
Abstract
Testosterone (TTe) and free testosterone (FTe) are clinically important indicators for the diagnosis of androgen disorders, so accurate quantitative determination of them in serum is clinically of paramount significance. Currently, there is no available method suitable for routine and simultaneous measurement of TTe and FTe. Here, we developed a new UPLC-MS/MS method to quantify serum TTe and FTe simultaneously and accurately. Rapid equilibrium dialysis was used to obtain FTe in serum followed by derivatization with hydroxylamine hydrochloride. With these strategies, TTe and FTe could be measured in single injection. After optimizing the extraction and derivatization conditions, the performance of LC–MS/MS was evaluated and applied to quantify the levels of TTe and FTe in clinical samples from 42 patients. The assays were linear for TTe within the range of 0.2–30 ng/mL and for FTe within the range of 1.5–1000 pg/mL. This improved method provided a limit of quantification for TTe of 0.2 ng/mL and for FTe of 1.5 pg/mL. The intra- and inter-run CVs were less than 4.3% and 3.6% for TTe and less than 8.2% and 6.7% for FTe, respectively. The intra- and inter-run accuracies for both TTe and FTe were in the range of 96.1–108.1%. Interference, carryover effect, and matrix effect were in acceptable range. In conclusion, our new LC–MS/MS method is simple to perform and can serve as a reliable method for simultaneous determination of TTe and FTe in clinical practice, providing important information for diagnosis, treatment, and monitoring of androgen-related diseases.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
As steroid hormone from the androgen group, circulating testosterone (TTe) in the blood exists in the free and protein-bound forms, with high affinity to sex hormone-binding globulin (SHBG) and with lower affinity to albumin [1]. TTe is reported to affect multiple functions including pubertal growth, bone, muscle mass, erythropoiesis, autoimmunity, and cognition [2,3,4,5,6]. Analysis of TTe is routinely used to assess polycystic ovary syndrome, male hypogonadism, and menopausal women. Free testosterone (FTe), accounting for 0.5–4% of the TTe, is referred to as metabolically active fraction because only free form can penetrate the cell membrane to interact with the androgen receptor and hence is of physiological significance [7,8,9]. It has been reported that FTe could improve precision in assessment of androgenic status than TTe, especially in female hyperandrogenemia and androgen-deficient male [10, 11]. Considering that TTe and FTe levels are clinically meaningful, developing specific methods with adequate sensitivity and accuracy to determine them is necessary.
Based on the antigen–antibody reaction, immunoassays gave desirable results for the TTe concentrations in men and were extensively used in clinical lab to measure TTe [12, 13]. But they lacked the required sensitivity and specificity to precisely measure the TTe in women and children due to the lower TTe levels of women and children than the levels of men. To be specific, immunoassays were highly susceptible to interferences, mostly due to the cross-reactivity of other similar steroids, resulting in overestimation of TTe levels in women and children [14,15,16]. For FTe, it is still a challenging task for immunoassays to directly determine FTe levels. Thus, its concentrations were calculated based on the quantitative results for TTe, albumin, and SHBG by the use of mathematical models. However, the results of different mathematical models always showed big differences, preventing their widespread implementation in clinical use [17,18,19].
With the development of liquid chromatography tandem mass spectrometry (LC–MS/MS) technique, LC–MS/MS has been demonstrated to be more accurate than immunoassays and has been recognized as the most reliable method for measuring sex hormones [20,21,22,23,24,25,26]. So far, several papers have been published for the measurement of TTe or FTe in serum with LC–MS/MS using different techniques for sample preparation [7, 27,28,29,30]. In these studies, TTe and FTe were tested separately, which would certainly extend the testing time of each sample. To our best knowledge, there is currently no report available for the simultaneous measurement of serous TTe and FTe. In order to reduce the turnaround time, improve the analytical throughput, and provide more informative results in each run, it is desirable to develop methods for simultaneous assessment of serous TTe and FTe.
Here, we developed and validated a simple, sensitive, and accurate LC–MS/MS method for simultaneously measuring TTe and FTe in serum. By using hydroxylamine reagent to react with FTe, we successfully measured TTe and FTe in a single run, which could not only shorten the detection time, but also provide more clinically valuable information for the diagnosis, treatment, and monitoring of androgen-related diseases.
Materials and methods
Materials and serum samples
Testosterone (purity > 98%), the isotope internal standard (IS) testosterone-13C3, dehydroepiandrosterone (DHA), methoxyamine hydrochloride (MOA), hydroxylamine hydrochloride (HOA), formic acid (FA), acetonitrile (ACN, LC/MS grade), and methanol (MeOH, LC/MS grade) were bought from Sigma-Aldrich (St. Louis, MO, USA). Androsterone, ( +)-epiandrosterone, androstanolone, and etiocholanolone were purchased from Beijing Tanmo Quality Inspection Technology Co., Ltd. 1-Testosterone was bought from Shanghai Acmec Biochemical Co., Ltd.
