
Abstract
This study reports the development and validation of a new analytical method for simultaneous speciation analysis of Se and Hg in fish muscle. For this purpose, four Se species (selenite/Se(IV), selenate/Se(VI), selenomethionine/SeMet, and selenocysteine/SeCys) and two Hg species (inorganic mercury/iHg and methylmercury/MeHg) were extracted simultaneously by microwave-assisted enzymatic hydrolysis and then separated by HPLC in less than 15 min by using a column with both anion and cation exchange mechanisms and a mobile phase consisting of a mixture of methanol 5% (v/v), 45 mM HNO3, 0.015% 2-mercaptoethanol, and 1.5 mM sodium 3-mercapto-1-propanesulfonate. The separated species of Hg and Se were detected online by inductively coupled plasma-mass spectrometry (ICP-MS). The speciation analysis method was validated by means of the accuracy profile approach by carrying out three series of measurements in duplicate on three different days over a time-span of 3 weeks. The limits of quantification (LOQ) are in the range of 0.010–0.013 mg/kg wet weight (ww) for all selenium species, except for Se(IV) (0.15 mg/kg ww), while the coefficient of variation in terms of intermediate reproducibility (CVR) was < 7%. The LOQ for MeHg was 0.006 mg/kg ww, while the CVR was 3%. The method was successfully applied to the analysis of muscle samples from four different fish species: rainbow trout, tuna, swordfish, and dogfish.
Graphical Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
It is widely recognised that selenium (Se) and mercury (Hg) have antagonistic effects; i.e., Se shows protective properties against the bioaccumulation and toxicity of Hg [1, 2]. However, the behaviour of methylmercury (MeHg) in fish and fishery products in relation to Se species is poorly understood. This is explained partially by the challenges related to the analytical chemistry, mainly the difficulty of simultaneous speciation of MeHg and the multiples Se species present in fish tissues [3].
Speciation analysis of Se and Hg separately has been widely studied using different analytical techniques. Among them, the mostly employed are the hyphenated techniques using gas (GC) or high-performance liquid chromatography (HPLC) coupled to inductively coupled-plasma mass spectrometry (ICP-MS), which are powerful tools for this purpose. The most efficient separation method for Se speciation analysis is HPLC, while GC has been the dominant method for Hg species separation, with a derivatisation step [3,4,5]. However, there is a growing trend to use HPLC also for Hg species separation due to its simplicity [6, 7].
In the case of Se speciation analysis, three chromatographic methods are frequently used, such as molecular exclusion, ion exchange, or reversed phase with ion-pairing reagents such as heptafluorobutanoic acid (HFBA) and among these approaches, ion-exchange chromatography was initially the preferred separation technique to separate the ionic Se species [5]. For Hg speciation analysis, the mostly used chromatographic method is the reversed phase (RP) with a mobile phase generally consisting of ammonium acetate, methanol (MeOH), l-cysteine, and 2-mercaptoethanol (hereafter referred by its commonly used abbreviation BME) in various combinations [7].
It is worth to note that single-element speciation analysis methods are counterproductive since they require separate procedures for each element species. This involves multiple analyses of the same samples, making the process laborious and time-consuming. To meet the current demands for efficiency and time savings, the development of more advanced multi-elemental speciation analysis methods based on HPLC-ICP-MS is necessary. These methods can significantly improve the sample throughput, minimising the use of reagents and the amount of waste [7, 8]. However, simultaneous quantification of multiple Hg and Se species is still an analytical challenge mainly due to very different physio-chemical properties of Hg and Se species [3, 5], which hamper notably their simultaneous extraction and also their efficient (HPLC) separation [9]. Ideally, the species of all elements should behave similarly during the analysis, but this is not always the case [7, 8]. Therefore, the conditions must be optimised so that all multi-elemental species are quantitatively extracted, separated, and determined.
It is worth to highlight that simultaneous speciation analysis of Se and Hg by HPLC-ICP-MS has been scarcely carried out so far; Li et al. (2007) and Moreno et al. (2010 and 2013) focused on urine and serum, plasma, and water samples [6, 10, 11]. Li et al. achieved the separation of four Se species such as trimethylselenoniumiodide, selenocystine (SeCys2), selenourea, and selenomethionine (SeMet) and two Hg species, MeHg and inorganic Hg (iHg). For this purpose, the authors employed reversed-phase HPLC with gradient elution, consisting of a mobile phase “A” of HFBA and MeOH, and a mobile phase “B” of BME, ammonium acetate, and MeOH [6]. Moreno et al. (2010) also achieved the separation of Se(IV), Se(VI), d-SeMet, l-SeMet and SeCys, MeHg, and iHg by RP-HPLC using a gradient elution program (mobile phase A: tetraethylammonium chloride; mobile phase B: BME, ammonium acetate and MeOH) [10]. In 2013, Moreno et al. reported also the separation of iHg, MeHg, and Se(IV) by RP-HPLC using a gradient of MeOH and 0.14% (v/v) BME in 50 mM ammonium acetate at pH ≅ 4.6 [11].
Simultaneous speciation analysis of Hg and Se species in complex biological matrices, such as fish, is an important tool to assess and understand the fish exposure to MeHg in relation to Se, which has an antagonistic effect. To the best of our knowledge, (single run) simultaneous speciation of Se and Hg in fish samples by HPLC-ICP-MS has not yet been reported.
This study focuses on the development and validation of a novel method for simultaneous speciation analysis of Hg (MeHg and iHg) and Se (inorganic species and selenoproteins, via their seleno-amino acids) by HPLC-ICP-MS in fish samples following microwave-assisted enzymatic extraction. For this purpose, several separation mechanisms were investigated and the mixed anion-cation exchange HPLC separation turned out to be the most efficient. A very robust method validation based on the accuracy profile approach was carried out. Finally, the novel method proposed in this study was applied to the simultaneous speciation analysis of Hg and Se in various fish species (marine and fresh water) hence demonstrating its capability to accurately assess the levels of Hg and Se species in such complex samples allowing to better understand their antagonistic features.
