
Abstract
The pathogenesis of virus-associated acute encephalopathy (VAE) involves brain edema caused by disruption of the blood–brain barrier (BBB). We aimed to develop an in vitro VAE model using an in vitro BBB model, to evaluate the dynamics of vascular dysfunction caused by tumor necrosis factor (TNF)-α. A co-culture model, consisting of Transwell®-grown human brain microvascular endothelial cells and pericytes, was treated with serially diluted TNF-α. Transendothelial electrical resistance (TER) was measured using cellZscope®. A permeability assay, using fluorescein isothiocyanate-conjugated sodium or dextran, was performed. Changes in claudin-5 localization and expression after TNF-α treatment were observed using immunofluorescence staining and western blot analysis. The TER decreased and permeability increased after TNF-α treatment; recovery time was dependent on TNF-α concentration. Claudin-5 was delocalized after TNF-α treatment and recovered in a TNF-α concentration-dependent manner. The expression of claudin-5 decreased 24 h after the TNF-α treatment and completely recovered 48 h after TNF-α treatment. Claudin-5 delocalization was likely associated with vascular hyperpermeability. To conclude, we evaluated vascular endothelial cell permeability and injury in VAE using an in vitro BBB model treated with TNF-α. This system can be useful for developing novel therapeutic strategies for VAE and designing treatments that target vascular permeability.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Virus-associated acute encephalopathy (VAE) is an acute brain dysfunction preceded by viral infections, such as the influenza virus, human herpes virus, and rotavirus (Asano et al. 1992; Mizuguchi 1997; Kasai et al. 2000; Karampatsas et al. 2018). It is characterized by impaired consciousness and signs of increased intracranial pressure and may be accompanied by convulsions or seizures. In children, it is associated with severe neurological defects and high mortality rates. Pathological studies of the brain at necropsy showed that severe cases of VAE are characterized by extensive brain edema and plasma extravasation, indicating disruption of the blood–brain barrier (BBB) (Mizuguchi et al. 2007). In VAE, elevated concentrations of several cytokines, such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6, have been observed in blood serum (Ichiyama et al. 1998; Hosoya et al. 2005; Morita et al. 2005). TNF-α is a key mediator of VAE. Production of IL-1β is induced by TNF-α, which shares many of its activities, including its ability to induce IL-6 production (Clark et al. 2010). Some studies have reported that IL-6 protects neurons against various types of damage (Ali et al. 2000; Yamashita et al. 2005). It has been hypothesized that the pathophysiology of the cytokine storm is attributable to the excessive production of proinflammatory cytokines that induce vascular endothelial cell dysfunction, resulting in the deterioration of vascular permeability.
Human brain microvascular endothelial cells (HBMECs), possessing well-developed tight junctions (TJs), play a critical role in BBB function; however, the surrounding components, such as pericytes, astrocytic endfeet, and basement membranes, comprising various extracellular matrix proteins, also contribute to the BBB’s establishment (Almutari et al. 2016). Materials cross endothelial cellular sheets via the transcellular and paracellular pathways (Tsukita et al. 2001). TJs are the most apical type of cell–cell contact in the lateral membrane between adjacent cells, appearing as a set of continuous, anastomosing intramembranous particle strands that are responsible for the control of paracellular pathways. TJ strands include proteins, such as occludin, claudin, and junctional adhesion molecules (Furuse et al. 1993, 1998; Tsukita et al. 2001). Claudins are major constituents of TJs and can form strands in the absence of other TJ proteins (Furuse et al. 1998). Mammals express 24 claudins, and tissue-specific combinations of claudins result in tissue-specific barrier characteristics (Krause et al. 2008). Hewitt et al (2006) demonstrated that the brain has a distinct claudin expression profile, with high expression of claudin-2 and -5. Claudin-5, -3, and -12 are localized at the BBB (Zlokovic 2008; Neuhaus et al. 2008). Multiple claudins can interact in both a homotypic and heterotypic cis- and trans-fashion to form TJ strands (Tsukita et al. 2001; Hewitt et al. 2006; Krause et al. 2008). Among the members of the claudin family, claudin-5 is indispensable for BBB function (Morita et al. 1999; Nitta et al. 2003), while the significance of claudin-3 and claudin-12 expression at the BBB TJs remains unclear. Recently, the absence of claudin-3 expression at the BBB endothelium TJs was confirmed in a study by Castro et al. (2019a). Furthermore, the expression of claudin-12 at the BBB has been reported, but this was determined to not be essential for the establishment or maintenance of the BBB TJ integrity (Castro et al. 2019b).
Experimentally, the TJ barrier function has been assessed by measuring the transepithelial or transendothelial electrical resistance (TER) and evaluating the ability of TJs to restrict the passage of small molecules, such as dextran, through the paracellular space. Previously, we demonstrated a dynamic change in vascular permeability induced by TNF-α using the TER measurement and a permeability assay of human umbilical vein endothelial cells (HUVECs) (Miyazaki et al. 2017). However, HUVECs are not brain-derived. Therefore, it is important to use a BBB model to develop an in vitro model of the VAE pathophysiology. This study aimed to elucidate the dynamics of vascular dysfunction caused by TNF-α in an in vitro BBB model using HBMECs and pericytes.
Materials and methods
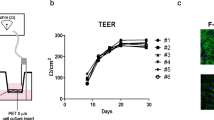
The procedures using the in vitro VAE model are demonstrated in Fig. 1.

