While QCSI was suggested to be the gold standard to quantify bone marrow involvement in GD [39], it is currently available in only one center world-wide, and is not free of pitfalls. Therefore, it would be impractical to expect to use it as such [40, 41].
Several scoring systems [Düsseldorf Gaucher Score, bone mineral burden (BMB), vertebral disc ratio (VDR), Spanish-MRI score (S-MRI), Terk Classification, Rosenthal Score] have been established in order to quantify the severity and extent of bone involvement, and these can be regarded as good alternatives to QCSI in daily routine [39]. These scoring systems appear to be useful in evaluating the extent of bone involvement in GD patients prior to therapy and during follow-up. However, it would be useful if a panel of experts would recommend the preferential use of one of the above imaging techniques in order to allow comparative studies of very large cohorts of patients with varying demographic characteristics and disease status.
99 mTc-methylene diphosphonate (99 mTc-MDP) bone scintigraphy, as an alternative to MRI, can be used in the discrimination of osteomyelitis and AVN if performed 72 h after clinical onset [42]. Bone scintigraphy can also be used for the investigation of occult fractures or the evaluation of loosening of hip joint prostheses, in which case 3-phase bone scintigraphy should be applied [42]. The lipophilic tracer 99 mTc-MIBI identifies glycolipid deposits due to Gaucher cells, and thus is useful for the quantification of bone marrow infiltration prior to therapy and during follow-up on ERT. 99mTc-MIBI scintigraphy has been suggested to have a role in children in whom bone marrow undergoes a developmental conversion from red to yellow marrow in the appendicular skeleton, as well as to be used as an alternative to MRI in those patients who cannot undergo MRI imaging [42]. However, these radioisotope studies are associated with a significant amount of radiation, which is not advisable for patients—mainly children, but also adults—who may be prone to malignancies due to the nature of their metabolic disorder, and therefore are not considered relevant for routine follow up practices.
BMD measured by DXA is the gold standard for the quantification of bone loss and the diagnosis of osteoporosis [32].
Bone marrow aspiration and biopsies (when there is a dry tap) are sometimes performed for differential diagnosis of GD with idiopathic thrombocytopenic purpura (ITP), acute leukemia, other malignant lesions, or to evaluate changes in bone metabolism [24]. However, bone marrow aspiration, as already mentioned, should be avoided for the sole purpose of GD diagnosis, given the existence of non- invasive and more accurate enzymatic tests. A paper by Mistry et al. from 2011 provides an algorithm for the diagnosis of GD in patients with thrombocytopenia and, while bone marrow aspiration is not justified prior to performance of the enzyme assay in Ashkenazi Jewish patients, in whom GD is more common than haematological disorders, it is acceptable in non-Jewish patients in whom the likelihood of malignancies is much greater than that of GD [43].
Finally, it would be interesting to analyze the impact of devastating bone disease on quality of life, while the SF-36 survey has been used in clinical trials, the visual analog scale (VAS) or narrative analog scale (NAS) may be more practical and relevant for routine follow-up of GD patients as part of the evaluation of bone pain [24].
Bone Metabolism and Biochemical Markers in GD
Bone remodeling is the continuous process regulated by the balanced activities of bone-resorbing osteoclasts and bone-forming osteoblasts to maintain normal physiological structure and mineral content. The activation of different bone cells in the bone remodeling process is orchestrated by multiple pathways such as receptor activator of nuclear factor (NF)-κB ligand (RANKL) and Wnt signaling pathways [44]. Changes in osteoclast and osteoblast activity can be measured by markers of bone metabolism. The usefulness of bone turnover markers (markers of bone formation and bone resorption) in GD is still unclear, and their utility in monitoring the effects of ERT on bone metabolism has not been investigated exhaustively.
A few studies [45–49] have evaluated various markers of bone turnover in treatment-naïve patients, or on ERT, and also in patients switching from ERT to substrate reduction therapy (SRT) (Table 1).
Markers of bone formation in treatment-naïve patients appeared to be normal or decreased, whereas markers of bone degradation were mainly normal or increased [45, 46, 48]. According to this finding, Stowens DW et al. [47] reported a decrease in bone resorption on seven patients who underwent bone biopsies. A study measured serum osteocalcin (OC), a marker of bone formation, and type I collagen C-terminal telopeptide (ICTP), a marker of bone resorption, in 16 patients with type 1 GD and in 29 age-matched controls, and the results indicated a significant decrease of both OC and ICTP values in patients with GD compared to the unaffected controls [46]. Recently, Van Dussen et al. found that OC was decreased in 50 % of 40 type 1 GD patients with no significant change in ICTP and N-terminal propeptide of type 1 procollagen (PINP) and concluded that imbalances in bone turnover result primarily from a decrease in bone formation [48]. In addition, OC concentration was negatively correlated to measures of overall disease severity and positively correlated with imaging data, suggesting a relation with disease severity [48]. Furthermore, a recent study in GBA gene-deficient mice demonstrated the predominance of altered osteoblastic function in GD [49].
Ciana et al. described the efficacy of ERT on bone involvement in a group of 12 type 1 GD patients by monitoring biochemical indices of bone resorption-formation and lumbar BMD measured by DXA [50]. These parameters were measured at baseline, after 6 and 12 months, and then every year for a mean ERT follow-up period of 4.5 years [50]. A significant decrease of carboxyterminal propeptide of type I procollagen (PICP), markers of bone formation, was detected in the patient group at baseline, while ICTP was remarkably higher. No changes in bone formation indices were observed during the follow-up period, while urinary calcium excretion increased significantly. A significant BMD improvement was also detected after an average ERT period of 4.5 years. The authors concluded that these findings suggested the ineffectiveness of the biochemical markers used in monitoring ERT in type 1 GD skeletal involvement, whereas DXA was demonstrated to be a reliable method with which to follow up BMD improvement [50]. Sims et al. evaluated the changes on ERT versus baseline values, and during ERT bone formation increased by 60 % and bone resorption decreased by 20–40 % [51]. Fiore et al. evaluated serum levels of OC, bone-specific alkaline phosphatase (BAP), urinary excretion of pyridinoline (Pyr/Cr) and deoxypyridinoline (D-Pyr/Cr) cross-links in 12 patients with type 1 GD (mean age 33 ± 13 years), 10 of whom had received enzyme treatment for 2-8 years. OC and BAP concentrations were within the normal range; Pyr/Cr and D-Pyr/Cr were, instead, significantly higher than in controls [52].
The limits of these studies are: the small number of patients, and in some reports the use of less-sensitive bone markers, which may explain the lack of consistent results concerning bone metabolism in GD [24]. However, it seems that in GD patients there are both increased bone degradation and impaired bone formation, leading to osteoporosis [24].
Patients treated consecutively with ERT and SRT, OC and alkaline phosphatase (ALP) were in the lower part of reference ranges at the time of the change from ERT to the start of SRT, and decreased during follow-up of 12-28 months (statistically significant for ALP) [53]. There was no statistically significant change in markers of bone resorption C-terminal cross linked telopeptide (CTX), and Tartrate-resistant acid phosphatase 5b (TRAP5b). Imaging parameters of bone disease (MRI, DXA) generally remained stable throughout follow up, although one patient experienced worsening bone parameters observed by MRI. This patient also experienced increasing bone pain and demonstrated a significant increase in TRAP5b [53].
Mikosch P et al. showed also that CatK, which mediates bone matrix destruction, is two to three fold increased in sera from GD patients as compared to healthy controls [53]; after treatment with ERT serum, CatK activities decreased significantly. Osteoporosis or lytic bone lesions in GD patients may be caused, at least in part, by high CatK [35].
The changes of bone metabolism seem not to be mediated via the OPG (osteoprotegerin)/RANK (Receptor Activator of NF-kB)/RANKL system, since osteoprotegerin (OPG) levels in GD-patients were comparable to controls [54].
There are few studies on vitamin D in GD-patients. Parisi et al. found a vitamin D deficiency in nine young GD patients, with 25-OH vitamin D levels <30 ng/mL in all patients [55]. Mikosch et al. evaluated 25-OH vitamin D concentrations in 60 type 1 GD patients. The authors observed varying degrees of 25-OH vitamin D deficiency in the majority of these patients, and BMD was positively correlated to the vitamin D values measured during the seasonal nadir of vitamin D (December–May) [53].