Materials and methods
Chemicals and reagents
All solutions were prepared with analytical reagent grade chemicals and ultrapure water (18.2 MΩ cm), Millipore Milli-Q™ (Merck Millipore, Saint Quentin in Yvelines, France).
Protease from Streptomyces griseus, Type XIV, ≥ 3.5 units/mg solid, ammonium phosphate buffer (prepared by the addition of ammonium dihydrogen phosphate to a concentration of 1 mM and adding a small volume of ammonia until pH ≅ 7 is reached), and BME, which were employed for extraction were purchased from Sigma-Aldrich (Saint-Quentin-Fallavier, France).
The mobile phases were prepared with MeOH and HNO3 (67%, v/v) purchased from VWR chemicals (Fontenay sous-Bois, France), sodium 3-mercapto-1-propanesulfonate (MPS), BME and trifluoroacetic acid (TFA, ≥ 99%) from Sigma-Aldrich, and the l-cysteine HCl.H2O (99%, m/m), from Alfa Aesar (Kandel, Germany).
Methylmercury chloride (CH3HgCl) standard solution (1001 μg/L) was purchased from VHG Labs (LGC, Molsheim, France) and 1000 mg/L Se standard solutions of the individual Se species were prepared from sodium selenite (≥ 99%), Se(IV), sodium selenate (≥ 95%), Se(VI), seleno-dl-methionine (≥ 99%), SeMet, and Se-methylselenocysteine (≥ 95%), SeCys), all being purchased from Sigma-Aldrich.
Calibration standard solutions were prepared daily by diluting the appropriate amount of intermediate working solutions with ultrapure water in 50-mL volumetric tubes and adding appropriate amounts of ammonium phosphate buffer, BME, and HNO3.
The tuning solution for the daily optimisation of the ICP-MS (Tune B iCAP Q/RQ) consisted of barium (Ba), bismuth (Bi), cerium (Ce), cobalt (Co), indium (In), lithium (Li), and uranium (U) at 1.0 μg/L in HNO3 2% (v/v) and HCl 0.5% (v/v) and it was purchased from Thermo Fisher Scientific (Courtaboeuf, France).
The certified reference materials (CRMs) used in this study were ERM-CE101 (trout muscle) and ERM-BB422 (fish muscle) from the European Joint Research Centre (JRC) with total Hg (tHg) certified level of 0.0219 ± 0.0027 mg/kg and 0.601 ± 0.030 mg/kg, respectively, and total Se (tSe) certified level of 0.113 ± 0.011 mg/kg and 1.33 ± 0.13 mg/kg, respectively. NMIJ 7402-a (codfish tissue) CRM from the National Metrology Institute of Japan (NMIJ) has a certified level of 0.61 ± 0.02 mg/kg for tHg, of 0.58 ± 0.02 mg/kg for MeHg, and of 1.8 ± 0.2 mg/kg for tSe. Finally, DOLT-5 (dogfish liver tissue) from the National Research Council Canada has certified levels of 0.44 ± 0.18 mg/kg for tHg, of 0.119 ± 0.058 mg/kg for MeHg, and of 8.3 ± 1.8 mg/kg for tSe. The CRMs NMIJ and DOLT-5 were purchased from LGC Standards (Molsheim, France).
For the determination of total Hg and Se by ICP-MS, mono-elemental stock standard solutions at 1000 mg/L of Hg, Se, and gold (Au) and internal standards, rhenium (Re) and yttrium (Y), purchased from SCP Science (Courtaboeuf, France), were used to prepare working solutions daily in 6% (v/v) HNO3. A solution containing Au at 10 mg/L in 6% HNO3 (v/v) was used throughout for ICP-MS washing between the analyses to reduce the memory effects of Hg.
Instrumentation
The sample preparation was performed using a closed microwave system (Multiwave PRO) equipped with two rotors (ROTOR 8 NXF/NXQ and ROTOR 64MG5), all purchased from Anton Paar (Courtaboeuf, France), an UN75 oven (Memmert GmbH + Co. KG, Schwabach, Germany), and a rotary shaker (Heidolph reax 2, Merck KGaA, Darmstadt, Germany). A sensor was installed by the manufacturer specifically for this experimental set-up to accurately control the extraction temperature.
The separation of Hg and Se species was performed using a metal-free ICS 5000 HPLC system equipped with a 20-µL PEEK injection loop. Four different columns were compared, namely a Dionex IonPac CS5A column and its guard column (IonPac CG5A) and a Dionex IonPac™ AS7 column, both from Thermo Fisher Scientific, and one small-bore reversed-phase (C18) HPLC column (Poroshell), purchased from Agilent Technologies, Courtaboeuf, France.
An ICP-MS iCAP Q (Thermo Fisher Scientific, Courtaboeuf, France) was used in tandem with HPLC for Hg and Se species detection. The ICP-MS was equipped with a PFA standard nebuliser and a quartz spray chamber. Speciation analysis was performed using the ICP-MS in standard mode (no gas), while kinetic energy discrimination (KED) using helium (He) as a collision gas was employed for the determination of total Hg and Se. The ICP-MS measurement conditions were optimised daily by short-term stability tests run both in standard and KED modes.
The nebuliser of the ICP-MS was connected to HPLC column using 80-cm-long PEEK tubing (0.18 mm i.d.).
Data were processed using Thermo Fisher Scientific Qtegra software.
The optimum ICP-MS conditions and the instrumental settings (HPLC and microwave) for simultaneous speciation analysis are given in Table 1.