Methyl tert-butyl ether (MTBE) was purchased from Macklin Biochemical Co. Ltd. (Shanghai, P. R. China). Ultrapure water was made in-house using a Milli-Q Advantage ultrapure water system (Millipore, MA, USA). The hormone-free human serum was purchased from Shanghai Pufeng Biotechnology. The HEPES buffer used for equilibrium dialysis was adapted from literature [27]. Rapid equilibrium dialysis (RED) devices with a molecular weight cutoff of 8 kDa were purchased from Thermo Fisher Scientific (Rockford, IL, USA). The serum samples for method validation were the remnants of anonymized and previously analyzed serum samples in the Center for Medical Genetics & Molecular Diagnosis, Shenzhen Second People’s Hospital. All the procedures performed in the studies involving clinic specimens were in accordance with the ethical standards and were approved by the Ethics Committee of Shenzhen Second People’s Hospital (approval number 2023–026-01PJ).
Sample preparation
Preparation of stock and working solutions
The calibrators were prepared from MeOH stock solution of a concentration of 100 µg/mL for all the chemical standards and were stored at − 20 °C. Subsequent dilution with hormone-free human serum gave a series of working calibrators (0.2, 0.5, 2, 5, 10, 20, and 30 ng/mL) for TTe. For FTe, 1.5 pg/mL, 5 pg/mL, 10 pg/mL, 25 pg/mL, 100 pg/mL, 250 pg/mL, and 1000 pg/mL were prepared with HEPES buffer. Quality control (QC) samples for TTe were achieved by spiking 100 μL of hormone-free human serum with the working solution mixtures: high-level QC (HQC, 22.5 ng/mL), medium-level QC (MQC, 12 ng/mL), low-level QC (LQC, 3.5 ng/mL), and ultralow-level QC (ULQC, 0.35 ng/mL). QC samples for FTe were achieved by spiking 150 μL of HEPES with the calculated amount of the working solution mixtures: HQC (650 pg/mL), MQC (420 pg/mL), LQC (40 pg/mL), and ULQC (3 pg/mL). The IS working solution was prepared with MeOH at the following concentrations: 15 ng/mL T-13C3 for TTe, 300 pg/mL T-13C3 for FTe [31].
Sample preparation of total testosterone
One hundred microliters of working calibrators, QC samples, or serum samples was transferred into a 1.5-mL transparent polypropylene tube, and then, 500 μL of protein precipitation solution containing 50% ACN, 50% MeOH, and the internal standard was added respectively to all tubes, followed by vortexing for 5 min for protein precipitation. Subsequently, the mixture was centrifuged (14,000 g, 10 min, 4 °C), and then, 500 μL of supernatant was transferred to clean 1.5-mL transparent polypropylene tubes and dried in a vacuum concentrator at 30 °C for 1 h. The residue for each sample was reconstituted in 120 μL of 50% MeOH.
Sample preparation of free testosterone
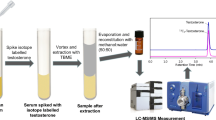
Three hundred microliters of calibrators, QCs, and human serum samples were added to the sample chamber of the RED device inserts and dialyzed for 4 h at 37 °C against 550 μL of modified HEPES buffer in buffer chamber. Then, 150 μL of dialysate in buffer chamber and 10 μL IS were added to each tube, respectively. The process of derivatization reaction for free testosterone was similar to previously published protocol but with little modification [27]. Briefly, standards, QCs, and samples were derivatized with 0.04 M HOA on a vortex mixer at 65 °C for 1 h. Then, samples were cooled for 10 min at 4 °C to stop the derivatization reaction. After that, samples were diluted with 200 μL of ddH2O before extracting with 900 μL MTBE. Following centrifugation at 14,000 g for 10 min at 4 °C, 750 μL of the organic phase was transferred to a 1.5-mL clean tube and dried down in vacuum centrifugal at 30 °C for 20 min. The residues were redissolved in 110 μL of the TTe solutions obtained in this section. After centrifuging at 14,000 g for 10 min at 4 °C, 100 μL supernatant was transferred to a clean 96-well plate for analysis by LC–MS/MS. Figure 1 shows the schematic procedure of sample multiplexing for TTe and FTe quantification.