Currently, the most reliable and commonly used biomarker in GD is chitotriosidase. The measurement of chitotriosidase concentrations, during and after therapy, is useful, since reduction in chitotriosidase activity is associated with clinical improvement [56]. Increased chitotriosidase activity in untreated, mildly affected patients should lead to closer monitoring and, when indicated, also to consider treatment [22]. The genetic polymorphisms of the chitotriosidase gene leading to lack of enzymatic activity in ~6 % of the world population, as well as various technical issues/difficulties, hamper the use of chitotriosidase activity as a sole index of disease burden [57]. Chemokine (C–C motif) ligand 18 CCL-18 (PARC2) is a substitute option in the case of a genetic lack of chitotriosidase [57]. A new and potentially more sensitive and more specific biomarker is the lyso-Glucosylsphingosine (lysoGb1) [58].
Also common in GD: low concentration of vitamin B12, lipoproteins (mainly HDL), and elevated levels of ferritin [59].
Differential Diagnosis
Diagnosis of GD by enzyme testing is unequivocal, but the rarity of the disease and the non-specific and heterogeneous nature of its symptoms may impede consideration of this disorder in the differential diagnosis. GD may go undiagnosed for many years, leading to severe complications that could be preventable or even reversible by ERT (i.e., AVN, severe bleeding, chronic bone pain, life-threatening sepsis, pathologic fractures, growth failure, and liver pathology] [60]. As with many other rare disorders, the first disease manifestations may be associated with more common disorders such as hepatosplenomegaly or low blood count, which often raise suspicion of infections (such as Epstein–Barr virus or Cytomegalovirus) or haematological malignancies.
Once the diagnosis of GD is considered, there are a few other LSDs which may present with hepatosplenomegaly and neurologic degeneration, such as GM1 gangliosidosis and Niemann-Pick disease types A and C. The presentation with a bone crisis quite often leads to the misdiagnosis of bacterial osteomyelitis, usually in young patients who undergo surgical drainage of the inflammatory bone, which becomes contaminated and eventually leads to chronic bacterial osteomyelitis [61]. Finally, in children, Legg-Calve-Perthes disease, an AVN of the femoral head could be confused with GD, given the identical clinical and radiological findings of these two disorders [62].
Mistry et al. conducted surveys of patients and Hematology-Oncology specialists to assess the frequency of diagnostic delays [60]. They reported a series of patients who suffered diagnostic delays and as a result developed disabilities, including potentially life-threatening manifestations of GD [60]. Of 136 patients surveyed, the average time from first appearance of GD symptoms to final diagnosis was 48.7 ± 123.6 months. More than two-thirds were evaluated and managed by a hematologist-oncologist [60]. Diagnostic delays led to complications that are potentially preventable or reversible with ERT
GD should be considered in the differential diagnosis of a patient with bone manifestations in the case of bone pain, osteopenia, osteoporosis, osteolytic lesions, bone infarcts, avascular bone necrosis, fractures, and rarely acute osteomyelitis, especially in young patients and in the Ashkenazi Jewish population, associated with one of the “typical” GD characteristics such as splenomegaly, bleeding tendency, low platelet counts, family history, and any of the associated diseases.
Therapeutic Approaches
The therapeutic approaches for GD include: splenectomy (in the pre-ERT era, rarely today), orthopedic surgery, ERT, substrate reduction therapy (SRT), and (in the near future) pharmacological chaperones (PC). The latter three are disease specific therapies, whereas the first two are treatments of complications. There are futuristic modalities like non-myeloablative stem cell transplantation and gene therapy [63], but these will not be reviewed.
In the past, prior to the availability of ERT, total or partial splenectomy was performed in patients with massive splenomegaly, or severe pancytopenia [18]. The beneficial effects of splenectomy included rapid normalization of platelet counts, decreased fatigue and bleeding tendencies, amelioration of abdominal discomfort and early satiety, and growth spurt in children [22]. However, the removal of the spleen led to progressive deterioration in liver and skeletal involvement, and therefore nowadays it is only performed in extremely unusual circumstances. However, it can be performed, even in the presence of huge splenomegaly, by laparoscopic surgery, thereby shortening the recovery period and decreasing patient’s discomfort [22].
Orthopedic surgery has an important role for patients who have developed irreversible joint damage (osteonecrosis) before the availability of ERT and, infrequently, despite ERT [64]. In case of femoral head osteonecrosis, total hip arthroplasty eliminates pain, preserves ambulation, and should allow for satisfactory daily life. In the future, newer types of implants would allow longer revision-free periods [64]. Lebel et al. have assessed the correlation of patient demographics, including ERT with bone histology, to facilitate decisions of whether and when to perform hip replacement surgery in patients with GD [65]. The histology of surgically removed femoral heads and categorized findings according the presence or extent of osteonecrosis, Gaucher cell infiltration, and bone regeneration qualifiers using a tripartite histology-based scoring system, have been examined. Histologic findings of Gaucher cell infiltration and bone regeneration qualifiers did not correlate with demographics or with exposure to ERT, and most specimens unexpectedly showed good regenerative response to osteonecrosis despite heavy Gaucher cell infiltration.
The first ERT (1991), the placenta-derived macrophage-targeted glucocerebrosidase, alglucerase (Ceredase™; Genzyme Corp), led to a revolution in the management of patients with GD [66]. ERT reduces hepatosplenomegaly, improves hypersplenism, decreases biomarkers, and ameliorates bone pain, together with a reliable safety profile [66]. Although the original regimen (“high-dose”) has been 60 units/kg body weight every other week, the authors (AZ, BB) have had good experience with lower doses, such as 15 U/kg every other week for adult patients and 30 U/kg every other week for children [66].
In 1994, Imiglucerase, the Chinese hamster ovary (CHO) cell–derived recombinant ERT (Cerezyme™; Genzyme Corp) was approved, and shortly thereafter it replaced alglucerase, based on a short-term high-dose comparative trial (imiglucerase vs alglucerase) which showed comparable safety and efficacy [67], and upon a second clinical trial that demonstrated similar safety and efficacy results of low-dose treatment (comparing 2.5 units/kg thrice a week vs. 15 units/kg every other week) [68]. ERT does not affect neurologic features (of type 3), because of its inability to penetrate the blood–brain-barrier, even in megadoses [69]. Recently, Weinreb et al. summarized the ICGG database in patients treated for 10 years, and confirmed the lasting effect alglucerase/imiglucerase treatment on haematological, visceral, and bone manifestations of GD type 1 [70].
In February 2010, velaglucerase alfa (VPRIV™, Shire, MA, USA), a gene-activated human glucocerebrosidase, was approved by the Food and Drug Administration (FDA) [71] and in 45 countries thereafter. Another new plant cell expressed human recombinant glucocerebrosidase taliglucerase alfa was approved by the FDA in 2012. Although considered “biosimilars”, these are not generic proteins, both have been developed as novel biologicals. Taliglucerase alfa has a high homology and a bioactivity similar to imiglucerase [72], and can be produced on a large scale at lower cost. Velaglucerase has a specific activity comparable to imiglucerase, but in contrast to imiglucerase and taliglucerase alfa, which have an amino substitution Histidine to Arginine at position 495, velaglucerase alfa has the wild type sequence, which may be a theoretical advantage, explaining booster effect in some of the patients switching from imiglucerase to velaglucerase alfa [73]. In addition, it appears that velaglucerase alfa is associated with fewer hypersensitivity reactions and fewer antibodies than the others, which could be related to the human cell lines used for its production [74]. Currently, 7 years of long-term follow-up with velaglucerase alfa has been achieved with the original cohort of patients participating in the phase I/II and extension clinical trial [75]. The most current analysis suggested improvement in bone parameters, both the MRI-based bone marrow burden scores and BMD, and this was despite a dose reduction from 60 to 30 units/kg body weight EOW after 15 months [74].
Taliglucerase alfa has also demonstrated significant improvement of bony parameters, studied by what is considered the most sensitive imaging tool of the bone marrow involvement in GD (albeit being available in only one center today in Amsterdam), quantitative chemical shift imaging (QCSI) [76].
Prospective (comparative) studies in larger cohorts are needed to validate these findings or to allow comparison of the different enzymes or new therapeutic modalities.