Analytical procedures
Determination of total Hg and total Se by ICP-MS
The levels of tHg and tSe in the fish muscles were determined by ICP-MS using a validated method described in a prior study [2]. Briefly, approximately 0.5 g of sample was digested using a closed microwave system and concentrated HNO3 (67%, v/v). The digests were diluted with ultrapure water after the addition of internal standards and then analysed by ICP-MS.
Single-run simultaneous speciation analysis of Hg and Se by HPLC-ICP-MS
Simultaneous extraction of Hg and Se species
Three enzymatic extraction procedures using protease XIV were employed to achieve simultaneous extraction of Hg and Se species. One procedure, adapted from the study by Sele et al. [12], was firstly employed. Briefly, 0.2 g of CRMs was accurately weighed into polypropylene tubes and then suspended in 2.5 mL of 1 mM ammonium phosphate buffer (pH ≅ 7) containing 8 g/L of protease XIV, followed by the addition of 500 µL of BME 10% (v/v) (final volume of 3 mL). The solutions were mechanically shaken by using an overhead shaker (Heidolph reax 2, Merck KGaA, Darmstadt, Germany) inside an oven at 37 °C during 12 h.
Further, using the same sample weight and the same reagents, microwave-assisted enzymatic extraction (MAEE) has been tested in an attempt to accelerate the enzymatic extraction process. A special rotor (64MG5) suitable for small volumes (maximum final volume of 5.0 mL and maximum sample amount of 0.2 g), allowing 64 simultaneous extractions in controlled temperature and stirring conditions (by magnetic stirring bars) was used for that purpose. Different durations (45 and 60 min) of extraction times in the conditions mentioned previously (3 mL solution containing 0.2 g of sample in 2.5 mL of 1 mM ammonium phosphate buffer at pH ≅ 7, containing 6.67 g/L of protease XIV and 500 µL of BME 10% (v/v)) were investigated. Final dilution volumes (25 and 50 mL) of the extracts were also compared.
pH adjustment tests of the extract solution after enzymatic hydrolysis were carried out to ensure that Se(IV) is present in a single chemical form. Actually, it must be noted that at the pH used for the extraction (≅ 7), Se(IV) can be present as HSeO3− and as SeO32−, whereas at the pH of the mobile phase (≅ 1.5), Se(IV) is present only as H2SeO3. The variation of the pH from ≅ 7 to ≅ 1.5 can induce a change in the speciation for Se(IV) solely, while all other Se species are present in a single chemical form at both pH values.
Simultaneous separation of Hg and Se species
Reversed-phase and ion-exchange HPLC separation mechanisms were tested to simultaneously separate Se and Hg species.
HPLC-ICP-MS analyses were carried out without the employment of the collision cell (standard detection mode) and 82Se and 202Hg isotopes were used for quantification.
Reversed-phase HPLC
For the RP-HPLC, a small-bore C18 column (Poroshell), known to be effective for Hg species separation [13], was tested with different combinations of mobile phases using TFA and MeOH. Ion pair reagents such as TFA are often used for Se speciation [5].
Ion exchange HPLC
Two ion-exchange columns (IonPac AS7 and IonPac CS5A), which are strong anion exchange stationary phases with hydrophobic properties, were also compared for the HPLC separation of Se and Hg. It is important to note that IonPac CS5A is a double-layer cation–anion column allowing anionic and cation exchange mechanisms to be performed on a single column. Different mobile phase combinations using MeOH, BME, HNO3, and MPS were employed for this purpose.
Other key parameters affecting the separation of Hg and Se species, such as the pH and the use of non-ionic eluent modifiers such as MeOH, were also investigated as proposed by Pohl et al. [14].
Method validation approach
The simultaneous speciation method was validated by means of the accuracy profile approach, following a French standard [15, 16].
The trueness (expressed here by bias, %), the repeatability (expressed here by the coefficients of variation for repeatability (CVr), %), and the intermediate precision of the method (expressed here by the coefficients of variation for intermediate reproducibility (CVR), %) were assessed using the accuracy profile approach by means of CRM analysis or by spike recovery experiments.
The theoretical validity domain was defined between 0.035 μg/L and the highest concentration used for the calibration curve, namely 2.5 μg/L and 5.0 μg/L in the analysed Hg and Se solutions, respectively, for all species, except for Se(IV), for which the theoretical validity domain was defined from 0.07 to 5.0 μg/L. The lower concentrations of the theoretical validity domain were defined by estimating the limit of quantification (LOQ) of Se species and MeHg by the analysis of solutions of low concentrations of each species, before the accuracy profile experiments. The MeHg validity domain consisted of eight levels of concentration, constructed with the analysis of two CRMs (DOLT-5 and NMIJ 7402-a) and also spiked samples. For Se species, the validity domain consisted of six levels of concentration being all levels constructed by using spiked samples because CRMs for Se species in fish are not available.
For the spike experiments, samples were spiked (with Se species and MeHg) prior to extraction using spiking levels covering the theoretical validity domain, i.e., corresponding to concentrations at ½ × the estimated LOQ, the estimated LOQ, 2 × the estimated LOQ, and, also, spiking at concentrations corresponding to various points of the calibration curve.
The final LOQ was determined by the accuracy profile being the intersection point of the acceptance (λ) and tolerance limits (β). The acceptance limit λ is set according to the criteria expected of repeatability and intermediate precision, while β is set according to the desired level of confidence in the method’s accuracy. The limit of detection (LOD) is equal to 3/10 × LOQ.
Finally, the method uncertainties take into account the accuracy and precision of the method.
Application of the method to simultaneous speciation analysis of Hg and Se species in fishery product samples
Three marine predatory fish species (swordfish, Xiphias gladius, Linnaeus, identified for the first time in 1758; tuna, Thunnus albacares, Bonnaterre, 1788; and dogfish, Scyliorhinus canicular, Linnaeus, 1758) purchased from local fish markets and one freshwater species (rainbow trout, Oncorhynchus mykiss, Walbaum, 1792) obtained from the aquaculture facilities of the ANSES laboratory in Plouzané (France) were analysed. Based on the vendors’ information, the swordfish were caught in the Pacific and Indian Oceans, tuna were caught in the Indian Ocean, while dogfish originated from France, except for one sample that was caught in the Netherlands.