The schematic procedure of sample preparation for TTe and FTe quantification
Liquid chromatography tandem mass spectrometry
Samples were analyzed on a Triple Quad 6500 + LC–MS/MS system connected to an Exion AD system (SCIEX, Singapore). The chromatographic separation was obtained on an Acquity BEH C18 column (2.1 × 50 mm id. 1.7 μm, Waters, USA) protected with a 1.7-μm Vanguard Pre-Column in front. The column temperature was kept at 40 °C. Mobile phases consisted of (A) water with 0.1% (v/v) FA and 5% (v/v) acetonitrile, and (B) MeOH. Gradient conditions at a flow rate of 0.2 mL/min were used as follows: 0.0–2.0 min, 40% B; 2.0–2.5 min, linear from 40 to 100% B; 2.5–4.8 min, 100% B; 4.8–5.1 min, linear from 100 to 10% B; 5.1–5.8 min, 10% B; 5.8–6.0 min, linear from 10 to 40% B; 6.0–7.0 min, 40% B. Thirty microliters of the final mixture was injected into LC–MS/MS. The electrospray ionization (ESI) source temperature was set at 550 °C and the spray voltage was + 5500 V. The curtain and collision gas were set at 35 and 10, respectively. The ion source gas 1 and gas 2 were held at 50 psi. For MRM, the dwelling time was set to be 35 ms. The MRM transitions parameters for TTe, FTe, and their internal standards are shown in Table 1. The first MRM transitions were used for quantitation while the second MRM transitions were utilized for identification. All the data were collected by Analyst® 1.7.2. and analyzed by SCIEX-OS software (version 2.0.1). Standard curves were obtained by plotting the peak area ratios of TTe/TTe-13C3 and FTe/FTe-13C3 against the homologous concentrations using weighted least-squares regression analysis with 1/x2.
Method validation
Based on the C62-A document generated from the Clinical and Laboratory Standards Institute (CLSI) [32], the method was validated for linearity, limit of quantitation, precision, accuracy, stability, interference check, matrix effect, recovery, and carryover as described in the following sections.
Linearity, limit of quantification, and limit of detection
The calibration graphs were obtained by the use of least-squares linear regression (y = ax + b), where x was the concentration in pg/mL of TTe and FTe, y was the ratio of peak area of analytes to those of the corresponding IS, a was the slope, and b was the intercept. Linear correlation coefficient r2 with weighting factor of 1/x2 was calculated to evaluate the linearity. The limit of detection (LOD) and the limit of quantification (LOQ) were determined based on signal-to-noise (S/N) ratio of 3:1 and 10:1, respectively [33]. Of note, LOQ should be determined with acceptance criteria of ± 20% precision and accuracy.
Intra- and inter-assay precision and accuracy
For determining precision and accuracy, QC samples prepared at four concentration levels were analyzed. To assess the intra-day and inter-day precision and accuracy of this method, QC samples at four levels were analyzed in five replicates on the same day and over three different days, respectively. Precision was expressed as the CV of experimentally measured concentrations. The mean value for precision should be less than 15%. Accuracy was expressed as percentage by measured concentration/actual concentration ratio.
Stability
Sample stability was evaluated using ULQC, LQC, MQC, and HQC levels under different storage or processing conditions. To assess the short-term stability of these QC samples, they were analyzed against fresh calibrators after being stored at room temperature for 24 h. Similar assessment method was employed to evaluate mid-term stability of QC samples when they were stored at 4 °C for 1 week. Freeze–thaw stability of QC samples was assessed for three cycles. For each of three cycles of freeze–thaw, QC samples were frozen at − 20 °C overnight and subsequently thawed at room temperature. For the assessment of long-term stability, QC samples were frozen at − 80 °C for 180 days. The measured concentration of QC samples was compared with the corresponding theoretical values. Post-treatment stability was determined by placing the TTe and the derivatized FTe at sample injection chamber at 4 °C for 84 h. The CVs of samples at each level should be less than 15%.
Interference check and matrix effect
Neat solutions of dehydroepiandrosterone, 1-testosterone, androsterone, ( +)-epiandrosterone, androstanolone, and etiocholanolone were prepared by dilution from stocks using 100% MeOH for interference checking. A pooled serum specimen from five females and five males with a known TTe (2809 pg/mL) and FTe (148 pg/mL) concentration was spiked with 30 ng/mL of each steroidal compound and was then assayed following the protocol described in the “Sample preparation of total testosterone” and “Sample preparation of free testosterone” sections. The calculated concentration of each spiked serum sample was then compared to that of the neat serum sample.
To verify the influence of the matrix on the determination of TTe and FTe, matrix effects (MEs) were evaluated by comparing the area response of analytes in post-extraction spiked hormone-free human serum or HEPES buffer with the area of analytes in neat MeOH at three concentration levels. The equation ME = B/A was used to calculate MEs, where B refers to the peak area ratios of the analytes to ISs in hormone-free human serum or HEPES buffer and A represents the peak area ratios of the analytes to ISs in samples prepared by MeOH. The ME values should be between 85 and 115%.
Recovery and carryover
The recovery for TTe was evaluated by spiking known testosterone standard into a serum specimen, while recovery for FTe was assessed by spiking testosterone with HEPES buffer. The low, medium, and high spiked levels for Te were 1.0 ng/mL, 6 ng/mL, and 15 ng/mL, respectively; and for FTe, they were 20 pg/mL, 100 pg/mL, and 400 pg/mL, respectively. When performing the recovery experiment, spiked and non-spiked samples were analyzed in five replicates, and recovery was expressed as a percentage by calculating the ratio of the measured spiked concentration to the actual added concentration. Recovery should be within the range from 85 to 115%. Carryover was assessed by calculating the ratio of the average response value of blank serum to the average response value of the highest calibration standard (30 ng/mL of TTe, 1000 pg/mL of FTe). It is worth to note that the blank serum should be injected directly after the highest calibrators.
