
Abstract
Gastrointestinal mesenchymal tumors, as the most common mesenchymal tumors in the gastrointestinal tract, are adjuvantly treated with multi-targeted tyrosine kinase inhibitors, such as imatinib and sunitinib, but there are problems of drug resistance and complex methods of monitoring therapeutic agents. The pathogenesis of this disease is related to mutations in tyrosine kinase (KIT) or platelet-derived growth factor receptor α, an important target for drug therapy. In recent years, the screening of relevant tyrosine kinase inhibitors from traditional Chinese medicine has become a hotspot in antitumor drug research. In the current study, the KIT-SNAP-tag cell membrane chromatography (KIT-SNAP-tag/CMC) column was prepared with satisfying specificity, selectivity, and reproducibility by chemically bonding high KIT expression cell membranes to the silica gel surface using the SNAP-tag technology. The KIT-SNAP-tag/CMC-HPLC-MS two-dimensional coupling system was investigated using the positive drug imatinib, and the results showed that the system was a reliable model for screening potential antitumor compounds from complex systems. This system screened and identified three potential active compounds of evodiamine (EVO), rutaecarpin (RUT), and dehydroevodiamine (DEVO), which possibly target the KIT receptor, from the alcoholic extract of the traditional Chinese medicine Evodia rutaecarpa. Then, the KD values of the interaction of EVO, RUT, and DEVO with KIT receptors measured using nonlinear chromatography were 7.75 (±4.93) × 10−6, 1.42 (±0.71) × 10−6, and 2.34 (±1.86) × 10−6 mol/L, respectively. In addition, the methyl thiazolyl tetrazolium assay validated the active effects of EVO and RUT in inhibiting the proliferation of high KIT-expressing cells in the ranges of 0.1–10 µmol/L and 0.1–50 µmol/L, respectively. In conclusion, the KIT-SNAP-tag/CMC could be a reliable model for screening antitumor components from complex systems.
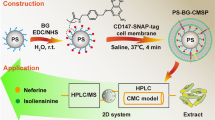
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the gastrointestinal tract, with an incidence of 1.5/100,000 per year and accounting for 1–2% of all gastrointestinal tumors. Most GISTs harbor tyrosine kinase (KIT) or platelet-derived growth factor receptor α (PDGFRA) gene mutations [1]. In particular, KIT mutations are detected in 75–80% of cancer cells from patients with GIST. KIT mutations affect cell proliferation and survival by altering the KIT protein activity through the shortening of the growth factor receptor KIT/CD117 protein on the surface of normal cells. The proto-oncogene KIT encodes a type III receptor tyrosine kinase whose ligand is SCF. SCF–KIT interaction is essential for the development of melanocytes, erythrocytes, germ cells, mast cells, and interstital cells of cajal (ICC) [2, 3].
The currently marketed KIT inhibitors are multitarget inhibitors. As a tyrosine kinase inhibitor (TKI), imatinib mesylate was the first-line targeted therapeutic agent for GIST approved by the U.S. Food and Drug Administration in 2002 and the State Drug Administration of China in 2013. Imatinib has high therapeutic efficacy and reduces cancer recurrence and metastasis. However, the drug still exhibits significant side effects and leads to lower quality of life in advanced patients. In addition, resistance to imatinib is one of the most important clinical challenges faced by patients with GIST [4], thereby increasing the demand for new therapies for advanced disease. The emergence of secondary mutations in KIT is the primary cause of imatinib resistance in ∼90% of KIT-mutant GISTs, and previous tumor tissue-based series revealed mutation clusters in the ATP-binding pocket (ATP-BP; encoded by exons 13 and 14) and the activation loop (A-loop; encoded by exons 17 and 18) [5]. Approved TKIs for treating imatinib-resistant GIST include sunitinib (second line), regorafenib (third line), and ripretinib (fourth line), each producing an inhibitory spectrum against certain secondary KIT mutations; however, these drugs offer only limited benefits [6,7,8,9]. Therefore, the development of novel therapeutic strategies remains essential for improving patient outcomes.
Cell membrane chromatography (CMC) is a new type of bioaffinity chromatography, in which the model uses specific cell membranes bound to silica gel as the chromatographic stationary phase. The technique has a wide range of applications in drug screening, protein interaction analysis, and precision medication monitoring [10]. With the optimization and development of CMC in recent years, cell membranes bind to carriers in three main ways: physical adsorption, chemical bonding, and biological protein tag linkage. However, traditional physically adsorptive CMC columns have a short column life, low screening efficiency, and limited application scenarios. Therefore, two main ideas have been put forth to improve this problem. The first idea involves the use of the protein tag technology for immobilizing proteins to improve the specificity of receptor–ligand binding. The second idea suggests improving the stationary phase carrier, increasing the membrane loading of the stationary phase, and expanding the application scope of cell membrane stationary phase via the nanosizing and surface modification of the stationary phase material. Using the SNAP-tag technology, the current group developed chemically bonded novel biomimetic materials to enhance the specificity of CMC, improve the stability and service life of the materials, and promote the application of CMC for screening lead compounds [11,12,13,14].
