
Abstract
The present study was intended to develop suitable methods for unambiguous identification and determination of ethyl (1-(diethylamino)ethylidene) phosphoramidofluoridate (known as A234-Novichok) biomarkers in urine and plasma samples. Multiple biomarkers were investigated for the first time, to verify intoxication by the A234-Novichok agent, using sensitive and accurate techniques including gas and liquid chromatography-tandem mass spectrometry (GC–MS/MS and LC–MS/MS). Like other nerve agents, in biological matrices, the A234-Novichok agent reacts with several proteins to form related adducts. Considering this, two different protein adduct biomarkers in blood samples were analyzed, and the regenerated A234 was determined. Two-dimensional chromatography and solid-phase extraction techniques were employed for blood sample preparation. Limits of detection for butyrylcholinesterase (BChE) adduct, the regenerated A234, and albumin covalent adduct were determined and reported as 1, 1, and 10 ng mL−1, while the related calibration curves were linear within the range of 2–100, 2–100, and 15–100 ng mL−1, respectively. The detection limit and linear range for the intact agent in the urine sample were determined as 0.1 and 1–100 ng mL−1, respectively. Since A234 and some other Novichok chemicals have been added to the Schedule 1 of the Chemical Weapons Convention (CWC), Annex on Chemicals, after UK incidents, the analytical methods developed in this work might be used for verification purposes, as well as OPCW Biomedical Proficiency Tests.
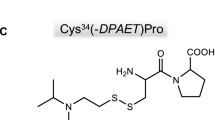
Graphical abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Extremely toxic organophosphorus G-type chemical nerve agents (NAs) such as tabun (GA), sarin (GB), cyclosarin (GF), and soman (GD), as well as phosphonothiolates (V-type nerve agents) consisting of VX, Russian VX, Chinese VX, etc., belong to the category of chemical warfare agents (CWAs). A trace amount of these agents might lead to severe physiological impacts and even death of the exposed person [1, 2]. In comparison with G-type NAs, the extremely low vapor pressure and more toxicity of V-type agents cause high persistency of such agents, and hence increased hazard via percutaneous route [3].
A novel category of NAs known as Novichok (A-series) was developed by the Soviet Union scientists during a top-secret program between the 1970s and 1990s [4]. The principal objective of this program was to introduce a new generation of NAs with higher toxicity, compared to V-agents, which would not be detected using standard NATO CWA detectors [5]. These compounds would be effective by absorption through the skin and mucous membranes, inhalation, and ingestion [3]. Based on information disclosed by Mirzayanov [6] and Hoenig [7], Novichok compounds are prepared from substituted phosphoroamidates (the first tested Novichok compound was N-2-diethylaminomethylacetoamido-methylphosphonofluoridate (Scheme S1-a) A230) and fluorophosphates (Scheme S1-b). According to Mirzayanov [6], Petr Kirpichev and his colleagues have synthesized and tested other Novichok chemicals including A232, A234, A242, and A262 (Scheme S1-a) [6].
Studies on the fourth generation of chemical warfare agents, Novichoks [5, 8, 9], have shown that A230 and A232 are almost 8 and 10 times more potent and deadlier compared to Russian VX and soman, respectively (as presented in Table S1, Supplementary Information) [9]. Though Novichok agents have never yet been used on battlefields, they have attracted lots of attention following the poisoning of Sergei Skripal and his daughter in Salisbury, and two others in Amesbury, the UK, in 2018 [1]. Novichok compound had never been declared under the Chemical Weapons Convention (CWC), and not appeared in CWC Annex on Chemicals, before UK incidents. On 27 November 2019, the 24th conference of state parties to the CWC approved amendment of the treaty’s Annex on chemicals, adding Novichok and carbamate NAs (1.A.13 to 1.A.16) [10], the first changes to the CWC Annex on Chemicals since the treaty came into force in 1997 [11, 12]. Another strange incident was the poisoning of Alexei Navalny (Russian political activist) in August 2020 by a cholinesterase inhibitor Novichok agent [13].