In contrast to ERT, which degrades the storage material within the lysosome thereby reducing its accumulation, SRT targets the biosynthetic cycle by partial inhibition of the glucocerebroside synthase, thereby reducing the load of substrate inside the lysosome, allowing the deficient enzyme to adequately hydrolyze the substrate and prevent storage. SRTs are small molecules that can be taken orally [22].
Miglustat (imino sugar N-butyl deoxynojirimycin) was the first approved SRT; it is taken orally three times a day, obviating the inconvenience of intravenous ERT. Due to its mode of action and less potent efficacy, it was approved for adult patients only with mild to moderate type 1 GD, for whom ERT was not suitable (according to the European Medicines Agency definition) or in whom ERT is not a therapeutic option (according to the FDA label); during the clinical trials the terminology used was “adult patients who are unable or unwilling to receive ERT” [77]. Clinical trials showed significant effects on key disease parameters [77], but the problematic safety profile, along with inferior efficacy [78], led to a small number of patients (estimated around 200 worldwide) who are using it for the management of GD. Although miglustat crosses the blood–brain barrier and should have been a prototype for therapeutic management of neuronopathic forms, the clinical trial with this SRT in type 3 GD failed to improve the primary end-points chosen to define neurologic benefits [79].
Eliglustat [Cerdelga; Sanofi-Aventis (Genzyme)] is a ceramide analog of the substrate, and has been recently approved by the FDA for treatment of adults with type 1 GD.
Unfortunately, eliglustat does not penetrate the blood–brain barrier and therefore does not provide any new hope for patients with type III GD. This drug has been the subject of several clinical trials for type 1 GD, including, most recently, three phase 3 trials, two of which have been submitted for registration of the drug, and are being continued in extension phases. The 4-year results of the phase 2 trial have recently been published, and they continue to show improvement in key clinical parameters (in 20 of 24 patients) including bones [80]. While it seems to be a better SRT, eliglustat requires long-term experience because of its complex cytochrome P450 metabolism, which may complicate the use of some medications, and because of potential nontrivial cardiotoxicity [81].
PCs therapy is a novel approach based on the ability of small molecules (the PCs) to interact with misfolded mutant proteins, preventing endoplasmic reticulum associated degradation (ERAD) in proteasomes and allowing trafficking to lysosomes, thereby increasing the amount of (mutant) enzyme that can hydrolyze the substrate [82]. In particular, this therapy is useful because only a modest increase in residual glucocerebrosidase should be sufficient to ameliorate the GD phenotype, and these small molecules should be able to cross the blood–brain barrier and impact the neuronopathic features of type III GD. While there is strong biological rationale and proof of concept from anecdotal studies [82] there has been only a single clinical trial with PC as monotherapy: isofagamine (Amicus Therapeutics, NL, USA), which failed phase 2, and its development had been stopped [83]. While new compounds are being tested pre-clinically, Don Mahuran from Canada has identified several existing drugs that can act as PC for mutant glucocerebrosidase [84], and there is growing evidence that one of them, Ambroxol, which is an over-the-counter mucolytic drug with more than 30 years of safe experience, available in various forms of administration and with many generic versions, can impact on both type 1 and type 3 GD. Paradoxically, its low cost has been a factor in preventing its development for patients with GD, and it is to be hoped that in the very near future clinical trials will take place either with or without the sponsorship of the pharmaceutic industry.
The Impact of Therapy on Bone
Several clinical trials have shown that ERT improves visceral, haematological and other disease parameters in type 1 GD within a relatively short period of 3–12 months [66, 85, 86]. The response to ERT of the bones, instead, appears to be much slower [85]. The ideal therapeutic effects in GD would be: prevention, stabilization, and reversal of bone disease progression. The degree of bone involvement prior to initiation of ERT, and possibly also the dose, are important variables. Early therapy could probably lead to a better bone outcome, but not all bone lesions are reversible in response to ERT. Moreover, extended treatment interruptions have been associated with poorer bone response, including the development of osteonecrosis, especially in children [86] and young adults.
Imiglucerase
Several studies have demonstrated the efficacy of imiglucerase therapy in reducing the burden of Gaucher cells in the bone marrow [87, 88]. However, non-homogeneous (type B) marrow packing, associated with irreversible lesions such as infarcts and necrosis, is less likely to be responsive to enzyme therapy [38].
Many reports describe improvement in BMD at the following sites: lumbar vertebrae, femora, tibial metaphyses, and distal ulnae. In a prospective non-randomized study of 33 type 1 GD patients treated with imiglucerase (60 U/kg/every 2 weeks), the mean Z-score for the spine increased from −0.72 (±1.302) at baseline to near normal levels by month 48 (p = 0.042), and for the femoral neck from −0.59 (±1.352) to −0.17 (±1.206) at 36 months (p = 0.035), an increase sustained also at 48 months [51]. Based on the data of the ICGG Gaucher Registry, Wenstrup et al. [89] reported in a large cohort of untreated patients with GD a low BMD of about 1SD below the reference population, which has remained unchanged over time. In contrast, patients receiving ERT showed a slow improvement of BMD with greater BMD increases in those patients treated with higher ERT dosages. After 8 years of ERT, the BMD approached that of the reference population [89]. A second study showed improvements of BMD in ERT treated patients with type 1 GD among all age groups, particularly in children and young adults, both of whom generally attained Z-scores higher than −1 [43]. On the other hand, one report described little or no gain of BMD in a small number of patients treated with a low dose of ERT [90].
Children compared to adults have a faster and consistent increase in BMD in response to treatment. In adults, an improvement in lumbar vertebra Z-score has required up to 4 years of imiglucerase therapy [51] compared with 2 years for children [91]. In a large cohort study of 884 children on long-term ERT, also based on the ICGG database, the median height approximated that of the normal population after 8 years of therapy [92]. No further bone crises occurred after 2 years of ERT, and BMD normalized after 6.6 years of ERT. In contrast, Drelichman et al. [93] described an interruption of ERT for 15–36 months in children, leading to recurrence of splenomegaly, hepatomegaly, worsening of blood count, growth retardation, and serious bone manifestations that did not resolve after re-initiation of ERT. Early treatment during childhood and adolescence may be crucial for patients, with an increased risk for future skeletal complications, such as “bad” genotypes or severe disease features of an older sibling, because most bone mineral is accrued before the age of 20 years, and there is only a small window of opportunity to achieve peak bone mass.
Data on the efficacy of imiglucerase therapy in preventing pathologic fractures are limited. An Israeli study from 2002 reported no fractures during a follow-up of more than 1 year in 10 children, four of whom had pathologic fractures prior to treatment, who were treated at 30 U/kg/month in 13 fractions per month (the old low-dose high-frequency regimen) [94].
Most patients with pre-existing bone pain generally report relief from bone pain within 1 year of initiating ERT [95]. In other studies, a reduction of bone pain and bone crisis could also be seen within a short time, further decreases of these symptoms could be observed during 2 years of ERT [96], and 70 % of patients had no or only very mild pain after 4 years of treatment [97]. In an ICGG Gaucher Registry analysis, the incidence of bone crisis fell from 17 % before treatment to 5 % in the first year after treatment, <1 % in the second year, and 3 % after the third year after initiation of therapy [98]. The effect of imiglucerase on delayed growth, examined in this cohort (42 % of 702 children were below the 5° percentile at baseline) showed, at baseline, the median height Z-score—1.4, which improved to 0.3 after 8 years of therapy, close to that of the non-Gaucher population [92].
A recent report assessed the achievement of predefined therapeutic goals (based on the ICGG) [99] in Israeli patients with GD receiving imiglucerase for four consecutive years on a low-dose regimen (constant dosage of 15 units/kg every other week for most of the adults and 30/kg every other week for most of the children). Among 164 patients, after four years low-dose ERT, there was a significant improvement in each of the therapeutic goal parameters (spleen and liver volumes, hemoglobin and platelet counts, and Z-scores for lumbar spine and femoral) from baseline. Children achieved improvement in linear growth and puberty. This survey highlights good overall outcome even with low-dose imiglucerase [99]. Higher doses of therapy, on the other hand, are controversial. Higher ERT doses (80 U/kg/4 weeks) demonstrated a more pronounced and quicker response of bone marrow involvement according to MRI as compared to lower ERT doses (15–30 U/kg/4 weeks) [29], however, even with high-dose therapy, in some clinical cases, bone disease persists [29]. This could be explained by the presence of sanctuary sites in the bone marrow where Gaucher cells escape the effects of treatment because of altered vascularization or fibrosis, or perhaps because sub-populations of Gaucher cells differ in their ability to take up enzyme [29]. On the other hand, a report has described little or no progression of skeletal disease among untreated adults with GD [100].