Schematic drawing of the procedure using the in vitro VAE model. Human brain vascular pericytes were seeded and cultured on the bottom side of the 24-well Transwell® inserts. The following day, HBMECs were seeded and cultured on the inside of the inserts. Under these conditions, the BBB models were established. TER measurement was started 4 days after seeding the HBMECs. After the TER became stable, serially diluted TNF-α was added to the medium in both the upper and lower compartments of the wells. Under these conditions, the VAE models were established. Each experiment was performed using the VAE model
Cell culture
HBMECs (Cell Systems, Kirkland, WA, USA) were cultured on rat-tail type I collagen-coated (5 μg/cm2; CULTREX®, Trevigen, Inc, Gaithersburg, MD, USA) CELLSTAR® Cell Culture Flasks (Greiner Bio-One GmbH, Frickenhausen, Germany) in Endothelial Cell Growth Medium MV with an Endothelial Cell Growth Medium MV2 kit (PromoCell GmbH, Heidelberg, Germany), containing 5 ng/mL epidermal growth factor, 10 ng/mL basic fibroblast growth factor, 20 ng/mL insulin-like growth factor, 0.5 ng/mL vascular endothelial growth factor, 1 μg/mL ascorbic acid, 0.2 μg/mL hydrocortisone, 5% fetal bovine serum (FBS), and penicillin–streptomycin (100 units/mL and 100 μg/mL, respectively; Thermo Fisher Scientific, Inc, San Jose, CA, USA) at 37 °C under humidified 5% CO2.
Human brain vascular pericytes (ScienCell Research Laboratories, Inc., San Diego, CA, USA) were cultured on poly-L-lysine-coated (2 μg/cm2; ScienCell Research Laboratories, Inc.) CELLSTAR® Cell Culture Flasks in pericyte medium (ScienCell Research Laboratories, Inc.) containing pericyte growth supplement, penicillin–streptomycin solution (100 units/mL and 100 μg/mL, respectively), and 2% FBS at 37 °C under humidified 5% CO2. At around 70% confluency the cells were trypsinized and stored at − 80 °C until use. These cells were used at passage 3–10.
Co-culture preparation
The co-culture models were prepared as reported previously (Nakagawa et al. 2007, 2009). To culture cells on the underside of a 24-well Transwell® insert (pore size, 0.4 μm; cell growth area, 0.33 cm2; Corning, Inc, NY, USA), the insert was placed inverted inside the chamber of a Falcon® 12-well Clear Multiwell Plate (Corning, Inc, NY, USA). Human brain vascular pericytes were seeded at 5 × 104 cells/cm2 on the bottom side of the poly-L-lysine-coated (2 μg/cm2) inserts and cultured in pericyte medium containing pericyte growth supplement, penicillin–streptomycin solution (100 units/mL and 100 μg/mL, respectively), and 2% FBS at 37 °C under humidified 5% CO2. To prevent evaporation, a 12-well plate cover was placed against the top of the inserts. The cells were allowed to adhere for 1 h prior to re-inversion and then allowed to adhere firmly overnight. The next day, HBMECs were seeded (1 × 105 cells/cm2) inside type I collagen-coated (5 μg/cm2) membranes on 24-well Transwell® inserts and cultured in Endothelial Cell Growth Medium MV with an Endothelial Cell Growth Medium MV2 kit containing 5% FBS and penicillin–streptomycin (100 units/mL and 100 μg/mL, respectively) at 37 °C under humidified 5% CO2. The medium was replaced the following day and every 2 days thereafter until the cells were confluent. Under these conditions, the in vitro BBB models were established.
Lyophilized recombinant human TNF-α (R&D Systems, Minneapolis, MN, USA) was reconstituted to 100 μg/mL using 0.1% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) and stored at − 20 °C until use. The specific activity of recombinant human TNF-α is approximately 7.6 × 104 IU/μg (7.6 × 102 IU/mL considering the TNF-α concentration as 10 ng/mL). Working solutions of TNF-α were prepared by dilution in the culture medium.
TER measurement
HBMECs and human brain vascular pericytes were cultured on 24-well Transwell® inserts, as mentioned previously, and the co-culture models were established. The medium was replaced the next day after seeding the HBMECs, and every two days thereafter until the cells were confluent. These inserts were set in the cellZscope® (nanoAnalytics GmbH, Münster, Germany), and TER measurement was started 4 days after seeding the HBMECs. After the TER became stable, serially diluted TNF-α was added to the medium in both the upper and lower compartments of the wells at final concentrations of 0 (control), 0.1, 1, or 10 ng/mL, respectively. We used the wells without added TNF-α as the controls to prevent the potential effects of cell metabolites on the barrier over the total experimental time course. Under these conditions, the in vitro VAE models were established. The TER level was automatically measured over time after HBMEC monolayer treatment at each concentration. TER levels correlate with the strength of TJs between adjacent cells; high and low TER values indicate low and high TJ permeability, respectively.