The dogfish was bought dressed (i.e., viscera, scales, head, tail, and fins removed) and its backbone was taken away. Swordfish and tuna were purchased as fillets (boneless sides of fish, without skin). Finally, rainbow trout was obtained in one piece (whole), and after cut into fillets. Five individuals of each fish species were obtained and each muscle was separately homogenised using a stainless steel free automatic mixer with titanium blades, giving a total of 20 fish muscle samples. Each sample was then freeze-dried and stored in the dark at room temperature until analysis. Subsamples of each fish muscle were taken to perform the different analytical procedures (total and speciation analysis).
Several internal quality controls (IQC) were put in place for the analytical quality assurance such as the five-point calibration standards to monitor linearity (r2 ≥ 0.995), a reagent blank to monitor possible cross-contamination or memory effects, daily analysis of two CRMs to check the method trueness, duplicates analysis to assess the repeatability, and the analysis of a standard solution every eight samples and at the end of the sequence to monitor instrument drift. Additionally, in order to assess the method applicability to the real samples, the samples were spiked at two levels with Se (0.5 and 1 µg/L) and with Hg species (0.15 and 0.2 µg/L).
Calculations
For mass balance (%) calculation, expressed as the sum of the Se or Hg quantified peaks relative to the tSe or tHg certified levels, the concentrations of unknown species were estimated using the calibration curve of the nearest eluting standard compound.
The extraction recovery (R, %) was calculated using the following equation:
where:
Cextract,i, level of the element “i” (Hg or Se) in the extract (mg/kg);
Ctotal certified,i, certified level of the element “i” (Hg or Se) in the CRM (mg/kg).
The identification of Se and Hg species in the samples was based on the comparison with the retention times of Se and Hg species in the standards and it was verified also by spike experiments.
The quantification of Hg and Se species was carried out by using the external calibration approach. All results are expressed in mg/kg of wet weight (ww). All the calculations were performed using Microsoft Excel. The chromatograms obtained by HPLC-ICP-MS were processed using OriginLab (OriginLab Corporation, Northampton, MA, USA).
Results and discussion
Optimisation of the extraction of Hg and Se species from fish muscle
The extraction recoveries of total levels of Hg and Se species from three CRMs (NMIJ 7402-a, ERM-CE101, and ERM-BB422) subjected to three different enzymatic hydrolysis procedures with 6.67 g/L of protease XIV in ammonium phosphate buffer and 500 µL of BME 10% (v/v) at 37 °C (final volume = 3 mL), collected in a final dilution volume of 25 mL, are shown in Fig. 1.

Extraction recoveries of Se and Hg in a final dilution volume of 25 mL, extracted by enzymatic hydrolysis using 6.67 g/L protease XIV in ammonium phosphate buffer and 500 µL of BME 10% (v/v) at 37 °C (the error bars represent the standard deviation, n = 2). Note: MW, microwave-assisted extraction
As can be seen in Fig. 1, enzymatic hydrolysis performed in oven (12 h) provided high extraction recoveries (> 90%) for total Se and Hg species in all CRMs tested, excepted for Hg in ERM-CE101 (63%). It is important to note that the BME addition will prevent SeMet oxidation and help Hg extraction, while mitigating Hg memory effects [5, 17].
MAEE was also successfully applied, achieving the best extraction recoveries within 60-min irradiation with 116% and 94% for total levels of Se and total Hg species in NMIJ 7402-a, 97% and 87% for Se and Hg in ERM-CE101, and 99% and 98% for Se and Hg in ERM-BB422. It is worth noting that some procedures performed with ERM-CE101 (Fig. 1) showed low reproducibility, probably due to the fact that this CRM is the only one that is fresh (and not freeze-dried as the others). Therefore, method optimisation was continued using only freeze-dried CRMs and samples.
As MAEE during 60 min gave the best extraction recoveries, tests to improve this procedure were carried out. Thus, the final dilution (with ultra-pure water) of the sample extract volume was optimised (25 and 50 mL) in order to achieve a maximum sensitivity while limiting the matrix effects: a final volume of 50 mL was found to be optimum in this respect, and improved repeatability has been achieved, as the column quickly saturated when using a more concentrated extract (25 mL). This simple approach improves sample throughput by eliminating the laborious standard addition technique [18]. In order to maintain the same BME concentration used in the 25-mL final dilution tests (0.2%), which has been shown to be effective in eliminating Hg memory effects [19], a small amount of BME was added after hydrolysis when using a 50-mL final dilution volume.
The pH of the extract solutions was adjusted at 1.5, by adding an adequate amount of HNO3 to achieve a HNO3 concentration of 3.59 g/L (570 µL HNO3 to 50 mL of final dilution volume), which assure that Se(IV) is present only as H2SeO3 form.
The final optimised conditions are therefore as follows: 0.02 g of sample extracted during 60 min by MAEE using 6.67 g/L protease XIV in ammonium phosphate buffer (pH ≅ 7) and 500 µL of BME in a final volume of 3 mL; after hydrolysis, the extracts’ pH is adjusted by addition of 570 µL of HNO3 and of 500 µL of BME (to obtain a final BME concentration of 0.2%) and finally, the extract mixture is completed with ultra-pure water to a final volume of 50 mL. The extraction recoveries for Se and Hg in NMIJ 7402-a (n = 2) obtained in these optimum conditions were 111% and 92%, respectively.