In forensic investigations, it would be required to identify the biomarkers of a toxic substance. Identification of the biomarkers allows for the understanding of the mechanisms of toxicity and selection of adequate treatments for victims of the agent. Analysis of biomedical samples looking for the known biomarkers might reveal the involved toxic chemicals in an incident and provide unambiguous evidence to be presented to a court of justice. Therefore, the main purposes of this study were (a) to introduce biomarkers for the Novichok (A234, 1.A.14) agent and (b) to develop an optimized procedure for the detection and determination of the known biomarkers in complex biological media such as urine and plasma.
Inspired by the reactions of other NAs in biological samples [14,15,16], we speculated that Novichoks would react with some proteins to form protein adduct metabolites. Phosphorylation and phosphonylation of proteins are two of the main metabolic pathways of these agents, which occur by releasing a leaving group from the agent so as to covalently bond to proteins, e.g., through serine OH-moiety in AChE and BChE, or tyrosine residues in human serum albumin.
To the best of our knowledge, until now, few scientific publications have provided experimental data regarding the identity of these toxic chemicals. Lee et al. studied the degradation products and fragmentation pathways of Novichok A234 in aqueous solutions at different pH levels by LC–MS/MS [17]. More recently, an anti-BChE antibody conjugated to magnetic beads was applied to the isolation of some Novichok agent BChE adducts before high-resolution mass spectrometry (HRMS) analysis as reported by Noort et al. [18] and Jeong et al. [19]. Besides BChE, albumin adduct is of essential interest for the verification of CWA exposure due to the high abundance of albumin in human blood, its relative ease of sampling, and its high stability in vivo (half-life of 20 days) [20].
There are several methods to confirm the exposure of NAs in biological samples:
-
1)
ChE activity measurement via Ellman assay [21, 22] (described in Supplementary Information);
-
2)
Regeneration of deactivated proteins using potassium fluoride: nucleophiles such as fluoride ions can react with phosphorylated proteins to release the agent. The released agent can be identified using GC–MS/MS or LC–MS/MS;
-
3)
Enzymatic digestion of the proteins and subsequently LC–MS/MS analysis of the resulting peptides, looking for phosphorylated moieties [23]; and
-
4)
Determination of intact agent or its corresponding hydrolysis metabolites in urine or plasma samples.
All the abovementioned strategies were considered in the present study, and appropriate methods were optimized and employed to verify A234 intoxication, while cheaper and more accessible instruments were utilized. Due to the low concentrations of the biomarkers in biomedical complex matrices, and to meet the identification criteria which are required for verification purposes at trace levels, MS/MS technique in multiple reaction monitoring (MRM) mode was utilized under optimized conditions for unambiguous identification and quantitative analysis of the selected biomarkers.
Experimental
Reagents and chemicals
All compounds used in this study were of analytical grade. Solvents including isopropanol, acetonitrile, ethyl acetate, formic acid (eluent additive for LC–MS), and acetone were supplied from Merck Co. (Darmstadt, Germany). HPLC-grade water (Merck, Darmstadt, Germany) was used for the preparation of aqueous solutions. Potassium fluoride (KF), ethylenediaminetetraacetic acid (EDTA), sodium acetate, ammonium bicarbonate (NH4HCO3), sodium azide, sodium sulfate, sodium hydroxide, S-butyrylthiocholine (BTC), Sepharose CL-4B, procainamide hydrochloride, ε-aminocaproic acid, and cyanogen bromide were procured from Sigma-Aldrich (St. Louis, MO, USA). 5,5′-Dithiobis (2-nitrobenzoic acid) (DTNB, Ellman’s reagent) was supplied from ACROS (NJ, USA). Anion exchanger Q-Sepharose fast flow gel was purchased from GE Healthcare Co. (Uppsala, Sweden).
Solid-phase extraction (SPE) Oasis HLB (400 mg, 3 mL) and DSC-18 (500 mg, 3 mL) cartridges were procured from Waters Co. (Milford, MA, USA) and SUPELCO (Bellefonte, PA, USA), respectively. Centrifugal ultrafilters (Amicon Ultra-10 and 50 kD) were purchased from Merck-Millipore (Bedford, MA).
Lyophilized human serum butyrylcholinesterase (HuBChE, B4186), pepsin (P6887) from porcine gastric mucosa, and pronase E (P6911) from Streptomyces griseus were purchased from Sigma-Aldrich (St. Louis, MO, USA). The enzyme assay was performed using a UV–Visible double-beam spectrophotometer (Perkin-Elmer, 550S, Waltham. MA, USA).