Results and discussion
LC–MS/MS development
To achieve the goal of determining TTe and FTe in serum at the same time, an alternative approach was to derivatize one of them. As FTe has a much lower concentration in human serum, we tried two derivative reagents HOA and MOA to enhance FTe’s sensitivity. As shown in Electronic Supplementary Material Fig. S1, HOA provided a relative better sensitivity of FTe than MOA; therefore, we chose HOA as the derivative reagent for the rest of experiments. After choosing HOA, we optimized the LC conditions by increasing the flow rate and changing the mobile phase gradients (Fig. S2). With this optimization, the retention times of Te and HOA-Te were decreased, while the undesirable carryover appeared when LC time was decreased to 5 min (Fig. S3). To balance the carryover and LC time, we finally chose 7 min as our LC time and 0.2 mL/min as the flow rate. As shown in Fig. 2, TTe and FTe can be clearly identified by mass spectrometry according to their corresponding mass-to-charge ratio even though they have the same retention time. Interestingly, distinct levels of TTe and FTe between the female and male patients could be shown from the chromatography profiles. Moreover, as only background noise was observed for TTe when injecting a blank sample with ISs (Fig. 2i), effect of ISs on the determination of TTe and FTe could be ignored under our experimental condition.

Representative chromatograms of TTe, FTe, and the corresponding ISs were shown in different samples. (a), (c), and (f) represent FTe in male serum, female serum, and standard solution, respectively. (b), (d), (h), and (i) represent TTe in male serum, female serum, standard solutions, and blank with ISs, respectively. (e) and (g) represent FTe-IS and TTe-IS in standard solutions
Optimization of derivatization procedures
To analyze the TTe and FTe simultaneously, the prerequisite condition was to mix the TTe and HOA-FTe before injection. But whether the remaining HOA in FTe solution would react with TTe and thus affected the quantitative detection of TTe and FTe remained to be investigated. When we directly mixed the TTe solution and FTe solution, the peak area of FTe in pooled serum samples was increased (Fig. 3a) while the peak area of TTe was decreased (Fig. 3b) with the increase of storage time at 4 °C. This indicated that the remaining HOA still reacted with TTe at this temperature though with a low reaction rate even at the condition of low HOA concentration (0.001 mol/L). It should be noted that stable peak area obtained for TTe at the condition of 0.001 mol/L HOA, suggesting that this level was insufficient for the derivation of FTe while higher HOA concentrations (0.01 mol/L and 0.1 mol/L) augmented the decrease of TTe peak area. When adding the procedure of removing excessive derivatization reagent after derivative reaction, both TTe and FTe were stable (CVs% < 8%) at storage time of up to 84 h for each HOA level as shown in Fig. 3c and d. Considering that relative better CVs of TTe and FTe were obtained at concentrations of 0.04 mol/L, this concentration was used in the following experiments.

Changes FTe and TTe peak area under different reaction conditions at 4 °C as function of storage times, mixing (a) FTe and (b) TTe directly and (c) mixing FTe and (d) TTe after removing excess HOA
Method validation
Linearity, LOD, and LOQ
In this study, seven levels of calibrators were used to generate the calibration curves for TTe and FTe. For the ranges of 0.2–30 ng/mL (TTe) and 1.5–1000 pg/mL (FTe), the linearity exhibited excellent correlation (r2 > 0.99) for the two analytes. The representative linear regressions are presented in Fig. 4a (TTe) and Fig. 4b (FTe). The LOD and LOQ of TTe and FTe were 0.05 ng/mL and 0.2 ng/mL and 0.5 pg/mL and 1.5 pg/mL, respectively (Fig. 5). Notably, the LOQs of TTe and FTe were reliably determined with ± 20% precision and accuracy. Moreover, LOQs of the present method were still superior or comparable to those of previously reported methods but with single injection [7, 27, 30], indicating that the LOQs of our newly developed method could well meet the requirement for clinical practice.

Calibration curves of peak area ratio versus concentration of (a) TTe and (b) FTe

Representative MRM transition chromatograms of (a) LOD and (b) LOQ for TTe and FTe
Precision and accuracy
As shown in Table 2, the precision of intra-day and inter-day was less than 15% for TTe and FTe at each concentration level. The intra-day and inter-day accuracy values for TTe and FTe ranged from 91.4 to 102.9% and from 96.1 to 114.7%, respectively. Therefore, both the precision and accuracy were well within acceptable limits, laying a basis for precise and accurate quantification of the TTe and FTe in human serum.