In the present study, a CMC stationary phase was prepared by binding the KIT-ST cell membrane to the SiO2-benzyl guanine (BG) surface using the SNAP-tag technology to construct a two-dimensional coupling system of KIT-SNAP-tag/CMC-high-performance liquid chromatography (HPLC)–mass spectrometry (MS). The model was first validated and then used to screen potential KIT receptor-targeting active compounds from the alcoholic extract of the traditional Chinese medicine (TCM) Evodia rutaecarpa. Thereafter, nonlinear chromatography (NLC) was used to investigate the interactions of potential compounds with KIT receptors. In addition, the inhibitory effects of the identified active compounds on cell proliferation were verified using the methyl thiazolyl tetrazolium (MTT) assay.
Materials and methods
Chemicals and reagents
Imatinib (HPLC >99%) was purchased from Shanghai Aladdin Biotech Co., Ltd., evodiamine (EVO, HPLC ≥98%) was bought from Sigma-Aldrich, Co. (product of USA), rutaecarpin (RUT, HPLC >99.5%) was obtained from Chengdu Must Biotech Co., Ltd. (Chengdu, China), and dehydroevodiamine (DEVO, HPLC >99.5%) was bought from Pufei De Biotech Co., Ltd. (Chengdu, China). E. rutaecarpa (produced in Xinjiang) was purchased from Beijing Tongren drug store in Xi’an and acetonitrile was obtained from ThermoFisher Scientific (Waltham, MA, USA). Terbutaline (HPLC >98%) was bought from Shanghai xianding Bio-Technology Co., Ltd. Trihexyphenidyl hydrochloride (HPLC >99%) was obtained from National Institutes for Food and Drug Control; dexamethasone (HPLC >99%) was purchased from Shanghai yuanye Bio-Technology Co., Ltd.
MTT and BCA protein concentration determination kits were purchased from Solarbio Science & Technology Co., Ltd. (Beijing, China). Purified water was prepared using the Mocell 1810b ultra-pure water system (Chongqing, China). All other reagents were at least of analytical grade.
Preparation of standard and sample solutions
E. rutaecarpa powder (0.4 g) was ultrasonicated in 10 mL of 70% methanol for 1 h. After centrifugation, the supernatant was collected and filtered using a 0.22-μm membrane filter before injection analysis. The stock solutions of imatinib, EVO, RUT, and DEVO were prepared for HPLC analysis at the concentration of 1 mg/mL and stored at 4°C in the dark. The reserve solutions of imatinib, EVO, RUT, and DEVO were prepared for in vitro assays at the concentration of 50 mmol/L in DMSO and stored at −20°C in the dark.
Cell lines
Human embryonic kidney 293 (HEK293) cells (Serial: SCSP-5209) were purchased from National Collection of Authenticated Cell Cultures. HEK293 cells overexpressing KIT-SNAP-tag (KIT-SNAP-tag cells, the SNAP-tag was inserted into the C-terminal of the KIT to produce a plasmid containing KIT-SNAP) and negative control HEK293 cells transfected with an empty plasmid (NC-HEK293) were prepared using the genetic engineering technology by Cyagen Biotech Co., Ltd. (Guangzhou, China). KIT expression at the mRNA and protein levels was detected using real-time polymerase chain reaction (qPCR) and western blot, respectively. The KIT-SNAP-tag cell and the NC-HEK293 cell lines were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum and 100 U/mL penicillin–streptomycin (1:1) with 0.5 µg/mL puromycin.
Preparation of KIT-SNAP-tag cell membrane stationary phase
KIT-SNAP-tag cell membrane suspensions were prepared according to a previous method with further modifications [11]. In brief, KIT-SNAP-tag cells were first harvested using trypsin. The cells were washed twice using normal saline via centrifugation (1000 × g, 4°C, 10 min). The resulting cell pellet was resuspended in 5 mL Tris-HCl (50 mM, pH = 7.4), sonicated for 30 min at 4°C, and ruptured using an ultrasonic cell breaker (Emerson Electric Co., USA) at 80 W for 3 s for a total of six cycles with 1-s intervals. The homogenate was centrifuged (1000 × g, 10 min), and the obtained supernatant was transferred into a clean tube and recentrifuged (12,000 × g, 20 min). Subsequently, the cell membrane (precipitate) was added to 5 mL physiological saline and ruptured using the ultrasonic cell crusher at 200 W for 3 s six times with 1-s intervals. Next, the cell membrane precipitate was washed using physiological saline and centrifuged (12,000 × g, 4°C, 20 min) to obtain the KIT-SNAP-tag cell membrane. The suspension of cell membranes (protein concentration was no less than 2.68 mg/mL, as determined using the BCA kit) was slowly added to and mixed with 10 mg BG-decorated silica gel (SiO2-BG, 5 μm, 100 Å, synthesized using the method described in a previous study [11]) and vortexed for 1 min, followed by incubation at 37°C (30 min). After centrifugation (1000 × g, 5 min), the pellet was collected and washed three times with normal saline, thereby preparing the KIT-SNAP-tag cell membrane stationary phase (KIT-SNAP-tag/CMSP). Different cell membrane concentrations (0.15, 0.24, 0.43, 0.85, 1.78, 2.68, and 3.34 mg/mL) were investigated at an immobilization time of 30 min.
The surface morphologies of SiO2-BG and KIT-SNAP-tag/CMSP were characterized by transmission electron microscope (TEM; JEM-2100, JEOL). SiO2-BG and KIT-SNAP-tag/CMSP in ethanol were separately added onto the copper meshes, and dried under infrared light before observation by TEM.