A234 was synthesized with the purity of 90% through an in-house method and was subsequently characterized using GC–MS (Fig. S1 and S2, and Scheme S2 in Supplementary Information) and LC–MS techniques. d1-A234 (Ethyl (1-(diethylamino) 2-deuterioethylidene)phosphoramidofluoridate) was prepared to be used as an internal standard.
GC-CI/MS/MS analysis
GC-CI/MS/MS analysis was performed using an Agilent 7890A gas chromatograph system connected to a 7000-A triple quadruple mass spectrometer. HP-5 (5% Phenyl-95% methylpolysiloxane J&W Scientific, Folsom, CA) capillary GC column (30 m, 0.32 mm i.d., and film thickness of 0.25 µm) was employed for chromatographic separation. The optimized GC temperature program was started from 60 °C and held for 0 min, and then, it was increased to 280 °C by a ramp of 20 °C min−1. The column temperature was finally held at 280° for 4 min. The aliquots (1 µL) were introduced through a splitless injection port at 250 °C. Helium was used as the carrier gas at a constant flow rate of 1.5 mL min−1.
The ion source and quadrupole analyzer temperatures were set at 230 and 150 °C, respectively. The mass spectrometer was operated using chemical ionization (CI) source and isobutane was used as the reagent gas in positive mode. GC–MS/MS analysis was carried out using nitrogen as the collision gas. The collision energy (CE) was varied within the range of 10–30 eV and optimized. Data acquisition was performed under both scanning (at m/z range of 35–500) and MS/MS conditions in MRM mode. Data acquisition and data analysis were performed using the Agilent Technologies MassHunter workstation software QQQ GC–MS reversion: B.04.01.
LC–MS/MS analysis
LC–MS/MS analysis was performed using a 1200 LC system (Agilent, Waldbronn, Germany) coupled to a 6410B triple quadrupole tandem mass spectrometer (Agilent Technologies, Santa Clara, CA, USA). The LC column was a rapid resolution Zorbax eclipse XDB-C18 (150 mm × 3.0 mm I.D., 3.5 µm) (Agilent Technologies, Santa Clara, CA, USA). The column temperature was set at 25 °C. Gradient elution was applied using 20 mM formic acid solution in 95:5 (v/v) water-acetonitrile mixture (mobile phase A) and 20 mM formic acid solution in 95:5 (v/v) acetonitrile–water mixture (mobile phase B). For each of the biomarkers, a special elution program was used, which is mentioned in Table S2. The eluent flow rate was set at 0.25 mL min−1. Due to different biomarker concentrations, different injection volumes within the range of 3–15 µL were applied, using an autosampler. The electrospray ionization (ESI) voltage was set at 4 kV. Ultra-high pure nitrogen was used as nebulizing and collision gas. The heated capillary temperature was maintained at 300 °C. The drying gas flow rate and nebulizer gas pressure were set at 9 L min−1 and 206.85 kPa, respectively. In order to achieve higher sensitivity, fragmentation voltages and collision energies were optimized for each biomarker. LC–MS/MS analyses were performed in product ion scan and MRM mode. Detection and identification of regenerated A234, intact A234 as a urinary biomarker, A234-Tyr, and A234-BChE adducts was performed in the positive ion MRM mode using transitions from the [M + H]+ precursor ion to the two or three most abundant product ions: regenerated A234 and intact A234 in urine (m/z 225 → 74 and 124; CE 15 eV, 225 → 197; CE 10 eV), A234-Tyr adduct (m/z 386.2 → 244.3 and 285.3; CE 15 eV), and A234-BChE adduct (m/z 1000.4 → 778; CE 30 eV, 1000.4 → 673 and 602; CE 40 eV). Data acquisition and data analysis were performed using the Agilent Technologies MassHunter workstation software reversion: B.02.01.
Safety considerations
A234 is a toxic chemical compound and should be handled only by trained personnel via appropriate safety procedures. Skin or eye contact and accidental inhalation or ingestion should be seriously avoided. Preparation of standard solutions and spiked plasma samples should be performed in a well-ventilated fume hood and all solutions containing A234 must be decontaminated after use.