Stability
As shown in Table 3, stability of the studied TTe and FTe in the QC samples was evaluated at four concentration levels. Stability was assessed at different storage conditions expressed as accuracy. Short-term stability for 24 h at room temperature was found to be within the range of 93.7–102.6% for TTe and 93.3–110% for FTe, respectively. Mid-term stability at 4 °C for 1 week was 96.7–100.3% for TTe and 89.3–107.8% for FTe. QC samples were found to be stable over three freeze–thaw cycles at − 20 °C with an accuracy between 91.8 and 111.6% for both TTe and FTe. Moreover, long-term stability at − 80 °C was found in the range of 92.0113% for TTe and FTe. In addition, post-treatment stability results suggested that the injection solution was stable for at least 84 h at 4 °C (Fig. 3c, d). All these data affirmed that QC samples were stable under the experimental conditions.
Interference check, matrix effect, recovery, and carryover
Of the 6 steroidal compounds tested, the accuracies in the concentrations of the neat and spiked sample were all in acceptable range (Table 4), suggesting that the interferences from 6 steroidal compounds were negligible, and the method was specific for both analytes. The matrix effect of samples at different concentration levels was assessed and the results are shown in Table 5. The MEs were 95–98% and 109–114% for TTe and FTe, respectively. The CVs% of the MEs for FTe and TTe were all less than 15%, indicating that the matrix effect was acceptable. The recovery results were within the range of 92.7–102.6% for TTe and 85.7–110.8% for FTe (Table 6). When analyzing a blank sample injected right after the highest calibrator sample, carryover was 0.06% for FTe and 0.09% for TTe, indicating that no carryover existed in the detection of a series of samples.
Application
Using this newly established method, we assessed the concentrations of both TTe and FTe in the residual serum samples collected from 23 males and 19 females. The differences between genders were compared and the results are shown in Fig. 6. As expected, TTe and FTe were significantly higher in the male groups compared to the female group (Fig. 6a, b), which was in accordance with the physiological conditions. Figure 6c exhibits that there was no significant difference in FTe% between the female and male groups but nevertheless the levels are in agreement with the expected range of approximately 0.5–4% in human. These results indicate that the established method is quite sensitive and can meet the requirements in simultaneously and accurately measuring TTe and FTe for clinical samples.

Serum levels of (a) TTe, (b) FTe, and (c) FTe% in male and female groups. Significance is controlled by the p-value (independent two-sample t test, two-sided) and minimum fold change indicated by the cutoff curve. The median is drawn as a horizontal line (***p < 0.0005)
Discussion
Accurate measurement of TTe and FTe concentrations is pivotal for the diagnosis of hypogonadism and other androgen disorders. For many years, traditional immunoassays have been the commonly used method for testing TTe to diagnose androgen disorders in clinical labs. However, these immunoassays are often criticized due to their poor precision and inaccuracy when assessing the level of TTe in hypogonadal men, women, and children. With its technical superiority in sensitivity, specificity, accuracy, and simultaneous analysis of multiple compounds, LC–MS/MS has been increasingly adopted by clinical laboratories and has become a highly recommended method for quantifying the sex steroids. According to the updated endocrine society clinical practice guideline, FTe has been recommended to be measured when serum TTe is close to or below the low limit of the physiological range or when the level of SHBG is abnormal [13, 34]. Moreover, FTe has been suggested to be preferred biomarkers than TTe in the diagnosis of androgen-based diseases [35, 36]. Based on this, we have developed a LC–MS/MS method for the simultaneous detection of TTe and FTe.
Compared with TTe, the detection of FTe usually requires ultrafiltration or equilibrium dialysis steps to obtain the free forms. Although pretreatment steps of ultrafiltration are time-efficient and straightforward, the gold standard procedure for FTe measurement is still equilibrium dialysis coupled to LC–MS/MS. This is because equilibrium dialysis can maximally mimic in vivo conditions and obtain the intrinsic protein interaction but without applying external forces (e.g., pressure, centrifugal force) [37]. However, the overnight pretreatment of conventional equilibrium dialysis method is time-consuming and has hurdled its wide application in the laboratory. Also, equilibrium dialysis does not match well with the high-throughput analytical performance of mass spectrometry when analyzing only one parameter (i.e., FTe) in single run. Although calculating free testosterone can be readily accessible, its accuracy and precision are affected by the accuracy of TTe and SHBG quantification [9, 38]. Therefore, it is conceivable that good correlation between calculated FTe and measured FTe for women can only be achieved when LC–MS/MS methods are used for measuring TTe [39]. Theoretically, the LC–MS/MS analytical throughput depends on the time spent on the LC separation. If TTe and FTe need to be analyzed separately, the analytical time almost needs to be doubled compared to measure them simultaneously in a single run.