Immunofluorescence
SiO2-BG and KIT-SNAP-tag/CMSP were first closed with 5% skimmed milk powder at 37°C for 30 min, then washed twice with PBS and incubated with KIT antibody (1% BSA diluted 200-fold) for 1 h at 37°C, then washed three times with PBS and incubated with green fluorescent secondary antibody (1% BSA diluted 200-fold) for 2 h at 37°C, protected from light, and then photographed as a stationary phase under a laser confocal scanning microscope.
System suitability of the KIT-SNAP-tag/CMC model
Traditional CMC stationary phases primarily rely on physical adsorption to coat cell membranes on the surface of silica carriers, which has the disadvantages of nonspecific adsorption interference, easy shedding of cell membranes, and other stability problems. SNAP-tag can be combined with BG in a highly specific manner, and the use of the protein tag technology can be used to chemically bind the target proteins on the surface of the carriers with a high degree of stability and specificity. These processes are expected to improve CMC stability and specificity. KIT-SNAP-tag/CMSP was loaded onto a column (inner diameter: 5 × 1.0 mm) to obtain the KIT-SNAP-tag/CMC column.
Various drugs acting on different receptors (β2-adrenergic receptor agonist, terbutaline; central anticholinergic, trihexyphenidyl hydrochloride; and adrenocorticotropic hormone drug, dexamethasone) were selected as negative ligands to verify the selectivity of the KIT-SNAP-tag/CMC column. The column specificity was investigated via the retention time of imatinib on KIT-SNAP-tag/CMC, NC-HEK293/CMC, and SiO2-BG/CMC columns. To validate the quality control procedure for KIT-SNAP-tag/CMC, different column batches were prepared and equilibrated in a HPLC system, and then, imatinib was injected. The retention time was used as the indicator of the repeatability of the KIT-SNAP-tag/CMC columns.
Establishment and validation of KIT-SNAP-tag/CMC online HPLC-MS system
The two-dimensional coupling system was assembled according to existing methods [11]. A conventional HPLC system (LC-2030AHT, Shimadzu, Kyoto, Japan) was used in the first dimension, and the column part was the KIT-SNAP-tag/CMC system for screening antitumor compounds acting on the KIT receptor from the complex TCM system. The following parameters were set: mobile phase A (100%, water); flow rate, 0.2 mL/min; chromatographic column temperature, 37°C; and UV detection wavelength, 266 nm (imatinib) and 342 nm (E. rutaecarpa methanol extract). For the two-dimensional system, an LC–MS and a matching LC–MS workstation (LCMS-IT-TOF, Shimadzu Corporation, Kyoto, Japan) were used. Complex samples were separated using Inertsil® ODS-3 column (150 mm × 4.6 mm I.D., 5 μm, GL Sciences Inc., Japan), with the mobile phases of 0.1% formic acid in water (A) and acetonitrile (B) at a flow rate of 0.8 mL/min using an optimized gradient program (0–9 min, 7–10% B; 9–14 min, 10–15% B, 14–30 min, 15–24% B; 30–35 min, 24–60% B; 35–45 min, 60% B; 45–60 min, 60–100% B). The MS settings were as follows: nebulizer gas (N2, purity > 99.999%) flow rate at 3 L/min and drying gas (N2, purity > 99.999%) pressure at 100 kPa; interface, ESI source; temperature of curve desolution line and heat block, 200℃; interface voltage, 4.5 kV; detector voltage, 1.57 kV; CID gas (Ar, purity > 99.999%); CID energy, 50%; ion accumulation, 30.0 ms; scan mode, positive ionization; automatic precursor ion selection; scan range, 100–1000 m/z; and detector voltage, 1.70 kV. The two dimensions were combined by a 6-port two-position switching valve (VICI Valco Instrument Co. Inc., Houston, TX, USA) and a Shim-pack VP-ODS pre-columns (10 mm × 2.0 mm i.d., 5 µm, Shimadzu Corporation, Kyoto, Japan).
For the KIT-SNAP-tag/CMC online HPLC-IT-TOF-MS system, imatinib was used as the marker ligand to verify the suitability of the two-dimensional system.
Application of online system
The two-dimensional online system was used to screen, analyze, and identify active compounds in the total extract of E. rutaecarpa. For further verification, the system was used to analyze the mixed standard solution of the retained compounds identified from E. rutaecarpa.
Interaction analysis
NLC was first developed by H.C. Thomas and extended to affinity chromatography by Wade et al. [15, 16]. It assumes that the contribution of some kinetic processes is negligible compared with analyte binding to and dissociation from the ligand. Different combination and dissociation rates are the main factors for a peak profile. The band broadening of a peak and the shape deviation often follow non-Gaussian functions. In particular, NLC is suitable for the analysis of tailed chromatographic peaks. The expression of NLC is modeled as follows:
where y represents the intensity of the measured signal and x is the reduced retention time. The T (u, v) function is a switching function to produce a peak skew. Formula (2) is the conversion function when the chromatographic column is overloaded and I0 and I1 are modified Bessel functions. The terms a0, a1, a2, and a3 are the best-fit parameters used to fit this equation to the experimental peak. The rate and equilibrium constants for the analyte–ligand interaction produced the following relationships: dissociation rate constant, kd = 1/a2t0; dissociation constant, KD = C0/a3; and binding rate constant, ka = kd /KD, where t0 is the column void time and C0 is the injected solute concentration multiplied by the ratio of sample volume to the column dead volume.
This method does not require column saturation with a large number of ligands, as it can effectively compensate for the shortcomings of frontier analysis and zonal elution in terms of the lengthy analysis time and high drug dosage [17].