In correlation with the reduction of bone symptoms, ERT has been shown to have a significant positive effect on the quality of life of GD patients with skeletal disease after 2 years of treatment [101]. Finally, Rudzki et al. [101] evaluated also bone marrow biopsies prior to and after 26–32 months of ERT. The effect was mainly achieved by the reduction of Gaucher cell burden, the disappearance of Gaucher cells, and, but only partially, by the shrinkage of Gaucher cells. In addition, hematopoiesis increased in most of the patients, and, the estimated trabecular bone volume decreased in the control biopsies after ERT as compared to the initial biopsies before ERT [30, 51, 89]. These findings suggest that ERT shows relevant effects on the disease burden within the bone and bone marrow, and possible complex changes of hematopoiesis, cytokine expression, with consequent changes of bone metabolism. Further studies including bone biopsies will be needed to elucidate these questions.
Velaglucerase Alfa and Taliglucerase Alfa
One study has examined the achievement of therapeutic goals of velaglucerase alfa for skeletal pathology, specifically including the improvement in trabecular BMD in at least 3–5 years, in addition to anemia, thrombocytopenia, hepatomegaly, and splenomegaly [102]. Eight patients, aged 18–62 years, were treated with velaglucerase alfa 60 U/kg/every other week, and the dose was reduced stepwise to 30 U/kg/every other week during the second year of therapy. In addition, three of the eight patients were also receiving bisphosphonates (BPs). All patients had an improvement of BMD in at least one of two skeletal locations within 48 months of the initiation of therapy [102]. Recently, Elstein et al. analyzed the impact of velaglucerase alfa on bone marrow burden score (BM). The authors described an improvement in bone marrow burden scores (assessed using T1- and T2-weighted MRI images of the lumbar spine) through 5 years, which was sustained through 7 years, despite dose reduction beginning at 15 months [103]. Prospective studies in a large cohort are needed to validate these findings.
The pivotal double-blind randomized phase III study analyzed bone marrow fat fraction, measured by QCSI, to evaluate bone marrow response to treatment with taliglucerase alfa in naïve GD patients [104]. Eight GD patients with intact spleens were treated with taliglucerase alfa 30 or 60 U/kg biweekly. This treatment led to significant increases in lumbar spine fat fractions, which indicates clearance of Gaucher cells from bone marrow [104].
Substrate Reduction Therapy
Results regarding the effects of SRT on bone disease in type 1 GD are limited to those reported during the clinical trials, given that few patients world-wide receive this therapy. Nevertheless, it seems that miglustat, similar to ERT, exhibits faster improvement in bone pain and the occurrence of bone crisis than improvement on imaging findings [105]. No AVN or bone fractures were reported during the 2-year observational period, and BMD Z-scores improved from baseline at both the lumbar spine and femoral neck at each time point (months 6, 12, and 24) [105]. A prospective open-label investigational study, which evaluated the therapy with miglustat 100 mg in patients with Type 1 GD, reported an improvement of bone marrow infiltration by 2.9 points measured by S-MRI score in 28 patients [106].
The reported side effects of miglustat are: loose stools, tremor, and peripheral neuropathy. Miglustat is a steady-small molecule, and this feature could allow good penetration ability into bone and bone marrow cavity, and prevent recurrent priming of macrophages and release of cytokines [78]. The association between miglustat and ERT has not shown added benefits [107].
In a phase 2 clinical trial, 2-year follow-up data involving 20 patients treated with eliglustat tartrate showed improvement in lumbar spine BMD [108]. For 16 patients, improvements were seen in both T-scores and Z-scores. None of these patients received BPs for at least 3 months before initiation or during the study. Eight patients also showed reduced bone marrow infiltration on MRI, while 10 patients remained stable. No bone crises or pathological fractures were reported and, of seven patients with bone infarcts at baseline, six remained stable and one improved [108]. Eliglustat tartrate was well tolerated, 7 mild transient adverse events in 6 patients were considered treatment-related [108]. Phase 3 clinical trials have recently been concluded to confirm the efficacy, safety, and pharmacokinetics of eliglustat tartrate in adult patients with type 1 GD [108].
Bisphosphonates
In GD patients with osteopenia or osteoporosis, BPs may be applied in addition to disease specific therapy with either ERT or SRT. Several studies have described the benefit of BPs in treating bone manifestations of GD [109–111]. Wenstrup et al. followed 34 adults with type 1 GD, receiving ERT, treated daily with either 40 mg of alendronate or placebo for a period of 24 months. BPs treatment resulted in a significant improvement in BMD after 18 months, without improvement in focal lesions [109]. Ciana et al. treated five type 1 adult GD patients with pamidronate (45 mg every 3 weeks for 3–5 months) and observed an increased BMD and a rapid decrease in severe bone pain [110]. In a study by Samuel et al. on five adolescent patients, aminohydroxy propylidene BPs resulted in a significant decrease in bone crisis episodes and pathological fractures [111]. These limited reports appear to indicate that BPs may have a role in the treatment of skeletal manifestations of GD.
Among the possible BPs-related side effects, osteonecrosis of the jaw (ONJ) must be considered with particular interest in GD, since it is one of the most severe bone complications of GD. Interestingly, up to now, no cases of ONJ have been reported in type 1 GD treated with BPs [22]. Recommended measures to reduce the risk of ONJ on bisphosphonate therapy are: stop smoking, limit alcohol intake, and maintain good oral hygiene.
Conclusions and Future Objectives
Diagnosis of GD and Bone Manifestations
-
Early diagnosis and subsequent treatment of GD with ERT is fundamental for a significant improvement in bone outcome. Diagnostic delays lead to complications that are often preventable or reversible with ERT.
-
In many GD patients, bone disease may occur in the absence of typical signs and symptoms; it may show progressive deterioration over years, and represent one of the most debilitating aspects of the disease.
-
GD should be considered in the differential diagnosis of any patient with skeletal manifestations such as: bone pain, osteopenia, osteoporosis, osteolytic lesions, bone infarcts, AVN, fractures, and acute osteomyelitis, when they are associated with one of the “typical” GD characteristics.
-
Until now, knowledge and scientific research regarding the haematological and visceral aspects has exceeded knowledge of its bone manifestations, despite the fact that bone involvement is an important and frequent manifestation of GD.
-
In the future, broader physician education regarding GD and its bone manifestations will increase the likelihood of prompt detection of the disease and thereby improve its management.
-
For clinical work, further refinement and standardization of diagnostic approaches concerning bone disease in GD are warranted.
Pathophysiology of Bone Manifestations
-
The pathophysiology of bone disease is not well understood and, consequently, neither definitive treatment nor predictive testing for their occurrence is possible. The pathophysiological mechanism involves several factors such as: alterations of bone marrow and vascularity, immune cells, inflammation, macrophage-derived factors, cytokines, and hormones. Further studies are needed to understand the complex bone pathophysiology in GD, in particular by the experts of bone metabolism.
-
If possible, the assessment of bone marrow biopsies prior to and after of treatment for GD would be important to elucidate the complex pathophysiologic mechanism.
Bone Biomarkers
-
Currently, reports involving bone biomarkers in GD show variable results which do not support their routine use for the clinical assessment of bone status as an indication for therapy initiation, or for monitoring the response to therapy.
-
Better bone biomarkers that will be related to the bone manifestations of GD are needed for the diagnosis, staging and monitoring of the skeletal lesions in GD, in conjunction with imaging methods and bone densitometry measurements.
-
In particular, a reliable bone-biomarker should identify patients at risk of developing AVN.
Imaging Studies
-
Imaging of bone and bone marrow in GD aim to evaluate the disease burden, detect the presence of bone complications, and monitor the disease progression and response to therapy. Early detection and monitoring of the extent of bone disease by various imaging modalities, preferentially MRI, give the basis for therapeutic decisions. Further studies for long-term monitoring of the bone disease progression with serial imaging studies would be useful.