The sensitivity of testosterone can be improved by derivatization, which lays a foundation for the simultaneous analysis of TTe and FTe through molecular weight difference [28]. Moreover, the introduction of sample multiplexing has effectively enhanced the detection throughput by analyzing multiple samples in a single run. To be specific, the sample multiplexing is achieved by combining different samples after deriving by differential derivatization reagents and then separated by LC–MS/MS via unique ion pairs. With the development of rapid equilibrium dialysis, the FTe forms can be obtained within less than 4 h [40], providing the possibility of increasing throughput and clinical application of FTe via LC–MS/MS method. To assure good sensitivity of FTe measurement, we dissolved TTe and FTe one after another with the same solution to avoid over-dilution. With the aid of this strategy, the detection limit of FTe can reach down to 0.5 pg/mL. As thus, we developed a high-throughput assay for accurate quantification of TTe and FTe in serum without offsetting analytical characteristics, which lays a technical foundation for the wide application for simultaneously measuring TTe and FTe in clinical laboratories.
Conclusion
In conclusion, the presented study focuses on the development and validation of an UPLC-MS/MS method that can simultaneously quantify TTe and FTe in human serum. With the aid of HOA derivatization, the sensitivity of FTe was improved, thus allowing a simultaneous detection of TTe and FTe solutions within acceptable range after combining TTe and FTe together. The results of 42 clinical serum samples showed that the performance of this new method is sufficient for clinical application. Our data provide a practical and clinical method to quantify total and free Te in serum at the same time, which would improve the accuracy for diagnosing testosterone-related diseases.
References
Van Uytfanghe K, Stöckl D, Kaufman JM, Fiers T, Ross HA, De Leenheer AP, Thienpont LM. Evaluation of a candidate reference measurement procedure for serum free testosterone based on ultrafiltration and isotope dilution–gas chromatography–mass spectrometry. Clin Chem. 2004;50(11):2101-2110. https://doi.org/10.1373/clinchem.2004.037358.
Basaria S. Male hypogonadism. The Lancet. 2014;383(9924):1250–63. https://doi.org/10.1016/S0140-6736(13)61126-5.
Handelsman DJ, Hirschberg AL, Bermon S. Circulating testosterone as the hormonal basis of sex differences in athletic performance. Endocr Rev. 2018;39(5):803–29. https://doi.org/10.1210/er.2018-00020.
Mirand EA, Gordon AS, Wenig J. Mechanism of testosterone action in erythropoiesis. Nature. 1965;206(4981):270–2.
Agretti P, Pelosini C, Bianchi L, Grosso AD, Saba A, Canale D, Sessa MR. Importance of total and measured free testosterone in diagnosis of male hypogonadism: immunoassay versus mass spectrometry in a population of healthy young/middle-aged blood donors. J Endocrinol Invest. 2021;44(2):321–6. https://doi.org/10.1007/s40618-020-01304-7.
Salameh WA, Redor-Goldman MM, Clarke NJ, Reitz RE, Caulfield MP. Validation of a total testosterone assay using high-turbulence liquid chromatography tandem mass spectrometry: total and free testosterone reference ranges. Steroids. 2010;75(2):169–75. https://doi.org/10.1016/j.steroids.2009.11.004.
Rhea JM, French D, Molinaro RJ. Direct total and free testosterone measurement by liquid chromatography tandem mass spectrometry across two different platforms. Clin Biochem. 2013;46(7):656–64. https://doi.org/10.1016/j.clinbiochem.2013.01.005.
Kanakis GA, Tsametis CP, Goulis DG. Measuring testosterone in women and men. Maturitas. 2019;125:41–4. https://doi.org/10.1016/j.maturitas.2019.04.203.
Goldman AL, Bhasin S, Wu FCW, Krishna M, Matsumoto AM, Jasuja R. A reappraisal of testosterone’s binding in circulation: physiological and clinical implications. Endocr Rev. 2017;38(4):302–24. https://doi.org/10.1210/er.2017-00025.
Grassi G, Polledri E, Fustinoni S, Chiodini I, Ceriotti F, D'Agostino S, Filippi F, Somigliana E, Mantovani G, Arosio M, Morelli V. Hyperandrogenism by liquid chromatography tandem mass spectrometry in PCOS: focus on testosterone and androstenedione. J Clin Med. 2020;10(1). https://doi.org/10.3390/jcm10010119.
Bhasin S, Cunningham GR, Hayes FJ, Matsumoto AM, Snyder PJ, Swerdloff RS, Montori VM. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocr Metab. 2010;95(6):2536–59. https://doi.org/10.1210/jc.2009-2354.
Huhtaniemi IT, Tajar A, Lee DM, O’Neill TW, Finn JD, Bartfai G, Boonen S, Casanueva FF, Giwercman A, Han TS, Kula K, Labrie F, Lean MEJ, Pendleton N, Punab M, Silman AJ, Vanderschueren D, Forti G, Wu FCW. Comparison of serum testosterone and estradiol measurements in 3174 European men using platform immunoassay and mass spectrometry; relevance for the diagnostics in aging men. Eur J Endocrinol. 2012;166(6):983–91. https://doi.org/10.1530/EJE-11-1051.