The different concentrations of EVO (0.1, 0.2, 0.3, 0.4, 0.6, and 0.8 mM), RUT (0.1, 0.2, 0.4, 0.6, and 0.8 mM), and DEVO (0.1, 0.2, 0.3, 0.4, 0.6, and 0.8 mM) were injected into the KIT-SNAP-tag/CMC system to assess their interaction with KIT. The data were fitted with the NLC function using PeakFit 4.12.
MTT cell viability assay
The MTT assay was used to evaluate the ability of imatinib and active compounds derived from E. rutaecarpa to inhibit KIT-SNAP-tag HEK293 cells. At the same time, the inhibition of NC-HEK293 using these drugs was used as a control. KIT-SNAP-tag HEK293 cells were seeded in a 96-well plate (3 × 103 cells per well) and incubated for 24 h. In each group of wells, 10 μL of the different concentrations of imatinib, EVO, RUT, and DEVO solutions was added. In the control wells, 10 μL of blank DMEM medium was added and incubated for 48 h. After incubation, the supernatant was removed and 0.5 mg/mL MTT solution was carefully added to each well without forming bubbles in a light-proof environment, followed by incubation for 4 h at 37°C. The supernatant was aspirated and 150 μL DMSO was added to each well, followed by incubation for 15 min with shaking at 100 rpm. The absorbance of each well was measured at 490 nm using a microplate reader (UMI, Hangzhou), and cell viability was compared with the negative control. Cell viability was calculated using the following formula: cell viability (%) = (absorbance of experimental group − absorbance of blank group) / (absorbance of control group − absorbance of blank group) × 100%.
Molecular docking
To further verify of the receptor-inhibitor binding interactions of EVO, RUT, and DEVO with KIT, a molecular docking assay was performed using the SYBYL-X 2.0 program (Tripos, MO, USA) and PyMOL 2.5 (DeLano Scientific LLC). The structural file of KIT (PDB ID: 1T46) was downloaded from PDB.org. 1T46 is structural basis for the autoinhibition and STI-571(Imatinib) inhibition of c-kit tyrosine kinase. The detailed operation steps were carried out according to the previous literature [18]. Then, the optimized molecules were visualized using PyMOL 2.5.
Statistical analysis
All chromatograms were plotted in GraphPad Prism 6.01 and Origin 2023b. To assess significance, two-way t-tests were performed using GraphPad Prism and p-value ≤ 0.05 was considered significant.
Results
Verification of high KIT expression on HEK293 cells
SNAP-tag has been successfully used for the targeted covalent immobilization of proteins [19]. In HEK293 cells, KIT was fused with the SNAP-tag at the C-terminus of the receptor. The expression of KIT on KIT-SNAP-tag cells was then verified using qPCR and western blotting, and the results are shown in Table S1 and Fig. 1. qPCR showed that KIT gene expression was significantly higher in KIT-SNAP-tag cells than in NC-HEK293 cells (Table S1). Compared with the control group, KIT was upregulated 48-fold in KIT-SNAP-tag HEK293 cells. As shown in Fig. 1, KIT protein expression in KIT-SNAP-tag HEK293 cells was also significantly higher than that in the control group (p < 0.001), indicating that KIT protein was successfully highly expressed in the constructed KIT-SNAP-tag HEK293 cells.

Western blot detection of KIT protein expression (a) and quantitative results (b). The data are presented as mean ± standard deviation (SD) (*p < 0.05, **p < 0.01, and ***p < 0.001)
Preparation of the KIT-SNAP-Tag/CMC stationary phase
To obtain a more effective KIT-SNAP-tag/CMC stationary phase, the cell membrane concentration was measured during cell membrane immobilization on SiO2-BG using a BCA protein quantification assay kit. Then, 0.5 mL cell membrane was incubated with 3 mg SiO2-BG stationary phase at 37°C; a total of six cell membrane concentrations (0.15, 0.24, 0.43, 0.85, 1.78, 2.68, and 3.34 mg/mL) were analyzed. Fig. S1 shows the optimization results of the cell membrane concentrations. The immobilized cell membrane concentration was positively associated with the cell membrane concentration increasing from 0.15 to 2.68 mg/mL. After 2.68 mg/mL, immobilized cell membrane concentration did not increase, and 1 g SiO2-BG absorbed up to approximately 153 mg membrane protein. Thus, the optimum cell membrane protein concentration was 2.68 mg/mL.
For observation of the surface morphology of the stationary phase, the SiO2-BG and prepared KIT-SNAP-tag/CMSP were characterized by TEM. Compared with SiO2-BG (Fig. 2c and d), the pores on the surface of silica gel were obviously covered (Fig. 2a and b), and a transparent membrane on the surface could be observed under 100,000 times magnification (Fig. 2b). The membrane layer on KIT-SNAP-tag/CMSP spherical surface was about 8–15 nm thicker than that of SiO2-BG.

TEM and immunofluorescence characterization results of SiO2-BG and KIT-SNAP-tag/CMSP. a and b are TEM images of KIT-SNAP-tag/CMSP with magnifications of 1K× and 100K×, respectively; c and d are TEM images of SiO2-BG with magnifications of 1K× and 100K×; e and h are the laser confocal scan images of KIT-SNAP-tag/CMSP and SiO2-BG; f and i are the corresponding images under bright field. The magnification is all 1K×
To verify the membrane wrapping effect of KIT-SNAP-tag/CMSP and confirm that the immobilized cell membranes contained the target receptor KIT, immunofluorescence was used to visualize KIT. As shown in Fig. 2, no green fluorescence was detected in SiO2-BG (Fig. 2h and i), but fluorescence was observed in KIT-SNAP-tag/CMSP (Fig. 2e and f). This result indicated that the prepared KIT-SNAP-tag/CMSP contained the target receptor KIT.