Optimisation of the HPLC separation of Hg and Se species in fish matrices
Reversed-phase HPLC
When employing RP-HPLC, the most efficient separation was obtained using a mobile phase consisting of 1% (v/v) TFA + 2% (v/v) MeOH in isocratic mode at a flow rate of 0.3 mL/min; MeHg (retention time = 2.0 min, not shown here) and all four Se species were efficiently separated but at a cost of a long analysis time in case of Se (≅ 60 min, see Fig. 2).

Chromatograms obtained for the analysis of NMIJ 7402-a and a standard mixture of Se(IV), Se(VI), SeMet, and SeCys at 0.5 μg/L (each species) by RP-HPLC-ICP-MS under the optimised conditions. U1 and U2 represent unknown species
When analysing NMIJ 7402-a, six Se species were separated and the average of total Se species mass balance (accounting the six Se species and comparing to the total certified Se level) was 57 ± 4% (n = 3). The main Se species found in this case were SeMet and Se(IV), but two unknown Se species were also separated (Fig. 2).
In case of the speciation analysis of Hg in NMIJ 7402-a, two Hg species were found, with MeHg being the major species. Comparing the certified Hg values of this CRM and the estimated quantification of the unknown Hg species, it can be assumed that the unknown Hg species is iHg. However, as stated also above, this RP-HPLC-based method has the disadvantage of being very time consuming, with a retention time for Se(IV) of ≅ 60 min.
Ion exchange HPLC
Two ion exchange columns, namely IonPac AS7 and IonPac CS5A, have also been tested with different mobile phase compositions containing MeOH, BME, HNO3, and MPS. The pH of the mobile phase used in this study was adjusted with 10 to 50 mM HNO3 solutions, tested in both ion-exchange columns. The optimum baseline separation was achieved with an IonPac CS5A column at 45 mM HNO3, which was used throughout the study. Using the IonPac AS7 column at this pH (45 mM HNO3, pH ≅ 1.5), the separation of the four target Se species was achieved, but MeHg and iHg were poorly separated. The addition of MeOH to the mobile phase at 1.0, 2.0, and 5.0% (v/v) was also tested. The use of 5% MeOH provided the best results, resulting in an increase of the Se and Hg detection sensitivity and better peak shapes. It is known that the addition of a carbon source, such as MeOH, can minimise or eliminate Se polyatomic interference up to a maximum concentration of 5%, above which it causes a loss of sensitivity due to a reduction in plasma energy [5, 7].
Further optimisations using IonPac CS5A were carried out for real sample analyses, including the use of a guard column, which slightly improves the resolution between unknown Se species found in the samples.
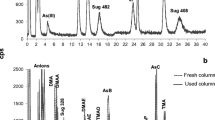
Figure 3 shows the chromatograms obtained for the analysis of a standard mixture of Se and Hg species and the NMIJ 7402-a CRM by the anion-cation exchange HPLC-ICP-MS method under the optimised conditions: the mobile phase consisted of a mixture of MeOH 5% (v/v), 45 mM HNO3 (pH ≅ 1.5), 0.015% (v/v) BME, and 1.5 mM MPS in isocratic mode, with a flow rate of 0.8 mL/min.

Anion-cation exchange HPLC-ICP-MS chromatograms obtained for the analysis of NMIJ 7402-a CRM and a standard mixture of MeHg and iHg at 5 μg/L and Se(IV), Se(VI), SeMet, and SeCys at 1 μg/L under optimised conditions
Good peak shapes and baseline separation were obtained with the developed anion-cation exchange HPLC method, except for iHg, which shows a broader peak (see Fig. 3). Regarding Hg mass balance, it was on average 103 ± 4% (n = 3). However, relatively low mass balances were achieved for Se species in the analysed CRMs: on average 20 ± 8% (n = 3) for NMIJ 7402-a due to the presence in fish of unknown Se species (U1 and U2 in Fig. 2), only detected by RP-HPLC-ICP-MS.
The analysis of the CRMs using two different chromatographic columns confirmed the results obtained with the anion-cation column for Se(IV) (0.242 ± 0.025 mg/kg) and SeMet (0.100 ± 0.013 mg/kg) as shown in Table 2. The levels of Se(VI) and SeCys were not quantified by either method.
Optimisation of the ICP-MS detection
The detection of Se by ICP-MS can be hindered by the formation of spectral interferences due to various matrix components, although it should be noted that when HPLC is used, the interfering species are generally separated from the target species, making the use of KED unnecessary as this approach degrades the overall sensitivity [20]. The 82Se isotope, which does not suffer from Ar-based interferences, was used in this study and a mathematical correction equation was employed to correct the potential isobaric interference of 82Kr+ naturally present in the plasma Ar. Unfortunately, 82Se can also be interfered by the presence of chloride (Cl), calcium (Ca), and bromine (Br) with the formation of polyatomic ions such as 12C35Cl35Cl+, which is the most significant [5].
In order to assess the possible interference arising from Cl on the chromatographic separation and ICP-MS detection, solutions of HCl in the range of 0, 0.05, 0.1, 0.5, 1.0, and 2.0% (v/v) were analysed in the same conditions as the samples. Under the optimum HPLC-ICP-MS conditions (detection of 82Se), one single peak, most probably due to 12C35Cl35Cl+ interference, was eluted at 0.6 min, which is significantly different than the retention times of the target Se species. Therefore, the 12C35Cl35Cl+ interference on the measurement of Se species does not hamper the analysis of real samples.
Novel HPLC-ICP-MS method for simultaneous speciation of Se and Hg in fish samples
The purposed novel HPLC-ICP-MS method, which allows the quantification of four Se species (Se(IV), Se(VI), SeMet, and SeCys) and of MeHg in fish samples, consists of the simultaneously extraction by MAEE in 60 min using 6.67 g/L protease XIV in ammonium phosphate buffer (pH ≅ 7) and 500 µL of BME (final volume of 3 mL). After hydrolysis, a pH adjustment in extracts and addition of 500 µL of BME should be performed, before dilution to a final volume of 50 mL. These solutions will be then separated and detected by HPLC-ICP-MS (in less than 15 min) by using one single column with both anion and cation exchange mechanisms (IonPac CS5A) and a mobile phase consisting of a mixture of methanol 5% (v/v), 45 mM HNO3, 0.015% BME, and 1.5 mM MPS.