Guzelce EC, Galbiati F, Goldman AL, Gattu AK, Basaria S, Bhasin S. Accurate measurement of total and free testosterone levels for the diagnosis of androgen disorders. Best Pract Res Cl En. 2022;36(4):101683. https://doi.org/10.1016/j.beem.2022.101683.
Welker KM, Lassetter B, Brandes CM, Prasad S, Koop DR, Mehta PH. A comparison of salivary testosterone measurement using immunoassays and tandem mass spectrometry. Psychoneuroendocrino. 2016;71:180–8. https://doi.org/10.1016/j.psyneuen.2016.05.022.
Moal V, Mathieu E, Reynier P, Malthièry Y, Gallois Y. Low serum testosterone assayed by liquid chromatography-tandem mass spectrometry. Comparison with five immunoassay techniques. Clin Chim Acta. 2007;386(1):12–19. https://doi.org/10.1016/j.cca.2007.07.013.
La’ulu SL, Kalp KJ, Straseski JA. How low can you go? Analytical performance of five automated testosterone immunoassays. Clin Biochem. 2018;58:64–71. https://doi.org/10.1016/j.clinbiochem.2018.05.008.
de Ronde W, van der Schouw YT, Pols HAP, Gooren LJG, Muller M, Grobbee DE, de Jong FH. Calculation of bioavailable and free testosterone in men: a comparison of 5 published algorithms. Clin Chem. 2006;52(9):1777–84. https://doi.org/10.1373/clinchem.2005.063354.
Van Uytfanghe K, Stöckl D, Kaufman JM, Fiers T, De Leenheer A, Thienpont LM. Validation of 5 routine assays for serum free testosterone with a candidate reference measurement procedure based on ultrafiltration and isotope dilution–gas chromatography–mass spectrometry. Clin Biochem. 2005;38(3):253–61. https://doi.org/10.1016/j.clinbiochem.2004.12.001.
Raverot V, Lopez J, Grenot C, Pugeat M, Déchaud H. New approach for measurement of non-SHBG-bound testosterone in human plasma. Anal Chim Acta. 2010;658(1):87–90. https://doi.org/10.1016/j.aca.2009.10.057.
Seger C, Salzmann L. After another decade: LC–MS/MS became routine in clinical diagnostics. Clin Biochem. 2020;82:2–11. https://doi.org/10.1016/j.clinbiochem.2020.03.004.
Desai R, Harwood DT, Handelsman DJ. Simultaneous measurement of 18 steroids in human and mouse serum by liquid chromatography–mass spectrometry without derivatization to profile the classical and alternate pathways of androgen synthesis and metabolism. Clin Mass Spectrom. 2019;11:42–51. https://doi.org/10.1016/j.clinms.2018.12.003.
Liu D, Zhao R, Zhao S, Wang Z, Liu R, Wang F, Gao Y. A developed HPLC-MS/MS method to quantitate 5 steriod hormones in clinical human serum by using PBS as the surrogate matrix. J Chromatogr B. 2021;1186:123002. https://doi.org/10.1016/j.jchromb.2021.123002.
Chen F, Cheng Z, Wang Z, Peng Y, Wang B, Guo W, Pan B. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) based assay for the simultaneous quantification of 18-hydroxycorticosterone, 18-hydroxycortisol and 18-oxocortisol in human plasma. J Chromatogr B. 2022;1188:123030. https://doi.org/10.1016/j.jchromb.2021.123030.
Gravitte A, Archibald T, Cobble A, Kennard B, Brown S. Liquid chromatography–mass spectrometry applications for quantification of endogenous sex hormones. Biomed Chromatogr. 2021;35(1):e5036. https://doi.org/10.1002/bmc.5036.
Yuan T-F, Le J, Cui Y, Peng R, Wang S-T, Li Y. An LC-MS/MS analysis for seven sex hormones in serum. J Pharmaceut Biomed. 2019;162:34–40. https://doi.org/10.1016/j.jpba.2018.09.014.
Yu S, Zou Y, Yin Y, Yu J, Li Q, Xie S, Luo W, Ma X, Wang D, Qiu L. Establishing and verifying a robust liquid chromatography-tandem mass spectrometry method to simultaneously measure seven androgens present in plasma samples. Separations. 2022;9(11). https://doi.org/10.3390/separations9110377.
Chen Y, Yazdanpanah M, Wang XY, Hoffman BR, Diamandis EP, Wong PY. Direct measurement of serum free testosterone by ultrafiltration followed by liquid chromatography tandem mass spectrometry. Clin Biochem. 2010;43(4–5):490–6. https://doi.org/10.1016/j.clinbiochem.2009.12.005.
Star-Weinstock M, Williamson BL, Dey S, Pillai S, Purkayastha S. LC-ESI-MS/MS analysis of testosterone at sub-picogram levels using a novel derivatization reagent. Anal Chem. 2012;84(21):9310–7. https://doi.org/10.1021/ac302036r.