System suitability of the KIT-SNAP-Tag/CMC model
After KIT-SNAP-tag/CMSP was packed into columns and connected to the liquid system, the KIT-SNAP-tag/CMC system was established. The selectivity, specificity, and reproducibility of the KIT-SNAP-tag/CMC column were investigated, and the results are shown in Fig. 3a and b. Terbutaline, which acts on the β2 receptor; trihexyphenidyl hydrochloride, which affects the central cholinergic receptor; and dexamethasone, which acts on the glucocorticoid receptor, were minimally retained on the KIT-SNAP-tag/CMC column, whereas the KIT receptor-positive drug imatinib was significantly retained on the column (tR = 28.57 min). The chromatograms of these four drugs on the KIT-SNAP-tag/CMC column are depicted in Fig. 4a, and the results indicated that the column selectively recognized active compounds that interact with the KIT receptor.

Results of selectivity (a) and specificity (b) examination of KIT-SNAP-tag/CMC. a Chromatograms of the positive drug and three drugs acting on other targets on the KIT-SNAP-tag/CMC. (I) Imatinib; (II) terbutaline; (III) trihexyphenidyl hydrochloride; (IV) dexamethasone. b Chromatograms of the positive drug imatinib on three cell membrane columns. (II) BG-SiO2-KIT-ST/CMC; (II) BG-SiO2-NC/CMC; (III) BG-SiO2/CMC

Chromatogram of imatinib on KIT-SNAP-tag/CMC-HPLC-MS 2D coupling system. a Chromatogram of imatinib on KIT-SNAP-tag/CMC system; b chromatogram of retained components switched into the HPLC-MS system by enrichment; c HPLC-MS chromatogram of imatinib analyzed by the two-dimensional system without connecting KIT-SNAP-tag/CMC column
Next, the retention of imatinib on a SiO2-BG blank column and NC-HEK293/CMC column was investigated, and the chromatograms are shown in Fig. 4b. Although imatinib was retained on the NC-HEK293/CMC column (tR = 1.76 min), the retention time was shorter than that on the KIT-SNAP-tag/CMC column. At the same time, compared with the SiO2-BG blank column (tR = 18.35 min), cell membrane-coated columns significantly attenuated the nonspecific adsorption that may exist on silica gel columns, suggesting that the KIT-SNAP-tag/CMC chromatographic model has a certain specificity. The results of the reproducibility experiments shown in Table S2 revealed that the RSD (%) of the intra-column and inter-column retention time was 2.15% and 1.65%, respectively, indicating that the stability of this CMC model was satisfactory.
Application of the online KIT-SNAP-Tag/CMC model
The established KIT-SNAP-tag/CMC system was coupled with HPLC-MS for the screening and identification of potential active compounds. Imatinib solution was used to validate the screening system, and the results are shown in Fig. 4. The retained fraction of imatinib in the KIT-SNAP-Tag/CMC system (Fig. 4a) was enriched into the pre-column via the switching of the six-way valve, eventually switching to the HPLC-MS system for analysis (Fig. 4b). The comparison and MS identification of the chromatograms obtained from the switching of the two-dimensional system and the chromatograms obtained by the direct entry of imatinib into the HPLC-MS system (Fig. 4c) revealed that they were consistent with each other. These results indicated that the screening system was suitable for the screening, separation, and identification of potential KIT-interacting active compounds.
TCM has been shown to reduce drug toxicity and increase drug sensitivity, stabilize tumors, and improve the mid- and long-term survival of patients with gastric cancer [20, 21]. Moreover, TCM and its extracts are considered an important source of potential anticancer drugs and new chemotherapeutic adjuvants. Studies have shown that E. rutaecarpa, as one of the commonly used clinical medicines in the spleen and stomach discipline of TCM, can disperse cold and relieve pain, reduce rebelliousness, stop vomiting, and halt diarrhea. There are numerous known chemical constituents in E. rutaecarpa, including alkaloids, picric acid, volatile oil, flavonoids, phenolic acids and their derivatives, anthraquinones, and other compounds [22]. Currently, there are few reports [23] on the treatment of GIST with TCM; in the study, the extract of E. rutaecarpa was used for the identification of potential active antitumor compounds using the two-dimensional screening system validated in this study, and the results are shown in Fig. 5. The peak R1 (3–12 min) valve obtained from the E. rutaecarpa extract run through the KIT-SNAP-tag/CMC system was switched to the HPLC-MS system for analysis, and MS detected DEVO and EVO. Likewise, the peak R2 (16–40 min) valve obtained from the extract was identified as DEVO and RUT by MS.