Preparation of standard solutions of A234
Stock solutions of A234 and d1-A234 (800 µg mL−1) were prepared in acetonitrile and stored at 4 °C in the dark. Working standard solutions (0.05 to 20 µg mL−1) were prepared freshly through sequential dilution of stock solutions with acetonitrile, and were spiked in volumes within the range of 20–100 µL which were always lower than 10% (v/v) into plasma and urine samples.
Incubation of human plasma and urine samples with A234
Human plasma and urine samples were provided by volunteers from the DCRL laboratory. Briefly, heparinized human blood samples were centrifuged (3000 g, 15 min). Plasma was separated from red blood cells and stored at − 18 °C. Since four different methods were developed in the present study for the analysis of urine and plasma samples, three procedures were followed to prepare spiked samples:
-
a)
Analysis of BChE adduct: to prepare 1, 2, 5, 10, 25, 50, and 100 ng mL−1 solutions of the spiked analytes in plasma, 40–100 µL of A234 and d1-A234 were added to 2 mL of plasma. The samples were incubated at 37 °C with end-over-end shaking for 2 h, and subsequently, the analytical processes were followed.
-
b)
Investigation of tyrosine adduct and fluoride regenerated agent: 20–100 µL of A234 and d1-A234 solutions were added to 1 mL of human plasma to prepare concentration levels within the range of 1–100 ng mL−1. Before analytical processes, the samples were incubated at 37 °C with end-over-end shaking for 48 h.
-
c)
A six-point calibration curve of A234 in the urine sample was constructed by preparation of 1, 5, 10, 25, 50, and 100 ng mL−1 A234 solutions in blank pooled human urine samples via the addition of 20–100 µL of A234 working standard solutions to 1 mL of urine. Samples were incubated at 37 °C with end-over-end shaking for 2 h.
All blank samples were prepared by addition of 100 µL of acetonitrile to 1 mL of urine and plasma samples (2 mL for analysis of BChE adduct). Samples were further processed as described below.
Enzyme activity assay
Cholinesterase activity of plasma samples was monitored using the Ellman method [21]. The reaction mixture containing 2.8 mL of phosphate buffer (100 mM, pH 7.4), 20 µL of plasma sample, 100 µL of DTNB (15 mM solution), and 100 µL of BTC (30 mM solution) was placed in a quartz cuvette with a path length of 1 cm. The absorption change of this solution was measured at 412 nm, after 5-min delay.
Fluoride regeneration
Two different methods were used for the removal of free or unbound A234 from plasma samples to minimize the risk of false-positive identification:
In the first method, 3 mL of acetone was added to 1 mL of spiked plasma sample to precipitate the proteins. The mixture was then vortexed for 30 s and centrifuged at 3000 g for 5 min at room temperature. The supernatant was gently removed without disturbing the protein pellet. For regeneration of bounded A234, the protein pellet was re-suspended in 1 mL of acetone. Then, the mixture was centrifuged at 3000 g for 5 min, and the supernatant was discarded. This process was repeated twice. Finally, the precipitate was dried under a mild nitrogen gas stream. The protein pellet was dispersed completely, followed by the addition of 3 mL of 0.6 M KF in acetate buffer solution (0.2 M, pH 4). Next, the mixture was shaken gently for 3 h at room temperature and was subsequently centrifuged at 3000 g for 5 min. The supernatant was loaded on a 400-mg Oasis HLB extraction cartridge, preconditioned by 6 mL of ethyl acetate, 6 mL of isopropanol, and 6 mL of 0.6 M KF in acetate buffer solution, respectively. The cartridge was then washed with 3 mL of 0.6 M KF in acetate buffer and was subsequently dried under a gentle stream of nitrogen for 1 min. The regenerated agent was eluted with 4 mL of ethyl acetate, and then, it was collected in a glass tube containing sodium sulfate. Afterward, the extract was concentrated under a mild nitrogen stream to a final volume of 50 µL, and 1 µL of the sample was injected into GC-CI/MS/MS inlet for further analysis. Moreover, after the exchange of the solvent to acetonitrile (50 µL), a volume of 3 µL of the sample was injected into LC–MS/MS instrument.