Permeability assay
HBMECs and human brain vascular pericytes were cultured in 24-well Transwell® inserts, as mentioned previously, and the co-culture models were established. The medium was replaced the next day after seeding the HBMECs, and every two days thereafter until the cells were confluent. At specific time points (12, 24, and 48 h) after TNF-α treatment, the media containing 50 μg/mL sodium-fluorescein isothiocyanate (FITC) (average MW: 376 Da; Sigma-Aldrich, St. Louis, MO, USA) and 100 μg/mL FITC-conjugated dextran (average MW: 3–5, 70, and 250 kDa; Sigma-Aldrich), were, respectively, loaded into each of the upper compartments of the inserts, for each molecular size. One hour after adding sodium-FITC and FITC-conjugated dextran, samples (0.1 mL) were removed from the lower compartment. The inserts were not re-used after the samples were removed. The upper and lower compartments represent the intravascular and extravascular spaces, respectively; thus, this model represents solute permeation from the intravascular to the extravascular space. One hour after adding sodium-FITC or FITC-conjugated dextran, the fluorescein intensity of the medium in the lower compartments was measured using a fluorescent microplate reader Fluoroskan® (Thermo Fisher Scientific, Inc.) at excitation and emission wavelengths of 485 nm and 538 nm, respectively. Sodium-FITC and FITC-conjugated dextran concentrations were calculated with the use of a standard curve. The ratio to control these concentrations in the lower compartment was used as the index for functional permeability.
Immunofluorescence staining
The HBMECs were fixed in ice-cold acetone for 15 min at − 20 °C, followed by permeabilization with ice-cold methanol for 20 min at − 20 °C. After washing with PBS, the cells were blocked in PBS with 3% BSA and 0.3% Triton-X 100 for 1 h at room temperature and subsequently incubated overnight at 4 °C with anti-claudin-5 mouse monoclonal antibody (mAb) (1:200, cat. no. sc-374221; Santa Cruz Biotechnology, Inc, Dallas, TX, USA) and anti-ZO-1 mouse mAb (1:200, cat. no. 33–9100; Thermo Fisher Scientific, Inc, Waltham, MA, USA) for TJ staining, as primary antibodies. Subsequently, the cells were incubated with secondary antibodies that included a donkey Cy3 AffiniPure F(ab’)2 Frag Donkey Anti-Mouse IgG (1:2,000, cat. no. 111673; Jackson Immuno Research Laboratories, Inc, West Grove, PA, USA) and an Alexa Fluor 488 Goat Anti-Rabbit IgG (1:2,000, cat. no. A-11034; Molecular Probes Inc, Eugene, OR, USA) at room temperature for 1 h. Both the primary and secondary antibodies were diluted in 1% BSA and 0.3% Triton-X 100. After washing the cells, the membranes were covered with a coverslip and mounted with ProLong Gold Antifade Reagent containing 4,6-diamidino-2-phenylindole (DAPI) (Thermo Fisher Scientific, Inc.) to detect nuclei. Images were captured using a confocal laser microscope (FV1000; Olympus, Tokyo, Japan). For confocal Z-stacks, images were collected over a depth of 10 μm with an interval of 0.5 μm between each stack. The stacks were subsequently projected into one image.
Western blot analysis
At specific time points (12, 24, and 48 h) after TNF-α treatment, the HBMECs were lysed in 100 μL RIPA Lysis Buffer (Santa Cruz Biotechnology, Inc.). Cells were scraped into the solution and centrifuged at 14,000 rpm for 5 min. The supernatants were mixed with 4 × Laemmli buffer (Bio-Rad Laboratories, Inc, Hercules, CA, USA) supplemented with 10% β-Mercaptoethanol (Santa Cruz Biotechnology, Inc.) and heated for 5 min at 95 °C. Protein concentration was determined using a Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, Inc.). Equivalent amounts of protein (30 μg) for each sample were resolved in 12% sodium dodecyl sulfate (SDS)-polyacrylamide gels for electrophoresis and run at 200 V for 30 min. After electrophoresis, the gels were overlaid with a polyvinylidene difluoride (PVDF) membrane (Trans-Blot® Turbo™ Mini PVDF Transfer Packs, Bio-Rad Laboratories, Inc.) under a constant voltage of 25 V for 3 min. The PVDF membrane was then blocked with 5% skimmed milk (Nacalai Tesque, Inc, Kyoto, Japan) for 1 h at room temperature and incubated overnight with the primary anti-claudin-5 rabbit Ab (1:1,000, cat. no. C0145; Assay Biotechnology Company, Inc, Sunnyvale, CA, USA) and anti-β-actin mouse mAb (1:10,000, cat. no. A 5316; Sigma-Aldrich) at 4 °C. Horseradish peroxidase-conjugated secondary anti-rabbit IgG (cat. no. 7074; Cell Signaling Technology, Tokyo, Japan) and anti-mouse IgG (cat. no. 7076; Cell Signaling Technology, Tokyo, Japan) were diluted in 5% skimmed milk at a dilution of 1:5000. Protein expression levels were determined using enhanced chemifluorescence (ECL Select Western Blotting Detection Reagent, GE Healthcare, Buckinghamshire, UK) and laser scanner Light Capture II (ATTO Corporation, Tokyo, Japan) for exposure. Western blot band intensity was determined using ImageJ software, version 1.50i (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Data are reported as mean ± standard error (SE). All possible pairwise comparisons among the groups were conducted using a one-way analysis of variance followed by the Tukey–Kramer post hoc test. P values < 0.05 were considered statistically significant. Analyses were performed using BellCurve for Excel (Social Survey Research Information Co. Ltd, Tokyo, Japan) and GraphPad Prism 8 software.