Method validation by means of the accuracy profile approach
In this work, the method was validated using the accuracy profile approach, whose principle was described in detail elsewhere [2, 15, 16, 21]. The accuracy profiles obtained for MeHg and the four Se species are provided in Fig. 4.

Accuracy profile for the validation of a method for the simultaneous speciation analysis of Se(IV) (a), SeCys (b), Se(VI) (c), SeMet (d), and MeHg (e) in fishery products by anion-cation exchange HPLC-ICP-MS following microwave-assisted enzymatic extraction
As already mentioned, eight levels of concentrations were tested for MeHg (by the use of CRMs and spiked samples) and six levels for all Se species (by the use of spiked samples).
The probability β was set to 80%, for both MeHg and Se species. This means that the risk of expected results falling outside these limits is < 20%. The λ values for MeHg, SeMet, and Se(VI) were set to ± 20%, which corresponds to the intermediate precision coefficient of variation estimated at 6.7% for a coverage factor (k) of k = 3 (p = 99%). For SeCys and Se(IV), λ was set to ± 15% and to ± 30%, respectively. As can be seen in Fig. 4, β values are comprised within the acceptability limits.
The LOQs corresponding to the intersection of the λ and the β were 0.853 μg/L for Se(IV), 0.074 μg/L for SeCys, 0.055 μg/L for Se(VI), 0.072 μg/L for SeMet, and 0.038 μg/L for MeHg.
The figures of merit of the purposed method are summarised in Table 3. It is worth to note that some figures of merit (bias, CVr, CVR, and uncertainties) are dependent on the analyte concentration level. The values reported correspond to the higher value obtained among the concentration levels validated.
The LOQs expressed in mg/kg ww for the analysis of fish samples with an average moisture content of 30% are 0.149 mg/kg ww for Se(IV), 0.013 mg/kg ww for SeCys, 0.010 mg/kg ww for Se(VI), 0.013 mg/kg ww for SeMet, and 0.006 mg/kg ww for MeHg.
The bias (%) corresponding to the validity domain ranged from − 11.3% to 2.8%, whereas the CVr varied from 0.5 to 3.0%, while the CVR ranged from 2.6 to 7.0%.
The expanded uncertainty (U) at a k = 2 (p = 95%), which is calculated using k and the combined uncertainty (uc) values and the results, ranged from 6.1% (MeHg) and 15.7% (Se(VI)).
The figures of merit including linearity, specificity (obtained by spiking experiments), and repeatability of the method were satisfactory for the determination of Se(IV), SeMet, SeCys, Se(IV), and MeHg in fish with the proposed method.
Application of the method to simultaneous speciation analysis of Hg and Se species in fishery product samples
The novel HPLC-ICP-MS method developed in this study for simultaneous speciation analysis of Hg and Se species in fishery products was applied to the analysis of a selection of fish muscle samples. Good recoveries (results within the confidence interval of the theoretical spiked standard value which corresponds to k × CVR, using a k = 3) were obtained for the determination of all species in the spiked samples. The tSe and tHg levels were also determined in these samples and both the speciation and total data are presented in Table 4.
Regarding Se species, Se(IV) and SeCys levels were below the LOQ in all samples analysed in this study. Se(VI) was quantified in all fish species in the range 0.005–0.009 mg/kg ww except in rainbow trout, whereas SeMet was quantified in all the analysed samples (0.011 to 0.054 mg/kg ww) as the most abundant species, followed by Se(VI) (see Table 4). Similar levels of Se(VI) were found in the three predatory fish species. The lowest Se species levels were found in the freshwater fish. Our results are in good agreement with those previously published, as various authors reported that Se is mainly found in SeMet form in fish muscle, while inorganic species, such as Se(IV) or Se(VI), are not usually present in fish tissues [12, 22, 23]. However, the SeMet levels found in this study are lower than of those reported in other studies [22, 24]. Several factors such as fish size, age, gender, habitat, and migration patterns make it unlikely to obtain fish of the same species with the same range of Se and Hg levels [25].
The mean levels of MeHg (Table 4) varied between 0.026 and 1.77 mg/kg, and MeHg was the only or major species present in all fish species analysed. The lowest MeHg concentration was found in the freshwater rainbow trout (0.026 mg/kg), while the highest levels were found in swordfish (1.77 mg/kg), exceeding the maximum level of 1 mg/kg established for total Hg in this fish species [26].
The highest MeHg levels are generally found at the top of the aquatic food chain, i.e., in predatory fish such as sharks, swordfish, and tuna [27,28,29,30]. The MeHg concentrations found in the other fish species are below the regulatory limit fixed for the different fish species and are in good agreement with previously published data [31, 32]. Inorganic Hg was quantified in only some samples of two species: swordfish and dogfish. Others authors have also identified iHg in these fish species [32, 33].
Conclusions
This study reports the development and validation of a new analytical method for simultaneous speciation analysis of Se and Hg in fish muscle. To overcome the long duration of conventional enzymatic hydrolysis (generally during several hours up to 24 h), the extraction was developed and validated using a closed microwave system using a specific design for enzymatic extractions (37 °C) and the magnetically stirring option during the enzymatic reaction hence achieving a quantitative extraction yield during maximum 1 h. In addition, the proposed system devoted to small volume samples allowed simultaneous analysis of 64 samples in a single batch hence providing a very high sample throughput.
An efficient HPLC separation (using a simple isocratic elution) was also achieved in less than 15 min by using a mixed anion-cation exchange mechanism.