Treatment Approaches
-
ERT and SRT have shown positive effects on bone disease, in particular rapid reduction of bone pain, frequency of bone crisis, and overall improvement in quality of life, although results regarding the effects of SRT on bone disease in type 1 GD are quite limited. In some cases, successful treatment of bone complications may require higher ERT doses and a longer duration of therapy than those sufficient to improve visceral and hematologic manifestations. However, higher doses of therapy, on the other hand, are controversial. Further studies should shed light on this issue.
-
The temporal relationship between improvement in bone marrow involvement and improvement in other bone manifestations is unclear, and this suggests the importance of monitoring all aspects of skeletal disease and making treatment decisions based on these findings.
-
Skeletal involvement seems to improve slowly during ERT, but only a few studies evaluating BMD changes during a long follow-up period have been reported. Moreover, data on the efficacy of imiglucerase therapy in preventing pathologic fractures are limited. Further studies on long-term monitoring of the bone disease progression in childrens and in adults, with assessment of BMD and reduction of fracture risk would be useful.
-
Some reports appear to indicate that BPs may have a role in the treatment of bone manifestations of GD. Further data on fracture risk reduction, changes of bone biomarkers, and BMD assessement in GD with BPs would be necessary, evaluating also the potential risk of ONJ, a severe bone complication of GD.
-
Gene therapy has not been discussed; however, stem cell therapy with a low-risk conditioning regimen may have a role in the future.
Multidisciplinary Approach
Since GD affects multiple organs, all patients with GD should be regularly monitored from a clinical and laboratory standpoint, preferably at a specialized center where a multidisciplinary team is available to assess the course of the disease and effects of therapy.
References
Rosenbloom BE, Weinreb NJ (2013) Gaucher disease: a comprehensive review. Crit Rev Oncog 18:163–175
Ron I, Horowitz M (2005) ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum Mol Genet 15(14):2387–2398
Brady RO (1972) Biochemical and metabolic basis of familial sphingolipidoses. Semin Hematol 9:273–284
Hruska KS, LaMarca ME, Scott CR, Sidransky E (2008) Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat 29:567–583
Ilan Y, Elstein D, Zimran A (2009) Glucocerebroside: an evolutionary advantage for patients with Gaucher disease and a new immunomodulatory agent. Immunol Cell Biol 87:514–524
Fairley C, Zimran A, Phillips M, Cizmarik M, Yee J, Weinreb N, Packman S (2008) Phenotypic heterogeneity of N370S homozygotes with type I Gaucher disease: an analysis of 798 patients from the ICGG Gaucher Registry. J Inherit Metab 31:737–744
Abrahamov A, Elstein D, Gross-Tsur V, Farber B, Glaser Y, Hadas-Halpern I, Ronen S, Tafakjdi M, Horowitz M, Zimran A (1995) Gaucher’s disease variant characterised by progressive calcification of heart valves and unique genotype. Lancet 346:1000–1003
Meikle PJ, Fuller M, Hopwood JJ (2007) Epidemiology and screening policy. In: Futerman AH, Zimran A (eds) Gaucher Disease. CRC Press, Boca Raton, pp 321–340
Sidransky E (2004) Gaucher disease: complexity in a “simple” disorder. Mol Genet Metab 83:6–15
Biegstraaten M, van Schaik IN, Aerts JM, Hollak CE (2008) ‘Non-neuronopathic’ Gaucher disease reconsidered. Prevalence of neurological manifestations in a Dutch cohort of type I Gaucher disease patients and a systematic review of the literature. J Inherit Metab Dis 31:337–349
Bodamer OA, Hung C (2010) Laboratory and genetic evaluation of Gaucher disease. Wien Med Wochenschr 160:600–604
Tatti M, Motta M, Di Bartolomeo S, Cianfanelli V, Salvioli R (2013) Cathepsin-mediated regulation of autophagy in saposin C deficiency. Autophagy 9:241–243
Human Gene Database. http://www.hgmd.cf.ac.uk/ac/index.php
Taddei TH, Kacena KA, Yang M, Yang R, Malhotra A, Boxer M, Aleck KA, Rennert G, Pastores GM, Mistry PK (2009) The underrecognized progressive nature of N370S Gaucher disease and assessment of cancer risk in 403 patients. Am J Hematol 84:208–214
Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, Pastores G, Rosenbloom BE, Scott CR, Wappner RS, Weinreb NJ, Zimran A (2000) The Gaucher registry: demographics and disease characteristics of 1698 patients with Gaucher disease. Arch Intern Med 160:2835–2843
Tybulewicz VL, Tremblay ML, LaMarca ME, Willemsen R, Stubblefield BK, Winfield S, Zablocka B, Sidransky E, Martin BM, Huang SP et al (1992) Animal model of Gaucher’s disease from targeted disruption of the mouse glucocerebrosidase gene. Nature 357:407–410
Martins AM, Valadares ER, Porta G, Coelho J, Semionato Filho J, Pianovski MA, Kerstenetzky MS, Montoril Mde F, Aranda PC, Pires RF, Mota RM, Bortolheiro TC; Brazilian Study Group on Gaucher Disease and other Lysosomal Storage Diseases (2009) Recommendations on diagnosis, treatment, and monitoring for Gaucher disease. J Pediatr. doi:10.1016/j.jpeds.2009.07.004
Lewin A, Orvisky E, Goker-Alpan O, LaMarca ME, Sidransky E (2004) Glucocerebrosidase mutations in subjects with parkinsonism. Mol Genet Metab 81:70–73
Cheung WY, Greenberg CR, Bernstein K, Schacter B, Fourie T, Seftel MD (2007) Type I Gaucher disease following chemotherapy for light chain multiple myeloma. Intern Med 46:1255–1258
Arends M, van Dussen L, Biegstraaten M, Hollak CE (2013) Malignancies and monoclonal gammopathy in Gaucher disease; a systematic review of the literature. Br J Haematol 161:832–842
Mistry PK, Sadan S, Yang R, Yee J, Yang M (2007) Consequences of diagnostic delays in type 1 Gaucher disease: the need for greater awareness among hematologists-oncologists and an opportunity for early diagnosis and intervention. Am J Hematol 82:697–701
Zimran A (2011) How I treat Gaucher disease. Blood 118:1463–1471
Zimran A, Elstein D (2010) Lipid storage diseases. In: Lichtman MA, Kipps T, Seligsohn U, Kaushansky K, Prchal JT (eds) Williams Hematology, 8th edn. McGraw-Hill, New York, pp 1065–1071
Mikosch P, Hughes D (2010) An overview on bone manifestations in Gaucher disease. Wien Med Wochenschr 160:609–624
Boyce BF, Yao Z, Xing L (2009) Osteoclasts have multiple roles in bone in addition to bone resorption. Crit Rev Eukaryot Gene Expr 19:171–180
Allen MJ, Myer BJ, Khokher AM, Rushton N, Cox TM (1997) Pro-inflammatory cytokines and the pathogenesis of Gaucher’s disease: increased release of interleukin-6 and interleukin-10. QJM 90:19–25
van Breemen MJ, de Fost M, Voerman JS, Laman JD, Boot RG, Maas M, Hollak CE, Aerts JM, Rezaee F (2007) Increased plasma macrophage inflammatory protein (MIP)-1alpha and MIP-1beta levels in type 1 Gaucher disease. Biochim Biophys Acta 1772:788–796
Lacerda L, Arosa FA, Lacerda R, Cabeda J, Porto G, Amaral O, Fortuna A, Pinto R, Oliveira P, McLaren CE, Sá Miranda C, de Sousa M (1999) T cell numbers relate to bone involvement in Gaucher disease. Blood Cells Mol Dis 25:130–138
de Fost M, van Noesel CJ, Aerts JM, Maas M, Pöll RG, Hollak CE (2008) Persistent bone disease in adult type 1 Gaucher disease despite increasing doses of enzyme replacement therapy. Haematologica 93:1119–1120
Grigorescu Sido P, Drugan C, Cret V, Al-Kzouz C, Denes C, Coldea C, Zimmermann A (2007) Outcome of enzyme replacement in patients with Gaucher disease type I. The Romanian experience. J Inherit Metab Dis 30:783–789
Faden MA, Krakow D, Ezgu F, Rimoin DL, Lachman RS (2009) The Erlenmeyer flask bone deformity in the skeletal dysplasias. Am J Med Gen A 149A:1334–1345
Pastores GM, Wallenstein S, Desnick RJ, Luckey MM (1996) Bone density in Type 1 Gaucher disease. J Bone Miner Res 11:1801Y1807.