Results
TER was decreased after TNF-α treatment and recovery times were dependent on TNF-α concentration
Sequential TER was measured in the BBB model to evaluate dynamic changes in the TJ barrier function after TNF-α treatment. Sequential TER was measured for 3 days after TNF-α treatment (Fig. 2a). TER values decreased and reached the minimum level ~ 10 h after TNF-α treatment, at all TNF-α concentrations (Fig. 2a, b). Thereafter, TER values at all TNF-α concentrations tended to recover. TER values at TNF-α concentrations of 0.1 and 1 ng/mL recovered to the control level at approximately 30 and 40 h after treatment, respectively; those at a concentration of 10 ng/mL recovered almost to the control level at ~ 70 h after treatment (Fig. 2c). The decreased TER values at TNF-α concentrations of 0.1 and 1 ng/mL had increased beyond control levels (Fig. 2a).

Change in TER in an in vitro BBB model after TNF-α treatment. a Mean TER percentage (%) changes over time after TNF-α treatment. Each line shows the mean TER % (n = 5) of each concentration (control; TNF-α at 0.1, 1, and 10 ng/mL); error bars indicate SE. The vertical axis shows the mean TER %, and the horizontal axis shows the time after TNF-α treatment (h). The time of addition is time zero. The mean TER of the no-cell-cultured inserts was 7.19 ± 0.20 Ω cm2 (n = 5; mean ± SE). Initial TER values (17.29 ± 0.13 Ω cm2; n = 5; mean ± SE) at time 0 were regarded as the 100% value. TER % is the ratio of the TER values to time 0. The graph is representative of three independent experiments. b The minimum level of TER % at 10 h after TNF-α treatment. Bars show the mean minimum TER % (n = 5) of each concentration (control; TNF-α at 0.1, 1, and 10 ng/mL); error bars indicate SE. The vertical axis shows the mean TER % at the minimum level, and the horizontal axis shows the TNF-α concentration (ng/mL). c Recovery time to control TER %. Bars show the time of recovery to control TER % from time zero (n = 5) of TNF-α concentrations at 0.1, 1, and 10 ng/mL; error bars indicate SE. The vertical axis shows the mean recovery time (h) and the horizontal axis shows the TNF-α concentration (ng/mL). Statistical significance was determined using a one-way analysis of variance followed by the Tukey–Kramer post hoc test. **p < 0.01. BBB blood–brain barrier, TER transendothelial resistance, TNF-α tumor necrosis factor, SE standard error
Paracellular permeability increased after TNF-α treatment and recovered to baseline levels in a TNF-α concentration-dependent manner
A permeability assay using FITC-conjugated sodium and FITC-conjugated dextran of different molecular weights was performed at specific time points after TNF-α treatment to evaluate the solute permeability of the BBB model over time. Paracellular permeability increased for all molecular sizes after TNF-α treatment, and the recovery time to baseline level was delayed in a TNF-α concentration-dependent manner (Fig. 3a–d). We also observed size-selectivity in paracellular permeability after TNF-α treatment. Although the permeability of all molecular sizes, 12 and 24 h after treatment with a TNF-α concentration of 10 ng/mL, was significantly increased, the permeability at 48 h for 70 and 250 kDa recovered to the control level, but not for 376 Da and 3–5 kDa (Fig. 3a-d). The paracellular permeability showed a similar pattern to that of sequential TER.

Paracellular permeability for FITC-conjugated sodium and dextran in an in vitro BBB model after TNF-α treatment. Each bar indicates the mean ratio of fluorescein intensity (n = 6): a FITC-conjugated sodium (average MW: 376 Da), b FITC-conjugated dextran (average MW: 3–5 kDa), c FITC-conjugated dextran (average MW: 70 kDa), and d FITC-conjugated dextran (average MW: 250 kDa). Error bars indicate standard error. The vertical axis shows the ratio to control of the FITC-conjugated sodium or dextran concentrations in the lower compartment and the horizontal axis shows TNF-α concentration (ng/mL). The mean FITC-conjugated sodium or dextran concentrations for no-cell-cultured inserts in the lower chamber were 4.97 ± 0.91, 27.0 ± 1.9, 18.9 ± 1.5, and 33.8 ± 4.1 μg/mL for MW 376 Da, 3–5 kDa, 70 kDa, and 250 kDa, respectively. The control concentrations were as follows: 376 Da, 0.92 ± 0.05, 0.90 ± 0.04, and 0.96 ± 0.06 μg/mL; 3–5 kDa, 1.10 ± 0.01, 0.96 ± 0.02, and 1.08 ± 0.02 μg/mL; 70 kDa, 0.28 ± 0.00, 0.29 ± 0.00, and 0.29 ± 0.01 μg/mL; 250 kDa, 0.49 ± 0.00, 0.49 ± 0.01, and 0.49 ± 0.01 μg/mL at 12, 24, and 48 after TNF-α treatment, respectively. White, gray, and black bars indicate 12, 24, and 48 h after TNF-α treatment, respectively. All possible pairwise comparisons among the groups were conducted using a one-way analysis of variance followed by the Tukey–Kramer post hoc test. *p < 0.05, **p < 0.01 vs. control at 12 h. †p < 0.05, †† p< 0.01 vs. control at 24 h. #p < 0.05, ##p < 0.01 vs. control at 48 h. The figure is representative of three independent experiments. BBB, blood–brain barrier; FITC, sodium–fluorescein isothiocyanate; MW, molecular weight; TNF-α, tumor necrosis factor
TNF-α treatment induced the delocalization of claudin-5, but not ZO-1, which recovered in a dose-dependent manner
Immunofluorescence staining of claudin-5 and ZO-1 was performed using the insert membrane in the BBB model to evaluate the changes in localization after TNF-α treatment. Based on confocal laser microscopy images, the delocalization of claudin-5 was observed 12 h after TNF-α treatment at all TNF-α concentrations (Fig. 4a). The delocalization of claudin-5 treated with TNF-α at 0.1 and 1 ng/mL recovered at 24 and 48 h after treatment, respectively, and that with TNF-α at 10 ng/mL partially recovered at 48 h after treatment (Fig. 4a). The localization of ZO-1 remained unchanged at all time points after TNF-α treatment at all concentrations (Fig. 4b).