Chromatograms of Evodia rutaecarpa methanol extract analyzed by KIT-SNAP-tag/CMC online coupled with HPLC-MS system. a Chromatogram of Evodia rutaecarpa methanol extract retained on KIT-SNAP-tag/CMC column; b HPLC-MS chromatograms of the retained fractions, R1 and R2; c HPLC-MS chromatograms of Evodia rutaecarpa methanol extract analyzed by the two-dimensional system without connecting KIT-SNAP-tag/CMC column
The cleavage modes of the three potential active compounds are shown in Fig. 6. Compound 1 had m/z 302.1279 [M+H]+ as the quasi-molecular ion peak and m/z 286.0988 and 258.1039 as the characteristic fragment ions, which were consistent with DEVO [24, 25]. Therefore, it was identified as DEVO. Compound 2 (t = 40.460 min) had m/z 304.1447 [M+H]+ as the quasi-molecular ion peak. Its MS2 plot contained the fragment ions 171.0930, 161.0716, 144.0813, and 134.0610, where the fragment ions 171.0930 and 134.0610 are the characteristic fragment ions produced by the RDA cleavage of EVO. As [M+H-C10H10N] (+ m/z 161.0716) and [M+H-C9H8N2O] (+ m/z 144.0813) are the characteristic fragment ions produced by the rearrangement of the intramolecular double bonds after the RDA cleavage of EVO, this compound was identified as EVO [26]. Finally, compound 3 had m/z 288.1128 [M+H]+ as its quasi-molecular ion peak, which was 16 less than that of compound 2, indicating the removal of a CH4 group. The fragment ion 244.0874 was detected in its MS2 plot, a characteristic fragment ion produced by RUT after RDA cleavage, indicating that compound 3 was RUT. All these inferences were combined with the maximum absorption wavelength and retention time of the corresponding peaks.

Ion fragmentation peaks of the three active components obtained by retained fraction enrichment switching into the HPLC-MS system. a, b, c EVO, RUT, and DEVO
The mixed control of the three potential active compounds was validated using the two-dimensional screening system, and the results were consistent with the chromatograms and mass spectra produced from the E. rutaecarpa extract via this system (Fig. S2). Thus, the potential KIT-interacting active compounds in E. rutaecarpa extract were DEVO, EVO, and RUT.
Interaction analysis of active compounds
To investigate the interactions between the above three compounds and KIT, the serial concentrations of DEVO, EVO, and RUT were analyzed individually using the KIT-SNAP-tag/CMC system. Figure 7 shows the chromatographic superposition of the serial concentrations of EVO (0.1–0.8 mmol/L), RUT, and DEVO on the KIT-SNAP-tag/CMC column, respectively, with the concentrations decreasing sequentially from the top to bottom. Owing to the constant number of KIT on the CMC column, the time required for receptor–ligand binding to reach saturation on the CMC column gradually decreased as the ligand drug concentration gradually increased.

Superposition of chromatographic curves and example of nonlinear fitting of a series of concentrations of EVO, RUT, and DEVO on KIT-SNAP-tag/CMC columns. a The concentrations of EVO were 0.1, 0.2, 0.3, 0.4, 0.6, 0.8 mmol/L. b The concentrations of RUT were 0.1, 0.2, 0.4, 0.6, 0.8 mmol/L. c The concentrations of DEVO were 0.1, 0.2, 0.3, 0.4, 0.6, 0.8 mmol/L. d, e, and f are example plots of nonlinear fits at 0.2 mmol/L for EVO, RUT, and DEVO, respectively
The nonlinear fitting of each chromatogram was performed using PeakFit. The binding kinetic parameters (KD, ka, and kd) were calculated from the average value of a1, a2, and a3 in the aforementioned concentration range (Table 1 and Table S3). The binding constant KD of EVO, RUT, and DEVO were 7.75 (±4.93) × 10−6 mol/L, 1.42 (±0.71) × 10−6 mol/L, and 2.34 (±1.86) × 10−6 mol/L, respectively, indicating that the three compounds interacted with KIT in the following order of affinity: EVO > DEVO > RUT.
Potential binding target of the imatinib and the three active components
The binding modes of the four compounds with the KIT were studied using molecular docking. As shown in Figure S3, imatinib, EVO, RUT, and DEVO matched the active pocket of the KIT well in the sphere space field model. As shown in Table S4, the scoring values of the four components imatinib, EVO, RUT, and DEVO in combination with KIT were 5.4715, 3.7348, 3.8165, and 4.2300. In addition, imatinib formed one hydrogen bond with ARG791 (Figure S3a), The remaining three active components interact with the KIT probably through weak interaction forces such as van der Waals forces. The docking results indicated that the potential antitumor constituents bind well to the KIT.
In vitro cellular assay
The three active compounds EVO, RUT, and DEVO were identified using the KIT-SNAP-tag/CMC-HPLC-MS system. To validate and explore the pharmacological activities of the active compounds, the MTT assay was used to investigate the effects of different concentrations of imatinib and the three active compounds on the proliferation of KIT-SNAP-tag HEK293 cells. Figure 8a shows the cell viability results of the positive drug imatinib on KIT-SNAP-tag HEK293 cells. Imatinib showed a dose-dependent decrease in cell survival in the concentration range of 5–50 μmol/L. Figure 8b and c show a dose-dependent decrease in cell survival across a concentration range of EVO (0.1–10 μmol/L) and RUT (0.1–50 μmol/L), respectively. Figure 8d shows that DEVO had no significant effect on cell survival in the 5–50 μmol/L concentration range. These results indicated that EVO and RUT inhibited the proliferation of KIT-SNAP-tag HEK293 cells in the low concentration range.