The HPLC-ICP-MS speciation method was validated by means of the accuracy profile approach, which is one of the most robust method validation approaches. Very low quantification limits and excellent repeatability and intermediate reproducibility were obtained for both Hg and Se species.
The method was successfully applied to simultaneous speciation analysis of Hg and Se in one freshwater fish (rainbow trout) and three marine/predatory species (swordfish, tuna, and dogfish) showing a high variation of the Hg and Se levels. This demonstrates that the proposed method allows rapid, simple, accurate, and precise simultaneous speciation analysis of Se and Hg in complex biological matrices; hence, it can be applied to further studies related to Hg-Se antagonism in various fish species.
References
Dang F, Wang WX. Antagonistic interaction of mercury and selenium in a marine fish is dependent on their chemical species. Environ Sci Technol. 2011. https://doi.org/10.1021/es103705a.
Ribeiro M, Zephyr N, Silva JA, Danion M, Guérin T, Castanheira I, Leufroy A, Jitaru P. Assessment of the mercury-selenium antagonism in rainbow trout fish. Chemosphere. 2022. https://doi.org/10.1016/j.chemosphere.2021.131749.
Gajdosechova Z, Mester Z, Feldmann J, Krupp EM. The role of selenium in mercury toxicity – current analytical techniques and future trends in analysis of selenium and mercury interactions in biological matrices. Trends Analyt Chem. 2018. https://doi.org/10.1016/j.trac.2017.12.005.
Clémens S, Monperrus M, Amouroux D, Guérin T, Donard O. Mercury speciation in seafood using isotope dilution analysis: a review. Talanta. 2012. https://doi.org/10.1016/j.talanta.2011.12.064.
Jagtap, R, Maher, W. Determination of selenium species in biota with an emphasis on animal tissues by HPLC–ICP-MS. 2016. https://doi.org/10.1016/j.microc.2015.07.014.
Li YF, Chen C, Li B, Wang Q, Wang J, Gao Y, Zhao Y, Chai Z. Simultaneous speciation of selenium and mercury in human urine samples from long-term mercury-exposed populations with supplementation of selenium-enriched yeast by HPLC-ICP-MS. J Anal At Spectrom. 2007. https://doi.org/10.1039/B703310A.
Favilli L, Giacomino A, Malandrino M, Inaudi P, Diana A, Abollino O. Strategies for mercury speciation with single and multi-element approaches by HPLC-ICP-MS. Front Chem. 2022. https://doi.org/10.3389/fchem.2022.1082956.
Marcinkowska M, Barałkiewicz D. Multielemental speciation analysis by advanced hyphenated technique - HPLC/ICP-MS: a review. Talanta. 2016. https://doi.org/10.1016/j.talanta.2016.08.034.
Maher M, Krikowa F, Ellwood W, Foster S, Jagtap R, Raber G. Overview of hyphenated techniques using an ICP-MS detector with an emphasis on extraction techniques for measurement of metalloids by HPLC–ICPMS. Microchem J. 2012. https://doi.org/10.1016/j.microc.2012.03.017.
Moreno F, García-Barrera T, Gómez-Ariza J. Simultaneous analysis of mercury and selenium species including chiral forms of selenomethionine in human urine and serum by HPLC column-switching coupled to ICP-MS. Analyst. 2010. https://doi.org/10.1039/c0an00090f.
Moreno F, García-Barrera T, Gómez-Ariza J. Simultaneous speciation and preconcentration of ultra trace concentrations of mercury and selenium species in environmental and biological samples by hollow fiber liquid phase microextraction prior to high performance liquid chromatography coupled to inductively coupled plasma mass spectrometry. J Chromatogr A. 2013. https://doi.org/10.1016/j.chroma.2013.02.083.
Sele V, Ørnsruda R, Sloth JJ, Amlund H, Berntssena MHG. Selenium and selenium species in feeds and muscle tissue of Atlantic salmon. J Trace Elem Med Biol. 2018. https://doi.org/10.1016/j.jtemb.2018.02.005.
Ghosn M, Chekri R, Mahfouz C, Khalaf G, Guérin T, Amara R, Jitaru P. Toward a routine methodology for speciation analysis of methylmercury in fishery products by HPLC coupled to ICP-MS following the validation based on the accuracy profile approach. Int J Environ Anal Chem. 2020. https://doi.org/10.1080/03067319.2020.1767095.
Pohl CA, Stillian JR, Jacks PE. Factors controlling ion-exchange selectivity in suppressed ion chromatography. J Chromatogr A. 1997. https://doi.org/10.1016/S0021-9673(97)00705-X.
AFNOR. NF V03-110 Analysis of Agri-Foodstuffs. Protocol of Characterization for the Validation of a Quantitative Method of Analysis by Construction of an Accuracy Profile. 2010. https://www.boutique.afnor.org/en-gb/standard/nf-v03110/analysis-ofagrifoodstuffs-protocol-of-characterization-for-the-validation-/fa159944/35346. Accessed 16 May 2023.
Mermet JM, Granier G. Potential of accuracy profile for method validation in inductively coupled plasma spectrochemistry. Spectrochim Acta B. 2012. https://doi.org/10.1016/j.sab.2012.06.003.
López I, Cuello S, Cámara C, Madrid Y. Approach for rapid extraction and speciation of mercury using a microtip ultrasonic probe followed by LC–ICP-MS. Talanta. 2010;82(2):594–9. https://doi.org/10.1016/j.talanta.2010.05.013.
Leufroy A, Noël L, Dufailly V, Beauchemin D, Guérin T. Determination of seven arsenic species in seafood by ion exchange chromatography coupled to inductively coupled plasma-mass spectrometry following microwave assisted extraction: method validation and occurrence data. Talanta. 2011. https://doi.org/10.1016/j.talanta.2010.10.050.