Confocal laser microscope images of immunofluorescent staining of TJ proteins, claudin-5 and ZO-1, in HBMECs after TNF-α treatment. The expression of (a) claudin-5 and (b) ZO-1 after TNF-α treatment at 0.1, 1, and 10 ng/mL at specific time points (12, 24, and 48 h). Claudin-5 (a) and ZO-1 (b) show continuous cell membrane staining at the cell–cell borders (red). Nuclei were counterstained with DAPI (blue). Scale bar (white) indicates 20 μm. DAPI 4,6-diamidino-2-phenylindole, TJ tight junction, TNF-α tumor necrosis factor, HBMEC human brain microvascular endothelial cell
The results of the delocalization of claudin-5 showed a similar pattern to those of TER and the permeability assay.
Claudin-5/β-actin protein expression decreased at a TNF-α concentration of 1 and 10 ng/mL but recovered
Western blot analysis was performed to evaluate the change in the expression of claudin-5 protein, as decreased claudin-5 protein expression is associated with vascular permeability. The western blot band for claudin-5 remained unchanged 12 h after TNF-α treatment at all concentrations. Claudin-5 decreased 24 h after TNF-α treatment at concentrations of 1 and 10 ng/mL, and completely recovered 48 h after TNF-α treatment (Fig. 5a, b). Results of the expression of claudin-5 showed a different pattern to those of TER, the permeability assay, and the delocalization of claudin-5.

Western blot analysis of the TJ protein claudin-5 in cell lysates from HBMECs after TNF-α treatment. a Western blot images of claudin-5 and β-actin in HBMECs after TNF-α treatment. The western blot images of HBMECs treated for 12, 24, and 48 h with different concentrations of TNF-α (n = 2) are shown. Protein lysates of HBMECs were dispensed to duplicate lanes and separated by electrophoresis and blotted; claudin-5 was detected with specific antibodies. The protein level of β-actin served as a loading control. This figure is representative of two independent experiments. b The western blot intensity ratio of claudin-5/β-actin 12, 24, and 48 h after TNF-α. Each bar shows the intensity ratio of claudin-5 to β-actin (n = 4) compared with the control; error bars indicate standard error. The vertical axis shows the intensity ratio of claudin-5 to β-actin compared with the control, and the horizontal axis shows TNF-α concentration (ng/mL). Each figure indicates 12, 24, and 48 h after TNF-α treatment, respectively. All possible pairwise comparisons among the groups were conducted using a one-way analysis of variance followed by the Tukey–Kramer post hoc test. ††p < 0.01 vs. control at 1 ng/mL of TNF-α concentration. ##p < 0.01 vs. control at 10 ng/mL of TNF-α concentration. TJ tight junction, HBMEC human brain microvascular endothelial cell, TNF-α tumor necrosis factor
Discussion
In the present study, we evaluated the vascular endothelial cell permeability in VAE using an in vitro BBB model with HBMECs and human brain vascular pericytes and investigated the pathophysiology of VAE by evaluating endothelial cell injury. We showed that paracellular tightness decreased after TNF-α treatment and recovered in a TNF-α concentration-dependent manner. Previously, we reported the pathophysiology of VAE by evaluating TJs using HUVECs as an in vitro endothelial cell injury model (Miyazaki et al. 2017). However, HUVECs are not brain-derived, and the constituents of TJs differs between cells derived from the brain and those derived from other organs. Moreover, HUVECs do not possess the specific barrier properties found in highly impermeable microvascular beds, such as the BBB. HBMECs, especially in in vitro BBB models, are thought to strengthen BBB function.