Arnheim E, Chicco G, Phillips M, Lebel E, Foldes AJ, Itzchaki M, Elstein D, Zimran A, Altarescu G (2008) Molecular aspects of osteopathy in type 1 Gaucher disease: correlation between genetics and bone density. Rheumatol Int 28:873–877
Deegan PB, Pavlova E, Tindall J, Stein PE, Bearcroft P, Mehta A, Hughes D, Wraith JE, Cox TM (2011) Osseous manifestations of adult Gaucher disease in the era of enzyme replacement therapy. Medicine (Baltimore) 90:52–60
Moran MT, Schofield JP, Hayman AR, Shi GP, Young E, Cox TM (2000) Pathologic gene expression in Gaucher disease: up-regulation of cysteine proteinases including osteoclastic cathepsin K. Blood 96:1969–1978
Zimran A, Morris E, Mengel E, Kaplan P, Belmatoug N, Hughes DA, Malinova V, Heitner R, Sobreira E, Mrsić M, Granovsky-Grisaru S, Amato D, vom Dahl S (2009) The female Gaucher patient: the impact of enzyme replacement therapy around key reproductive events (menstruation, pregnancy and menopause). Blood Cells Mol Dis 43:264–288
Mistry PK, Deegan P, Vellodi A, Cole JA, Yeh M, Weinreb NJ (2009) Timing of initiation of enzyme replacement therapy after diagnosis of type 1 Gaucher disease: effect on incidence of avascular necrosis. Br J Haematol 147:561Y570.
Poll LW, Koch JA, vom Dahl S, Loxtermann E, Sarbia M, Niederau C, Häussinger D, Mödder U (2000) Extraosseous manifestation of Gaucher’s disease type I: MR and histological appearance. Eur Radiol 10:1660–1663
Vom Dahl S, Poll L, Di Rocco M, Ciana G, Denes C, Mariani G, Maas M (2006) Evidence-based recommendations for monitoring bone disease and the response to enzyme replacement therapy in Gaucher patients. Curr Med Res Opin 22:1045–1064
Weinreb NJ1, Aggio MC, Andersson HC, Andria G, Charrow J, Clarke JT, Erikson A, Giraldo P, Goldblatt J, Hollak C, Ida H, Kaplan P, Kolodny EH, Mistry P, Pastores GM, Pires R, Prakash-Cheng A, Rosenbloom BE, Scott CR, Sobreira E, Tylki-Szymańska A, Vellodi A, vom Dahl S, Wappner RS, Zimran A; International Collaborative Gaucher Group (ICGG) (2004) Gaucher disease type 1: revised recommendations on evaluations and monitoring for adult patients. Semin Hematol 41:15–22.
van Dussen L, Akkerman EM, Hollak CE, Nederveen AJ, Maas M (2014) Evaluation of an imaging biomarker, Dixon quantitative chemical shift imaging, in Gaucher disease: lessons learned. J Inherit Metab Dis 101(4):898–904
Mikosch P, Kohlfürst S, Gallowitsch HJ, Kresnik E, Lind P, Mehta AB, Hughes DA (2008) Is there a role for scintigraphic imaging of bone manifestations in Gaucher disease? Nuklearmedizin 47:239–247
Mistry PK, Cappellini MD, Lukina E, Ozsan H, Mach Pascual S, Rosenbaum H, Helena Solano M, Spigelman Z, Villarrubia J, Watman NP, Massenkeil G (2011) A reappraisal of Gaucher disease-diagnosis and disease management algorithms. Am J Hematol 86:110–115. doi:10.1002/ajh.21888
Crockett JC, Rogers MJ, Coxon FP, Hocking LJ, Helfrich MH (2011) Bone remodelling at a glance. J Cell Sci 124:991–998
Drugan C, Jebeleanu G, Grigorescu-Sido P, Caillaud C, Craciun AM (2002) Biochemical markers of bone turnover as tools in the evaluation of skeletal involvement in patients with type 1 Gaucher disease. Blood Cells Mol Dis 28:13–20
Ciana G, Martini C, Leopaldi A, Tamaro G, Katouzian F, Ronfani L, Bembi B (2003) Bone marker alterations in patients with type 1 Gaucher disease. Calcif Tissue Int 72:185–189
Stowens DW, Teitelbaum SL, Kahn AJ, Barranger JA (1985) Skeletal complications of Gaucher disease. Medicine 64:310–322
van Dussen L, Lips P, Everts VE, Bravenboer N, Jansen ID, Groener JE, Maas M, Blokland JA, Aerts JM, Hollak CE (2011) Markers of bone turnover in Gaucher disease: modeling the evolution of bone disease. J Clin Endocrinol Metab 96:2194–2205
Mistry PK, Liu J, Yang M, Nottoli T, McGrath J, Jain D, Zhang K, Keutzer J, Chuang WL, Mehal WZ, Zhao H, Lin A, Mane S, Liu X, Peng YZ, Li JH, Agrawal M, Zhu LL, Blair HC, Robinson LJ, Iqbal J, Sun L, Zaidi M (2010) Glucocerebrosidase gene-deficientmouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage. Proc Natl Acad Sci USA 107(45):19473–19478. doi:10.1073/pnas.1003308107
Ciana G, Addobbati R, Tamaro G, Leopaldi A, Nevyjel M, Ronfani L, Vidoni L, Pittis MG, Bembi B (2005) Gaucher disease and bone: laboratory and skeletal mineral density variations during a long period of enzyme replacement therapy. J Inherit Metab Dis 28:723–732
Sims KB, Pastores GM, Weinreb NJ, Barranger J, Rosenbloom BE, Packman S, Kaplan P, Mankin H, Xavier R, Angell J, Fitzpatrick MA, Rosenthal D (2008) Improvement of bone disease by imiglucerase (Cerecyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48-month longitudinal cohort study. Clin Genet 73:430–440
Fiore CE, Barone R, Pennisi P et al (2002) Bone ultrasonometry, bone density, and bone turnover markers in type 1 Gaucher disease. J Bone Miner Metab 20:24–38
Mikosch P, Reed M, Baker R, Holloway B, Berger L, Mehta AB, Hughes DA (2008) Changes of bone metabolism in seven patients with Gaucher disease treated consecutively with imiglucerase and miglustat. Calcif Tissue Int 83:43–54
Magal I, Lebel E, Altarescu G, Itzchaki M, Rudensky B, Foldes AJ, Zimran A, Elstein D (2006) Serum levels of osteoprotegerin and osteoprotegerin polymorphisms in Gaucher disease. Br J Haematol 133:93–97
Parisi MS, Mastaglia SR, Bagur A, Goldstein G, Zeni SN, Oliveri B (2008) Body composition and bone metabolism in young Gaucher disease type I patients treated with imiglucerase. Eur J Med Res 13:31–38
Pastores GM, Weinreb NJ, Aerts H, Andria G, Cox TM, Giralt M, Grabowski GA, Mistry PK, Tylki-Szymańska A (2004) Therapeutic goals in the treatment of Gaucher disease. Semin Hematol 41:4–14
Boot RG, Verhoek M, de Fost M, Hollak CE, Maas M, Bleijlevens B, van Breemen MJ, van Meurs M, Boven LA, Laman JD, Moran MT, Cox TM, Aerts JM (2004) Marked elevation of the chemokine CCL18/PARC in Gaucher disease: a novel surrogate marker for assessing therapeutic intervention. Blood 103:33–39
Rolfs A, Giese AK, Grittner U, Mascher D, Elstein D, Zimran A, Böttcher T, Lukas J, Hübner R, Gölnitz U, Röhle A, Dudesek A, Meyer W, Wittstock M, Mascher H (2013) Glucosylsphingosine is a highly sensitive and specific biomarker for primary diagnostic and follow-up monitoring in Gaucher disease in a non-Jewish, Caucasian cohort of Gaucher disease patients. PLoS ONE 8:e79732. doi:10.1371/journal.pone.0079732
Stein P, Yu H, Jain D, Mistry PK (2010) Hyperferritinemia and iron overload in type 1 Gaucher disease. Amer J Hematol 85:472–476
Mistry PK, Sadan S, Yang R, Yee J, Yang M (2007) Consequences of diagnostic delays in type 1 Gaucher disease: the need for greater awareness among hematologists-oncologists and an opportunity for early diagnosis and intervention. Am J Hematol 82:697–701
Beutler E, Grabowski G (2000) The metabolic and molecular bases of inherited disease. In: Scriver C, Sly W, Childs B et al (eds) Gaucher Disease. McGraw-Hill, New York, pp 3663–3668