Effects of imatinib (a), EVO (b), RUT (c), and DEVO (d) on the proliferation of KIT-SNAP-tag cells. (Results are expressed as mean ± SD values, n = 3; *p < 0.05, **p < 0.01, ***p < 0.001 compared with control group)
To verify whether the three active compounds acted on KIT, the proliferation inhibition of NC-HEK293 cells without KIT expression was evaluated as control using imatinib, EVO, RUT, and DEVO. The results showed that the effect of imatinib on the proliferation inhibition of NC-HEK293 cells in the range of 5–10 μmol/L was not significantly different from that of cells with high KIT expression and that the inhibitory effect was significantly inferior to that of KIT-SNAP-tag HEK293 cells in the concentration range of 10–50 μmol/L. The inhibitory effects of the three active compounds EVO, RUT, and DEVO on NC-HEK293 cells were also less significant than those on SNAP-tag-KIT HEK293 cells.
The MTT assay revealed that EVO, RUT, and DEVO could dose-dependently inhibit the proliferation of KIT-SNAP-tag HEK293 cells within the ranges of 0.1–10 μmol/L, 0.1–50 μmol/L, and 50–100 μmol/L, respectively. The inhibitory effect, which was exerted by the inhibition of cell proliferation via the action on KIT, of EVO and RUT was superior to that of imatinib to a certain extent.
Discussion
The KIT-SNAP-tag/CMC system developed in this study exhibited good specificity and higher stability than the traditional CMC column, improving the screening efficiency of CMC, expanding the application scope of the protein tag technology and CMC to a certain extent, and providing a more powerful tool for the screening of antitumor drugs in TCM. However, this CMC system with silica gel as the carrier still has the problems of low cell membrane utilization and short column activity time. As a biomimetic material, CMC still presents with great challenges in improving stability and column activity time.
Targeted therapies offer hope to GIST patients, it is not only by finding new therapeutic targets [27, 28], but also by conducting more in-depth mechanistic studies of existing KIT/PDGFRA inhibitors and screening novel potential active ingredients. The potential active compounds EVO and RUT identified using the KIT-SNAP-tag/CMC system had certain inhibitory effects on the proliferation of high KIT-expressing HEK293 cells. The inhibitory effect was significantly different compared with that on the NC-HEK293 control group, suggesting, to a certain extent, that these two potential active compounds act on the KIT receptor and thus exert a series of pharmacological effects. Subsequent research on the pharmacological mechanism of these two compounds in the signaling pathway downstream of the KIT receptor may prove valuable.
A previous study [29] suggested that resistance to imatinib during GIST treatment is caused by genetic mutations. Thus, imatinib-resistant cell membranes can be used to construct a drug-resistant CMC model to identify TKIs for treating imatinib-resistant GIST, offering a novel solution for the treatment of patients with GIST on imatinib treatment exhibiting disease progression.
Taken together, the KIT-SNAP-tag/CMC system coupled with HPLC-MS was developed and validated to be an effective method for screening potential anti-GIST compounds from TCM. EVO and RUT were identified from E. rutaecarpa and confirmed as active anti-GIST compounds.
References
Iorio N, Sawaya R, Friedenberg F. Review article: the biology, diagnosis and management of gastrointestinal stromal tumours. Aliment Pharmacol Ther. 2014;39(12):1376–86. https://doi.org/10.1111/apt.12761.
Seiichi H, Koji I, Yasuhiro M, et al. Gain-of-Function Mutations of c-kit in Human Gastrointestinal Stromal Tumors. Sci. 1998;279:577–80. https://doi.org/10.1126/science.279.5350.577.
Corless C, Barnett C, Heinrich M. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11:865–78. https://doi.org/10.1038/nrc3143.
Eichler M, Pink D, Menge F, et al. Quality of life of GIST patients with and without current tyrosine kinase inhibitor treatment: Cross-sectional results of a German multicentre observational study (PROSa). Eur J Cancer Care (Engl). 2021;30:e13484. https://doi.org/10.1111/ecc.13484.
Liegl B, Kepten I, Le C, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008;216:64–74. https://doi.org/10.1002/path.2382.
Serrano C, Bauer S, Gómez-Peregrina D, et al. Circulating tumor DNA analysis of the phase III VOYAGER trial: KIT mutational landscape and outcomes in patients with advanced gastrointestinal stromal tumor treated with avapritinib or regorafenib. Ann Oncol. 2023;34(07):615–25. https://doi.org/10.1016/j.annonc.2023.04.006.
Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and Safety of Sunitinib in Patients with Advanced Gastrointestinal Stromal Tumour after Failure of Imatinib: A Randomised Controlled Trial. Lancet. 2006;368:1329–38. https://doi.org/10.1016/S0140-6736(06)69446-4.
Demetri GD, Reichardt P, Kang YK, et al. Efficacy and Safety of Regorafenib for Advanced Gastrointestinal Stromal Tumours after Failure of Imatinib and Sunitinib (GRID): An International, Multicentre, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet. 2013;381:295–302. https://doi.org/10.1016/S0140-6736(12)61857-1.
Amalya S, Monika A, Robin L, et al. Ripretinib for the treatment of adult patients with advanced gastrointestinal stromal tumors. Expert Rev Gastroent. 2023;17(02):119–27. https://doi.org/10.1080/17474124.2023.2167711.
Ma W, Wang C, Liu R, et al. Advances in cell membrane chromatography. J Chromatogr A. 2021;1639:461916. https://doi.org/10.1016/j.chroma.2021.461916.