In the present study, we demonstrated a dynamic change in vascular permeability induced by TNF-α using TER measurements and a permeability assay. TNF-α acts directly on brain microvascular endothelial cells and has been shown to directly impair TJ function in various endothelial cell lines (Mark and Miller 1999; Capaldo and Nusrat 2009). TNF-α perturbs endothelial barrier function by influencing the structure and function of TJs, thereby enhancing paracellular permeability. Nuclear factor-κB (NF-κB) activated by TNF-α results in decreased claudin-5 expression in brain endothelial cells by which the NF-κB subunit p65 inactivates claudin-5 promoter activity (Aslam et al. 2012). Moreover, Aslam et al. (2012) reported that a time dependent decrease in claudin-5 mRNA was observed in TNF-α-treated brain endothelial cells over a 24 h period, and that other cytokines did not reduce claudin-5 expression. TNF-α/NF-κB-dependent reduction in claudin-5 promoter activity could be a pathologically relevant molecular mechanism underlying defective microvascular permeability. Ma et al. (2004, 2005) proposed that TNF-α-induced NF-κB activation results in alteration of TJ protein localization and expression, and the functional opening of the TJ barrier by contraction of perijunctional actin-myosin filaments via activation of the myosin light chain kinase pathway. Thus, it is speculated that TNF-α produced in response to viral infections causes increased permeability due to BBB disruption by altering TJ protein expression and localization, although the regulation of these processes is not completely understood. The increases in permeability after TNF-α treatment appear to occur via the paracellular pathway. The main evidence supporting this is the observation that the effects of TNF-α were dependent on the size of the permeability marker used (Ma et al. 2004, 2005). Deli et al (1995) reported that a significant increase in the permeability of bovine brain capillary endothelial cell monolayers for both the 342 Da sucrose and 5 kDa insulin was induced 16 h after a 1-h challenge with TNF-α. Anda et al (1997) reported that the incubation of bovine brain microvascular endothelial cell monolayers with TNF-α at 50 and 500 U/mL led to a significant increase in the passage of small molecules, such as the 300 Da cisplatin, and that the passage of high molecular weight 50 kDa FITC-dextran was not influenced by TNF-α at the same concentrations. In the present study, we observed size-selectivity in paracellular permeability after TNF-α treatment in an in vitro BBB model with HBMECs and pericytes. In our experiments, FITC-dextran was added only to the upper chamber compartment, which may have resulted in an increased FITC concentration in the lower compartment due to an osmotic gradient. In addition, the free FITC in the FITC-dextran reagent may also have affected this osmotic gradient. Although the osmotic pressure gradient was not evaluated between compartments, the fact that the FITC concentration increased in a TNF-α concentration-dependent manner in the lower layer led us to conclude that TNF-α affects cell permeability. Moreover, clinical studies have reported TNF-α levels < 15–543 pg/mL in the serum of patients with VAE (Ichiyama et al. 1998; Kawada et al. 2003; Morita et al. 2005). Owing to the dilution effect from plasma, the amount of TNF-α at the localized site of inflammation or injury may actually be greater than the measured concentration.
In the present study, immunofluorescent staining revealed that claudin-5 was delocalized 12 h after treatment with TNF-α at 10 ng/mL and partially recovered 48 h after treatment. Western blot analysis revealed that claudin-5 expression decreased 24 h after treatment and fully recovered 48 h after treatment at the same concentration. TER partly recovered 48 h after treatment with TNF-α at 10 ng/mL, suggesting that paracellular permeability was mainly related to the localization of claudin-5. In contrast, the localization of ZO-1 remained unchanged after TNF-α treatment, suggesting that ZO-1 may have no direct relationship with paracellular permeability Nitta et al (2003) reported that claudin-5-deficient mice exhibit a size-selective loosening of the BBB against small molecules < 800 Da and die within 10 h after birth. TNF-α induces delocalization and decreased expression of claudin-5, resulting in vascular hyperpermeability (McKenzie and Ridley 2007; Capaldo and Nusrat 2009). Our results demonstrate that TNF-α induces delocalization and loss of claudin-5 expression, suggesting that it induces BBB dysfunction via TJ remodeling. Although western blotting results are indicative of whole-cell protein expression and immunofluorescent staining results of protein localization, the results of the two experiments did not agree with each other. Therefore, paracellular permeability might be related to the localization of claudin-5 as the delocalization of claudin-5 exhibits a similar pattern to that of the TER and the permeability assay.
There are some limitations to the present study. First, we used primary HBMECs derived from surgical material. It is unclear whether these cells can be regarded as normal tissue as the patient background is unknown. If the structure and tightness of TJs varies based on tissue or species, it is important to use HBMECs to develop an in vitro model of the VAE pathophysiology. Therefore, in this study, the use of HBMECs was required for investigating VAE using the BBB model and tissue-specific TJ proteins. Second, a co-culture model of HBMECs and human brain vascular pericytes were used in the BBB model. However, the BBB is not only composed of vascular endothelial cells and pericytes but also neighboring cells. Astrocytes induce the development of TJs in cerebral endothelial cells, thereby increasing paracellular barrier properties (Megard et al. 2002). Pericytes are in close contact with endothelial cells; therefore, a co-culture of endothelial cells with pericytes is a recommended in vitro model for the construction of an in vitro BBB model. Products from pericytes, such as the transforming growth factor-β, angiopoietin-1, and the extracellular matrix induce and up-regulate BBB functions (Hori et al. 2004; Dohgu et al. 2005; Hartmann et al. 2007). Nakagawa et al (2007) reported that brain microvessel pericytes strengthen the BBB function of endothelial cells and that in vitro co-culture models of brain microvascular endothelial cells and pericytes are more effective than those of astrocytes. Third, we assessed the TJ barrier function using TER measurements and a permeability assay. However, cellular edema, which is ascribed to the dysfunction of channels on the cell membrane, also affects brain edema. Therefore, in vitro BBB models may lack important features of cell–cell interactions, such as induction or signaling. Nevertheless, although our model might not be a complete in vitro VAE model, given its properties, we believe that it is appropriate for the purpose of this study. Fourth, the concentration-dependent effects of TNF-α are unclear as we did not assess the concentration-dependent influence on TNF-α target activation in our model. The concentration of TNF-α required to achieve acute breakdown may not be important as TNF-α is rapidly degraded and the TNF-α targets are deactivated. Finally, only TNF-α was used to induce hyperpermeability in HBMECs. However, other cytokines such as IL-6 and IL-1β, as well as various other factors may also affect vascular permeability (de Vries et al. 1996). Nevertheless, our system using the in vitro BBB model is thought to closely resemble the pathology underpinning the vascular hyperpermeability induced by TNF-α. Moreover, this system might be useful for elucidating the pathophysiology in other diseases associated with TNF-α in addition to VAE.