Colletti JD, Redor-Goldman MM, Pomperada AE, Ghoshal AK, Wu WW, McPhaul MJ, Clarke NJ. Sample multiplexing: increased throughput for quantification of total testosterone in serum by liquid chromatography-tandem mass spectrometry. Clin Chem. 2020;66(9):1181–9. https://doi.org/10.1093/clinchem/hvaa117.
Zhang X, Xu H, Zhou C, Yang L, Zhai S, Yang P, Zhao R, Li R. Magnetic solid phase extraction followed by in-situ derivatization with core–shell structured magnetic graphene oxide nanocomposite for the accurate quantification of free testosterone and free androstenedione in human serum. J Chromatogr B. 2022;1196:123188. https://doi.org/10.1016/j.jchromb.2022.123188.
Lie M, Thorstensen K. A precise, sensitive and stable LC-MSMS method for detection of picomolar levels of serum aldosterone. Scand J Clin Lab Inv. 2018;78(5):379–85. https://doi.org/10.1080/00365513.2018.1480060.
Chen F, Cheng Z, Peng Y, Wang Z, Huang C, Liu D, Wang B, Pan B, Guo W. A liquid chromatography-tandem mass spectrometry (LC-MS/MS)-based assay for simultaneous quantification of aldosterone, renin activity, and angiotensin II in human plasma. J Chromatogr B. 2021;1179:122740. https://doi.org/10.1016/j.jchromb.2021.122740.
He B, Di X, Guled F, Harder AVE, van den Maagdenberg AMJM, Terwindt GM, Krekels EHJ, Kohler I, Harms A, Ramautar R, Hankemeier T. Quantification of endocannabinoids in human cerebrospinal fluid using a novel micro-flow liquid chromatography-mass spectrometry method. Anal Chim Acta. 2022;1210:339888. https://doi.org/10.1016/j.aca.2022.339888.
Bhasin S, Brito JP, Cunningham GR, Hayes FJ, Hodis HN, Matsumoto AM, Snyder PJ, Swerdloff RS, Wu FC, Yialamas MA. Testosterone therapy in men with hypogonadism: an Endocrine Society* clinical practice guideline. J Clin Endocr Metab. 2018;103(5):1715–44. https://doi.org/10.1210/jc.2018-00229.
Li Y, Zhai Y, Li L, Lu Y, Su S, Liu Y, Xu Z, Xin M, Zhang Q, Cao Z. Divergent associations between serum androgens and ovarian reserve markers revealed in patients with polycystic ovary syndrome. Front Endocrinol. 2022;13:881740. https://doi.org/10.3389/fendo.2022.881740.
Cao Z, Lu Y, Cong Y, Liu Y, Li Y, Wang H, Zhang Q, Huang W, Liu J, Dong Y, Tang G, Luo YR, Yin C, Zhai Y. Simultaneous quantitation of four androgens and 17-hydroxyprogesterone in polycystic ovarian syndrome patients by LC-MS/MS. J Clin Lab Anal. 2020;34(12):e23539. https://doi.org/10.1002/jcla.23539.
de Boer T, Meijering H. Equilibrium dialysis, ultracentrifugation, and ultrafiltration in LC-MS bioanalysis. In: Sample preparation in LC-MS bioanalysis. 2019; pp 45–51. https://doi.org/10.1002/9781119274315.ch3.
Schuijt MP, Sweep CGJ, van der Steen R, Olthaar AJ, Stikkelbroeck NMML, Ross HA, van Herwaarden AE. Validity of free testosterone calculation in pregnant women. Endocr connect. 2019;8(6):672–9. https://doi.org/10.1530/EC-19-0110.
Antonio L, Pauwels S, Laurent MR, Vanschoubroeck D, Jans I, Billen J, Claessens F, Decallonne B, De Neubourg D, Vermeersch P, Vanderschueren D. Free testosterone reflects metabolic as well as ovarian disturbances in subfertile oligomenorrheic women. Int J Endocrinol. 2018;2018:7956951–7956951. https://doi.org/10.1155/2018/7956951.
Waters NJ, Jones R, Williams G, Sohal B. Validation of a rapid equilibrium dialysis approach for the measurement of plasma protein binding. J Pharm Sci. 2008;97(10):4586–95. https://doi.org/10.1002/jps.21317.
Funding
Financial support was provided by the National Natural Science Foundation of China (Grant No. 22004086), Shenzhen Science and Technology Innovation Commission (Grant No. JCYJ20210324115601005), and the Guangdong Province Basic and Applied Basic Research Regional Joint Fund-Youth Fund Project (Grant No. 2021A1515110849).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethics approval
The study was approved by the Ethics Committee of Shenzhen Second People’s Hospital.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Huang, R., Hong, Y., Wu, Y. et al. Simultaneous quantification of total and free testosterone in human serum by LC–MS/MS. Anal Bioanal Chem 415, 6851–6861 (2023). https://doi.org/10.1007/s00216-023-04963-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-023-04963-6