Kenet G, Hayek S, Mor M, Lubetsky A, Miller L, Rosenberg N, Mosheiff R, Itzchaki M, Elstein D, Wientroub S, Zimran A (2003) The 1226G (N370S) Gaucher mutation among patients with Legg-Calve-Perthes disease. Blood Cells Mol Dis 31:72–74
Ortolano S, Viéitez I, Navarro C, Spuch C (2014) Treatment of lysosomal storage diseases: recent patents and future strategies. Recent Pat Endocr Metab Immune Drug Discov 8:9–25
Lebel E, Ioscovich A, Itzchaki M, Zimran A, Elstein D (2011) Hip arthroplasty in patients with Gaucher disease. Blood Cells Mol Dis 15(46):60–65
Lebel E, Elstein D, Peleg A, Reinus C, Zimran A, Amir G (2013) Histologic findings of femoral heads from patients with Gaucher disease treated with enzyme replacement. Am J Clin Pathol 140:91–96
Grabowski GA, Barton NW, Pastores G, Dambrosia JM, Banerjee TK, McKee MA, Parker C, Schiffmann R, Hill SC, Brady RO (1995) Enzyme therapy in type 1 Gaucher disease: comparative efficacy of mannose-terminated glucocerebrosidase from natural and recombinant sources. Ann Intern Med 122:33–39
Grabowski GA, Kacena K, Cole JA, Hollak CE, Zhang L, Yee J, Mistry PK, Zimran A, Charrow J, vom Dahl S (2009) Dose-response relationships for enzyme replacement therapy with imiglucerase/alglucerase in patients with Gaucher disease type 1. Genet Med 11:92–100
Zimran A, Elstein D, Levy-Lahad E, Zevin S, Hadas-Halpern I, Bar-Ziv Y, Foldes J, Schwartz AJ, Abrahamov A (1995) Replacement therapy with imiglucerase for type 1 Gaucher’s disease. Lancet 345:1479–1480
Zimran A, Elstein D (2007) No justification for very high dose enzyme therapy for patients with type III Gaucher disease. J Inherit Metab Dis 30:843–844
Weinreb NJ, Goldblatt J, Villalobos J, Charrow J, Cole JA, Kerstenetzky M, vom Dahl S, Hollak C (2013) Long-term clinical outcomes in type 1 Gaucher disease following 10 years of imiglucerase treatment. Inherit Metab Dis 36(3):543–53. Erratum in: J Inherit Metab Dis (2014) 37:147
Zimran A, Loveday K, Fratazzi C, Elstein D (2007) A pharmacokinetic analysis of a novel enzyme replacement therapy with Gene-Activated human glucocerebrosidase (GA-GCB) in patients with type 1 Gaucher disease. Blood Cells Mol Dis 39:115–118
Shaaltiel Y, Bartfeld D, Hashmueli S, Baum G, Brill-Almon E, Galili G, Dym O, Boldin-Adamsky SA, Silman I, Sussman JL, Futerman AH, Aviezer D (2007) Production of glucocerebrosidase with terminal mannose glycans for enzyme replacement therapy of Gaucher’s disease using a plant cell system. Plant Biotechnol J 5:579–590
Elstein D, Altarescu G, Maayan H, Phillips M, Abrahamov A, Hadas-Halpern I, Tiomkin M, Zimran A (2012) Booster-effect with velaglucerase alfa in patients with Gaucher disease switched from long-term imiglucerase therapy: Early Access Program results from Jerusalem. Blood Cells Mol Dis 48:45–50
Ben Turkia H, Gonzalez DE, Barton NW, Zimran A, Kabra M, Lukina EA, Giraldo P, Kisinovsky I, Bavdekar A, Ben Dridi MF, Gupta N, Kishnani PS, Sureshkumar EK, Wang N, Crombez E, Bhirangi K, Mehta A (2013) Velaglucerase alfa enzyme replacement therapy compared with imiglucerase in patients with Gaucher disease. Am J Hematol 88:179–184. doi:10.1002/ajh.23382
Elstein D, Haims AH, Zahrieh D, Cohn GM, Zimran A (2014) Impact of velaglucerase alfa on bone marrow burden score in adult patients with type 1 Gaucher disease: 7-Year follow-up. Blood Cells Mol Dis 53:56–60
van Dussen L, Zimran A, Akkerman EM, Aerts JM, Petakov M, Elstein D, Rosenbaum H, Aviezer D, Brill-Almon E, Chertkoff R, Maas M, Hollak CE (2013) Taliglucerase alfa leads to favorable bone marrow responses in patients with type I Gaucher disease. Blood Cells Mol Dis 50:206–211
Elstein D, Hollak C, Aerts JM et al (2004) Sustained therapeutic effects of oral miglustat (Zavesca, N-butyldeoxynojirimycin, OGT 918) in type I Gaucher disease. J Inherit Metab Dis 27:757–766
Hollak CE, Hughes D, van Schaik IN, Schwierin B, Bembi B (2009) Miglustat (Zavesca) in type 1 Gaucher disease: 5-year results of a post-authorisation safety surveillance programme. Pharmacoepidemiol Drug Saf 18:770–777
Schiffmann R, Fitzgibbon EJ, Harris C et al (2008) Randomized, controlled trial of miglustat in Gaucher’s disease type 3. Ann Neurol 64:514–522
Lukina E, Watman N, Dragosky M, Pastores GM, Arreguin EA, Rosenbaum H, Zimran A, Angell J, Ross L, Puga AC, Peterschmitt JM (2014) Eliglustat, an investigational oral therapy for Gaucher disease type 1: phase 2 trial results after 4 years of treatment. Blood Cells Mol Dis. doi:10.1016/j.bcmd.2014.04.002
Kamath RS, Lukina E, Watman N, Dragosky M, Pastores GM, Arreguin EA, Rosenbaum H, Zimran A, Aguzzi R, Puga AC, Norfleet AM, Peterschmitt MJ, Rosenthal DI (2014) Skeletal improvement in patients with Gaucher disease type 1: a phase 2 trial of oral eliglustat. Skeletal Radiol 43:1353–1360
McEachern KA, Fung J, Komarnitsky S, Siegel CS, Chuang WL, Hutto E, Shayman JA, Grabowski GA, Aerts JM, Cheng SH, Copeland DP, Marshall J (2007) A specific and potent inhibitor of glucosylceramide synthase for substrate reduction therapy of Gaucher disease. Mol Genet Metab 91:259–267
Parenti G (2009) Treating lysosomal storage diseases with pharmacological chaperones: from concept to clinics. EMBO Mol Med 1:268–279
Maegawa GH, Tropak MB, Buttner JD, Rigat BA, Fuller M, Pandit D, Tang L, Kornhaber GJ, Hamuro Y, Clarke JT, Mahuran DJ (2009) Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J Biol Chem 284:23502–23516. doi:10.1074/jbc.M109.012393
de Fost M, Hollak CE, Groener JE, Aerts JM, Maas M, Poll LW, Wiersma MG, Häussinger D, Brett S, Brill N, vom Dahl S (2006) Superior effects of high-dose enzyme replacement therapy in type 1 Gaucher disease on bone marrow involvement and chitotriosidase levels: a 2-center retrospective analysis. Blood 108:850–855
Tóth J, Szücs FZ, Benkö K, Maródi L (2003) Enzyme replacement therapy in Gaucher disease: monitoring visceral and bone changes with MRI. Orv Hetil 144:749-55B
Hollak C, Maas M, Akkerman E, den Heeten A, Aerts H (2001) Dixon quantitative chemical shift imaging is a sensitive tool for the evaluation of bone marrow responses to individualized doses of enzyme supplementation therapy in type 1 Gaucher disease. Blood Cells Mol Dis 27:1005–1012
Rosenthal DI, Doppelt SH, Mankin HJ, Dambrosia JM, Xavier RJ, McKusick KA, Rosen BR, Baker J, Niklason LT, Hill SC et al (1995) Enzyme replacement therapy for Gaucher disease: skeletal responses to macrophage-targeted glucocerebrosidase. Pediatrics 96:629–637
Wenstrup RJ, Kacena KA, Kaplan P, Pastores GM, Prakash-Cheng A, Zimran A, Hangartner TN (2007) Effect of enzyme replacement therapy with imiglucerase on BMD in type 1 Gaucher disease. J Bone Miner Res 22:119–126
Schiffmann R, Mankin H, Dambrosia JM, Xavier RJ, Kreps C, Hill SC, Barton NW, Rosenthal DI (2002) Decreased bone density in splenectomized Gaucher patients receiving enzyme replacement therapy. Blood Cells Mol Dis 28:288–296