Conclusions
We evaluated the vascular endothelial cell permeability in VAE using an in vitro BBB model with HBMECs and human brain vascular pericytes treated with TNF-α. This system for evaluating VAE may be useful for investigating novel therapeutic strategies for VAE and other specific treatments that focus on vascular permeability. This model may also be useful for evaluating other diseases in addition to VAE, where TNF-α-induced breakdown occurs.
Code availability
Not applicable.
References
Ali C, Nicole O, Docagne F, Lesne S, MacKenzie ET, Nouvelot A, Buisson A, Vivien D (2000) Ischemia-induced interleukin-6 as a potential endogenous neuroprotective cytokine against NMDA receptor-mediated excitotoxicity in the brain. J Cereb Blood Flow Metab 20:956–966
Almutari MM, Gong C, Xu YG, Chang Y, Shi H (2016) Factors controlling permeability of the blood-brain barrier. Cell Mol Life Sci 73:57–77
Anda T, Yamashita H, Khalid H, Tsutsumi K, Fujita H, Tokunaga Y, Shibata S (1997) Effect of tumor necrosis factor-alpha on the permeability of bovine brain microvessel endothelial cell monolayers. Neuro Res 19:369–376
Asano Y, Yoshikawa T, Kajita Y, Ogura R, Suga S, Yazaki T, Nakashima T, Yamada A, Kurata T (1992) Fatal encephalitis/encephalopathy in primary human herpesvirus-6 infection. Arch Dis Child 67:1484–1485
Aslam M, Ahmad N, Srivastava R, Hemmer B (2012) TNF-alpha induced NFκB signaling and p65 (RelA) overexpression repress Cld5 promoter in mouse brain endothelial cells. Cytokine 57:269–275
Capaldo CT, Nusrat A (2009) Cytokine regulation of tight junctions. Biochem Biophys Acta 1788:864–871
Castro Dias M, Coisne C, Lazarevic I, Baden P, Hata M, Iwamoto N, Francisco DMF, Vanlandewijck M, He L, Baier FA, Stroka D, Bruggmann R, Lyck R, Enzmann G, Deutsch U, Betsholtz C, Furuse M, Tsukita S, Engelhardt B (2019) Claudin-3-deficient C57BL/6J mice display intact brain barriers. Sci Rep 9:203
Castro Dias M, Coisne C, Baden P, Enzmann G, Garrett L, Becker L, Hölter SM; German Mouse Clinic Consortium, Hrabě de Angelis M, Deutsch U, Engelhardt B (2019) Claudin-12 is not required for blood-brain barrier tight junction function. Fluids Barriers CNS 16:30
Clark IA, Alleva LM, Vissel B (2010) The roles of TNF in brain dysfunction and disease. Pharmacol Ther 128:519–548
de Vries HE, Blom-Roosemalen MC, van Oosten M, de Boer AG, van Berkel TJ, Breimer DD, Kuiper J (1996) The influence of cytokines on the integrity of the blood-brain barrier in vitro. J Neuroimmunol 64:37–43
Deli MA, Descamps L, Dehouck MP, Cecchelli R, Joó F, Abrahám CS, Torpier G (1995) Exposure of tumor necrosis factor-alpha to luminal membrane of bovine brain capillary endothelial cells cocultured with astrocytes induced a delayed increase of permeability and cytoplasmic stress fiber formation of actin. J Neurosci Res 41:717–726
Dohgu S, Takata F, Yamauchi A, Nakagawa S, Egawa T, Naito M, Tsuruo T, Sawada Y, Niwa M, Kataoka Y (2005) Brain pericytes contribute to the induction and up-regulation of blood-brain barrier functions through transforming growth factor-beta production. Brain Res 1038:208–215
Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, Tsukita S (1993) Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol 123:1777–1788
Furuse M, Sasaki H, Fujimoto K, Tsukita S (1998) A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occluding in fibroblasts. J Cell Biol 143:391–401
Hartmann C, Zozulya A, Wegener J, Galla HJ (2007) The impact of glia-derived extracellular matrices on the barrier function of cerebral endothelial cells: an in vitro study. Exp Cell Res 313:1318–1325
Hewitt KJ, Agarwal R, Morin PJ (2006) The claudin gene family: expression in normal and neoplastic tissues. BMC Cancer 6:186
Hori S, Ohtsuki S, Hosoya K, Nakashima E, Terasaki T (2004) A pericyte-derived angiopoietin-1 multimeric complex induces occluding gene expression in brain capillary endothelial cells through Tie-2 activation in vitro. J Neurochem 89:503–513
Hosoya M, Nunoi H, Aoyama M, Kawasaki Y, Suzuki H (2005) Cytochrome c and tumor necrosis factor-alpha values in serum and cerebrospinal fluid of patients with influenza-associated encephalopathy. Pediatr Infect Dis J 24:467–470
Ichiyama T, Nishikawa M, Yoshitomi T, Hayashi T, Furukawa S (1998) Tumor necrosis factor-alpha, interleukin-1 beta, and interleukin-6 in cerebrospinal from children with prolonged febrile seizures. Comparison with acute encephalitis/encephalopathy. Neurology 50:407–411
Karampatsas K, Osborne L, Seah ML, Tong CYW, Prendergast AJ (2018) Clinical characteristics and complications of rotavirus gastroenteritis in children in east London: A retrospective case-control study. PLoS ONE 13:e0194009