Harrington CF, Merson SA, D’ Silva TM. Method to reduce the memory effect of mercury in the analysis of fish tissue using inductively coupled plasma mass spectrometry. Anal Chim Acta. 2004. https://doi.org/10.1016/j.aca.2003.10.046.
Bednar AJ, Kirgan R, Jones W. Comparison of standard and reaction cell inductively coupled plasma mass spectrometry in the determination of chromium and selenium species by HPLC–ICP–MS. Anal Chim Acta. 2009. https://doi.org/10.1016/j.aca.2008.10.050.
Chevallier E, Chekri R, Zinck J, Guérin T, Noel L. Simultaneous determination of 31 elements in foodstufs by ICP-MS after closed-vessel microwave digestion: method validation based on the accuracy profile. J Food Compos Anal. 2015;41:35–41.
Moreno P, Quijano MA, Gutiérrez AM, Pérez-Conde MC, Cámara C. Study of selenium species distribution in biological tissues by size exclusion and ion exchange chromatagraphy inductively coupled plasma–mass spectrometry. Anal Chim Acta. 2004. https://doi.org/10.1016/j.aca.2004.02.029.
Jagtap R, Maher W, Krikowa F, Ellwood MJ, Foster S. Measurement of selenomethionine and selenocysteine in fish tissues using HPLC-ICP-MS. Microchem J. 2016. https://doi.org/10.1016/j.microc.2016.04.021.
Vicente-Zurdo D, Gómez-Gómez B, Pérez-Corona MT, Madrid Y. Impact of fish growing conditions and cooking methods on selenium species in swordfish and salmon fillets. J Food Compost Anal. 2019. https://doi.org/10.1016/j.jfca.2019.103275.
Burger J, Gochfeld M. Selenium and mercury molar ratios in saltwater fish from New Jersey: individual and species variability complicate use in human health fish consumption advisories. Environ Res. 2012;114:12–23. https://doi.org/10.1016/j.envres.2012.02.004.
European Commission. Commission regulation (EU) 2023/915 of 25 April 2023 on maximum levels for certain contaminants in food and repealing Regulation (EC) No 1881/2006. Official Journal of the European Union. 2006. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A02023R0915-20230810. Accessed 16 May 2023.
Liao W, Wang G, Zhao W, Zhang M, Wu Y, Liu X, Li K. Change in mercury speciation in seafood after cooking and gastrointestinal digestion. J Hazard Mater. 2019. https://doi.org/10.1016/j.jhazmat.2019.03.093.
Deng DF, Teh F, Teh SJ. Effect of dietary methylmercury and seleno-methionine on Sacramento splittail larvae. Sci Total Environ. 2008. https://doi.org/10.1016/j.scitotenv.2008.08.028.
Kumari S, Amit Jamwal R, Mishra N, Singh DK. Recent developments in environmental mercury bioremediation and its toxicity: a review. Environ. Nanotechnol. Monit Manag. 2020. https://doi.org/10.1016/j.enmm.2020.100283.
Mateu MB, Llovet MI, Bonancia BM, Roig JLD, Linares-Vidal V. Effects of cooking process on the concentrations of mercury, selenium and GPx activity in Tuna (Thunnus thynnus). Toxicol Lett. 2015. https://doi.org/10.1016/j.toxlet.2015.08.228.
FDA. Mercury concentrations in fish from the FDA Monitoring Program (1990–2010). 2010. https://www.fda.gov/food/environmental-contaminants-food/mercury-food-and-dietary-supplements. Accessed 26 June 2023.
Zmozinski AV, Carneado S, Ibáñez-Palomino C, Sahuquillo A, López-Sánchez JF, Silva MM. Method development for the simultaneous determination of methylmercury and inorganic mercury in seafood. Food Control. 2014. https://doi.org/10.1016/j.foodcont.2014.05.054.
Shao LJ, Gan WE, Su QD. Determination of total and inorganic mercury in fish samples with on-line oxidation coupled to atomic fluorescence spectrometry. Anal Chim Acta. 2006. https://doi.org/10.1016/j.aca.2006.01.039.
Acknowledgements
This study is a contribution to the Ph.D. research project (MERSEL-FISH) and it was jointly funded by ANSES (France) and INSA (Portugal).
Author information
Authors and Affiliations
Contributions
Conceptualisation: Mariana Ribeiro, José Armando Luísa da Silva, Isabel Castanheira, Axelle Leufroy, and Petru Jitaru. Data curation: Mariana Ribeiro, Isabel Castanheira, Axelle Leufroy, and Petru Jitaru. Formal analysis: Mariana Ribeiro and Eleonora Galli. Funding acquisition: Isabel Castanheira, Axelle Leufroy, and Petru Jitaru. Investigation: Mariana Ribeiro, Isabel Castanheira, and Petru Jitaru. Methodology: Mariana Ribeiro, Eleonora Galli, José Armando Luísa da Silva, Isabel Castanheira, Axelle Leufroy, and Petru Jitaru. Project administration: Isabel Castanheira and Petru Jitaru. Resources: Thierry Guérin, Isabel Castanheira, and Petru Jitaru. Supervision: Isabel Castanheira, Axelle Leufroy, and Petru Jitaru. Validation: Mariana Ribeiro, Eleonora Galli, José Armando Luísa da Silva, Isabel Castanheira, Axelle Leufroy, and Petru Jitaru. Roles/writing—original: Mariana Ribeiro. Writing—review and editing: Thierry Guérin, Isabel Castanheira, Axelle Leufroy, and Petru Jitaru.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ribeiro, M., Galli, E., Guérin, T. et al. Simultaneous speciation analysis of Hg and Se in fish by high-performance liquid chromatography and inductively coupled plasma-mass spectrometry following microwave-assisted enzymatic hydrolysis. Anal Bioanal Chem 415, 7175–7186 (2023). https://doi.org/10.1007/s00216-023-04984-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-023-04984-1