Bembi B, Ciana G, Mengel E, Terk MR, Martini C, Wenstrup RJ (2002) Bone complications in children with Gaucher disease. Br J Radiol 75:A37–A44
Andersson H, Kaplan P, Kacena K, Yee J (2008) Eight-year outcomes of long-term enzyme replacement therapy for 884 children with Gaucher disease type 1. Pediatrics 122:1182–1190
Drelichman G, Ponce E, Basack N, Freigeiro D, Aversa L, Graciela E, Kohan R (2007) Clinical concequences of interrupting enzyme replacement therapy in children with type 1 Gaucher disease. J Pediatr 151:197–201
Cohen IJ, Katz K, Kornreich L, Horev G, Frish A, Zaizov R (1998) Low-dose high frequency enzyme replacement therapy prevents fractures without complete suppression of painful bone crises in patients with severe juvenile onset type I Gaucher disease. Blood Cells Mol Dis 24:296–302
Goker-Alpan O (2011) Therapeutic approaches to bone pathology in Gaucher disease: past, present and future. Mol Genet Metab 104:438–447
Weinreb NJ, Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, Pastores G, Rosenbloom BE, Scott CR, Wappner RS, Zimran A (2002) Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am J Med 113:112–119
Weinreb N, Taylor J, Cox T, Yee J, vom Dahl S (2008) A benchmark analysis of the achievement of therapeutic goals for type 1 Gaucher disease patients treated with imiglucerase. Am J Hematol 83:890–895
Charrow J, Dulisse B, Grabowski GA, Weinreb NJ (2007) The effect of enzyme replacement therapy on bone crisis and bone pain in patients with type 1 Gaucher disease. Clin Genet 71:205–211
Tukan I, Hadas-Halpern I, Altarescu G, Abrahamov A, Elstein D, Zimran A (2013) Achievement of therapeutic goals with low-dose imiglucerase in Gaucher disease: a single-center experience. Adv Hematol 2013:151506
Beutler E, Demina A, Laubscher K, Garver P, Gelbart T, Balicki D, Vaughan L (1995) The clinical course of treated and untreated Gaucher disease. A study of 45 patients. Blood Cells Mol Dis 21:86–108
Rudzki Z, Okoń K, Machaczka M, Rucińska M, Papla B, Skotnicki AB (2003) Enzyme replacement therapy reduces Gaucher cell burden but may accelerate osteopenia in patients with type I disease: histological study. Eur J Haematol 70:273–281
Elstein D, Cohn GM, Wang N, Djordjevic M, Brutaru C, Zimran A (2011) Early achievement and maintenance of the therapeutic goals using velaglucerase alfa in type 1 Gaucher disease. Blood Cells Mol Dis 46:119–123
Elstein D, Haims AH, Zahrieh D, Cohn GM, Zimran A (2014) Impact of velaglucerase alfa on bone marrow burden score in adult patients with type 1 Gaucher disease: 7-year follow-up. Blood Cells Mol Dis 53:56–60
van Dussen L, Zimran A, Akkerman EM, Aerts JM, Petakov M, Elstein D, Rosenbaum H, Aviezer D, Brill-Almon E, Chertkoff R, Maas M, Hollak CE (2013) Taliglucerase alfa leads to favorable bone marrow responses in patients with type I Gaucher disease. Blood Cells Mol Dis 50:206–211
Pastores GM, Elstein D, Hrebícek M, Zimran A (2007) Effect of miglustat on bone disease in adults with type 1 Gaucher disease: a pooled analysis of three multinational open-label studies. Clin Ther 29:1645–1654
Giraldo P, Alfonso P, Atutxa K, Fernández-Galán MA, Barez A, Franco R, Alonso D, Martin A, Latre P, Pocovi M (2009) Real-world clinical experience with long term miglustat maintenance therapy in type 1 Gaucher disease: the ZAGAL project. Haematologica 94:1771–1775
Elstein D, Dweck A, Attias D, Hadas-Halpern I, Zevin S, Altarescu G, Aerts JF, van Weely S, Zimran A (2007) Oral maintenance clinical trial with miglustat for type I. Blood 110:2296–2301
Lukina E, Watman N, Arreguin EA, Dragosky M, Iastrebner M, Rosenbaum H, Phillips M, Pastores GM, Kamath RS, Rosenthal DI, Kaper M, Singh T, Puga AC, Peterschmitt MJ (2010) Improvement in hematological, visceral, and skeletal manifestations of Gaucher disease type 1 with oral eliglustat tartrate (Genz-112638) treatment: two-year results of a phase 2 study. Blood 116:4095–4098
Wenstrup RJ, Bailey L, Grabowski GA, Moskovitz J, Oestreich AE, Wu W, Sun S (2004) Gaucher disease: alendronate disodium improves bone mineral density in adults receiving enzyme therapy. Blood 104:1253
Ciana G, Cuttini M, Bembi B (1997) Short-term effects of pamidronate in patients with Gaucher’s disease and severe skeletal involvement. N Engl J Med 337:712
Samuel R, Katz K, Papapoulos SE, Yosipovitch Z, Zaizov R, Liberman UA (1994) Aminohydroxy propylidene bisphosphonate (APD) treatment improves the clinical skeletal manifestations of Gaucher’s disease. Pediatrics 94:385–389
Acknowledgments
The IOF/ESCEO symposium and work were supported by F.I.R.M.O. Fondazione Raffaella Becagli, a no profit organization fully devoted to research in bone and mineral disorders. The skeletal images are courtesy of Dr. Ehud Lebel, Department of Orthopedic Surgery, Shaare Zedek Medical center, Jerusalem, Israel.
Author Contributions
The paper was drafted by GM and MLB was principally responsible for the literature review and analysis of the data. All authors contributed to conception of the work, critically revising the work for intellectual content and approved the final version of the paper. All authors confirm that they are accountable for all aspects of the work and undertake to ensure that questions relating to the accuracy and integrity of the work are investigated and resolved.
Conflict of Interest
AZ: Consultant/advisory role, stoke ownership and remuneration: Protalix Biotherapeutics, Carmiel Israel; Genzyme Sanofi and Shire; Pfizer, Shire, Genzyme Sanofi. JYR: Consulting fees or paid advisory boards: Servier, Novartis, Negma, Lilly; Wyeth, Amgen, Glaxo Smith Kline, Roche, Merckle, Nycomed Takeda, NPS, IBSA Genevrier, Theramex, UCB, Asahi Kasei; Lecture fees when speaking at the invitation of a commercial sponsor: Merck Sharp and Dome, Lilly, Rottapharm, IBSA, Genevrier, Novartis, Servier, Roche, Glaxo Smith Kline, Merckle, Teijn, Teva, Analis, Theramex, Nycomed, NovoNordisk, Ebewee Pharm, Zodiac, Danone, Will Pharma. Grant support from industry: Bristol Myers Squibb, Merck Sharp and Dome, Rottapharm, Teva, Roche, Amgen, Lilly, Novartis, Glaxo Smith Kline, Servier, Pzifer, Theramex, Danone, Organon, Therabel, Boeringer, Chiltern, Galapagos. RR: Merck Sharp and Dohme, Eli Lilly, Amgen, GSK, Servier and Danone. CC: AMGEN, GSK, Alliance for Better Bone Health, MSD, Eli Lilly, Pfizer, Novartis, Servier, Merck, Medtronic and Roche. MLB: Conflict of interest as consultant and grants recipient are from: Amgen, Eli Lilly, MSD, Novartis, Roche and Servier.
Human and Animal Rights and Informed Consent
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Marcucci, G., Zimran, A., Bembi, B. et al. Gaucher Disease and Bone Manifestations. Calcif Tissue Int 95, 477–494 (2014). https://doi.org/10.1007/s00223-014-9923-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-014-9923-y