Kasai T, Togashi T, Morishima T (2000) Encephalopathy associated with influenza epidemics. Lancet 355:1558–1559
Kawada J, Kimura H, Ito Y, Hara S, Iriyama M, Yoshikawa T, Morishima T (2003) Systemic cytokine responses in patients with influenza-associated encephalopathy. J Infect Dis 188:690–698
Krause G, Winkler L, Mueller SL, Haseloff RF, Piontek J, Blasig IE (2008) Structure and function of claudins. Biochim Biophys Acta 1778:631–645
Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, Said HM (2004) TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am J Physiol Gastrointest Liver Physiol 286:G367–G376
Ma TY, Boivin MA, Ye D, Pedram A, Said HM (2005) Mechanism of TNF-{alpha} modulation of Caco-2 intestinal epithelial tight junction barrier: role of myosin light-chain kinase protein expression. Am J Physiol Gastrointest Liver Physiol 288:G422–G430
Mark KS, Miller DW (1999) Increased permeability of primary cultured brain microvessel endothelial cell monolayers following TNF-alpha exposure. Life Sci 64:1941–1953
McKenzie JA, Ridley AJ (2007) Roles of Rho/Rock and MLCK in TNF-alpha-induced changes in endothelial morphology and permeability. J Cell Physiol 213:221–228
Megard I, Garrigues A, Orloski S, Jorajuria S, Clayette P, Ezan E, Mabondzo A (2002) A co-culture-based model of human blood-brain barrier: application to active transport of indinavir and in vivo-in vitro correlation. Brain Res 927:153–167
Miyazaki K, Hashimoto K, Sato M, Watanabe M, Tomikawa N, Kanno S, Kawasaki Y, Momoi N, Hosoya M (2017) Establishment of a method for evaluating endothelial cell injury by TNF-α in vitro for clarifying the pathophysiology of virus-associated acute encephalopathy. Pediatr Res 81:942–947
Mizuguchi M (1997) Acute necrotizing encephalopathy of childhood: a novel form of acute encephalopathy prevalent in Japan and Taiwan. Brain Dev 19:81–92
Mizuguchi M, Yamanouchi H, Ichiyama T, Shiomi M (2007) Acute encephalopathy associated with influenza and other viral infections. Acta Neurol Scand 115:45–56
Morita K, Sasaki H, Furuse M, Tsukita S (1999) Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol 147:185–194
Morita H, Hosoya M, Kato A, Kawasaki Y, Suzuki H (2005) Laboratory characteristics of acute encephalopathy with multiple organ dysfunctions. Brain Dev 27:477–482
Nakagawa S, Deli MA, Nakao S, Honda M, Hayashi K, Nakaoke R, Kataoka Y, Niwa M (2007) Pericytes from brain microvessels strengthen the barrier integrity in primary cultures of rat brain endothelial cells. Cell Mol Neurobiol 27:687–694
Nakagawa S, Deli MA, Kawaguchi H, Shimizudani T, Shimono T, Kittel A, Tanaka K, Niwa M (2009) A new blood-brain model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochem Int 54:253–263
Neuhaus W, Wirth M, Plattner VE, Germann B, Gabor F, Noe CR (2008) Expression of claudin-1, claudin-3 and claudin-5 in human blood-brain mimicking cell line ECV304 is inducible by glioma-conditioned media. Neurosci Lett 446:59–64
Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M, Tsukita S (2003) Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol 161:653–660
Tsukita S, Furuse M, Itoh M (2001) Multifunctional strands in tight junctions. Nat Rev Cell Biol 2:285–293
Yamashita T, Sawamoto K, Suzuki S, Suzuki N, Adachi K, Kawase T, Mihara M, Ohsugi Y, Abe K, Okano H (2005) Blockade of interleukin-6 signaling aggravates ischemic cerebral damage in mice: possible involvement of Stat3 activation in the protection of neurons. J Neurochem 94:459–468
Zlokovic BV (2008) The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57:178–201
Acknowledgements
We would like to thank Naoki Tomikawa for the useful discussion and advice. We are also grateful to the laboratory’s technical staff, Ken Honzumi, Sakurako Norito, and Mieko Tanji, for their excellent technical skills.
Funding
The study was supported by a grant from the Grants-in-Aid for Scientific Research (KAKENHI, Grant No. 17K10120).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Hajime Maeda, Kyohei Miyazaki, Hayato Go, Koichi Hashimoto, and Mitsuaki Hosoya. The first draft of the manuscript was written by Hajime Maeda and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Additional information
Communicated by Sreedharan Sajikumar.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Maeda, H., Hashimoto, K., Go, H. et al. Towards the development of a human in vitro model of the blood–brain barrier for virus-associated acute encephalopathy: assessment of the time- and concentration-dependent effects of TNF-α on paracellular tightness. Exp Brain Res 239, 451–461 (2021). https://doi.org/10.1007/s00221-020-05985-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00221-020-05985-7