Fu J, Jia Q, Liang P, et al. Targeting and covalently immobilizing the EGFR through SNAP-tag technology for screening drug leads. Anal Chem. 2021;93:11719–28. https://doi.org/10.1021/acs.analchem.1c01664.
Jia Q, Fu J, Gao C, et al. MrgX2-SNAP-tag/cell membrane chromatography model coupled with liquid chromatography-mass spectrometry for anti-pseudo-allergic compound screening in Arnebiae Radix. Anal Bioanal Chem. 2022;414:5741–53. https://doi.org/10.1007/s00216-022-04167-4.
Zhou H, Fu J, Jia Q, et al. Magnetic nanoparticles covalently immobilizing epidermal growth factor receptor by SNAP-Tag protein as a platform for drug discovery. Talanta. 2022;240:123204. https://doi.org/10.1016/j.talanta.2021.123204.
Zhang L, Yi X, Wang S, et al. Construction of graphene quantum dots-decorated EGFR cell membrane chromatography for screening active compounds from Peucedanum praeruptorum Dunn. Anal Bioanal Chem, 2021:413: 1917-1927. https://doi.org/10.1007/s00216-021-03161-6.
Thomas HC. Heterogeneous ion exchange in a flowing system. J Am Chem Soc. 1944;66:1664–6. https://doi.org/10.1021/ja01238a017.
Wade J, Bergold A, Carr P. Theoretical description of nonlinear chromatography, with applications to physicochemical measurements in affinity chromatography and implications for preparative-scale separations. Anal Chem. 1987;59:1286–95. https://doi.org/10.1021/ac00136a008.
Li Q, Wang J, Zheng Y, et al. Comparison of zonal elution and nonlinear chromatography in determination of the interaction between seven drugs and immobilised β2-adrenoceptor. J Chromatogr A. 2015;1401:75–83. https://doi.org/10.1016/j.chroma.2015.05.012.
Shi Y, Sun W, Pan X, et al. Establishment of thrombin affinity column (TAC)-HPLC-MS/MS method for screening direct thrombin inhibitors from Radix Salviae Miltiorrhiae. J Chromatogr B. 2020;1139. https://doi.org/10.1016/j.jchromb.2019.121894.
Engin S, Trouillet V, Franz C, et al. Benzylguanine thiol self-assembled monolayers for the immobilization of SNAP-tag proteins on microcontact-printed surface structures. Langmuir. 2010;26(09):6097–101. https://doi.org/10.1021/la904829y.
Zhong Y, Wang P, Hao D, et al. Traditional Chinese medicine for transformation of gastric precancerous lesions to gastric cancer: A critical review. World J Gastrointest Oncol. 2023;15(01):36–54. https://doi.org/10.4251/wjgo.v15.i1.36.
Huang W, Ruan S, Wen F, et al. Multidrug Resistance of Gastric Cancer: The Mechanisms and Chinese Medicine Reversal Agents. Cancer Manag Res. 2020;12:12385–94. https://doi.org/10.2147/CMAR.S274599.
Kong Y, Qi Y, Cui N, et al. Research progress on modern chemical constituent and pharmacological effects of Evodia rutaecarpa. J Tradit Chin Med, 2023:40(05):79-83+89. https://doi.org/10.19656/j.cnki.1002-2406.20230513.
Hu X, Yang W, Kang Z, et al. Inhibition proliferation and promotion of apoptosis of GIST-T1 cells by the down-regulation of c-kit expression. J Army Med Univ. 2021;43(13):1235–42. https://doi.org/10.16016/j.1000-5404.202012232.
Huang X, Zhang Y, Yang X. Indoloquinazoline alkaloids from Evodiarutaecarpa and their cytotoxic activities. J Asian Nat Prod Res. 2011;13(11):977–83. https://doi.org/10.1080/10286020.2011.602015.
Su X, Yin M, Xu S, et al. Analysis of chemical constituents in Evodiarutaecarpa by UPLC-Q-TOF-MS. Chin Tradi Patent Med. 2017;39(06):1223–7.
Li M, Yuan J, Zhang X, et al. Analysis of chemical constituents from Evodiarutaecarpa by UHPLC-Q-TOF-MS/MS. J Tradit Chin Med. 2023;42(02):212–7.
Delvaux M, Hagué P, Craciun L, et al. Ferroptosis Induction and YAP Inhibition as New Therapeutic Targets in Gastrointestinal Stromal Tumors (GISTs). Cancers. 2022;14:5050. https://doi.org/10.3390/cancers14205050.
Wang S, Wang Z, He M, et al. Progress in the pathogenesis and targeted therapy of gastrointestinal stromal tumors. J Chin Med Univ. 2023;52(06):543–8.
Hayashi Y, Nguyen VTT. A narrative review of imatinib-resistant gastrointestinal stromal tumors Gastrointestinal Stromal Tumor, 2021;4:6. https://doi.org/10.21037/gist-21-1.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Number: 81973278, 81930096, and 82104118) and Natural Science Foundation of Shaanxi Province (grant number: 2023-YBSF-173 and 2023-YBSF-246).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Feng, J., Jiang, Y., Liu, S. et al. KIT-SNAP-tag/cell membrane chromatography model coupled with liquid chromatography–mass spectrometry for anti-GIST compound screening from Evodia rutaecarpa. Anal Bioanal Chem 416, 1457–1468 (2024). https://doi.org/10.1007/s00216-024-05148-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-024-05148-5