In the second method, the free agent was separated from the protein-bound A234 by an initial filtration using a cutoff filter. For this purpose, 1 mL of the plasma sample was diluted with three volumes of 50 mM NH4HCO3 solution. A 4 mL of the diluted plasma was concentrated to approximately 500 µL using a 10-kD molecular weight cutoff (MWCO) centrifugal filter, which had been conditioned by 2 × 4 mL of 50 mM NH4HCO3 solution. The residue sample solution which was remained on the top of the filter was washed using 5 × 4 mL of 50 mM NH4HCO3 solution, then concentrated to 500 µL, and was subsequently transferred to a new vial. The filter was rinsed with 3 × 0.5 mL of 50 mM NH4HCO3 solution. The collected solution was added to the content of the previous vial. The solution was then treated with 3 mL of sodium acetate buffer solution in the presence of fluoride ion (at the final concentration of 0.6 M KF) to regenerate A234. The mixture was shaken gently for 3 h at room temperature, and then, it was centrifuged at 4000 g for 5 min. The supernatant was loaded on a 400-mg Oasis HLB cartridge. The SPE process was followed using the same procedure as that of the first method, and the resulting sample was analyzed using GC-CI/MS/MS and LC–MS/MS.
Purification of A234 and d1-A234-BChE adducts
BChE was purified from human plasma using two-dimensional chromatography: anion exchange at pH value of 4.0, followed by affinity chromatography on the procainamide-Sepharose gel.
Preparation of procainamide-Sepharose CL-4B affinity gel
Procainamide-Sepharose CL-4B affinity gel was prepared according to the procedure of Ralston et al. [24], with minor modifications to obtain a high content of procainamide-bound gel (described in Supplementary Information).
BChE isolation
A 5 mL of Sepharose Q fast flow gel was inserted in a 50-mL polypropylene test tube. The Sepharose Q fast gel was conditioned by 3 × 15 mL of acetate buffer solution (20 mM sodium acetate, 1 mM EDTA, pH 4). The buffer solution was removed by centrifuging the tube (2000 g for 5 min). A 2 mL of spiked plasma sample was diluted with 18 mL of acetate buffer and added to Sepharose Q fast. The mixture was incubated with gentle end-over-end mixing at 4 °C for 20 h. Then, it was centrifuged at 2000 g for 4 min, and the supernatant was removed. The Sepharose Q fast was washed using 4 × 20 mL of acetate buffer solution to reach the point that 280 nm absorbance of the eluent was less than 0.1. After that, the Sepharose Q fast was eluted with 2 × 8 mL of acetate buffer solution containing 200 mM sodium chloride. The supernatant was collected by centrifuging (2000 g for 2 min). The collected solution was loaded on a 50-kD MWCO centrifugal filter (15 mL) which had been conditioned by 2 × 15 mL of acetate buffer solution (20 mM sodium acetate, 1 mM EDTA, pH 4) and concentrated to 500 μL. The filter was washed with 2 × 2.5 mL of phosphate buffer solution (20 mM, 1 mM EDTA, pH 7) and concentrated to 1 mL. The remained solution on top of the filter was subsequently transferred to a vial. The filter was rinsed with 2 × 1 mL of phosphate buffer solution (20 mM, 1 mM EDTA, pH 7), and the collected solution was added to the content of the previous vial.
Affinity chromatography
A 3 mL of procainamide-Sepharose gel was taken in a 15-mL polypropylene tube and conditioned using 2 × 10 mL of phosphate buffer solution (20 mM, 1 mM EDTA, pH 7). The BChE solution obtained from BChE isolation (previous section) was immediately loaded onto the equilibrated procainamide-Sepharose gel and gently mixed at 4 °C for 1.5 h. The gel was washed with 2 × 8 mL of phosphate buffer solution (20 mM, 1 mM EDTA, pH 7) containing 150 mM NaCl. The buffer solution was removed by centrifuging (2000 g for 5 min). Finally, the gel was eluted with 2 × 8 mL of 2 M NaCl in phosphate buffer (20 mM, 1 mM EDTA, pH 7) and 16 mL of the eluate was collected. The collected solution was concentrated to about 500 μL using a 15 mL 50 kD MWCO centrifugal filter which has been already conditioned by 2 × 15 mL of phosphate buffer (20 mM, 1 mM EDTA, pH 7). The solution of the residue sample in the filter was washed with 4 × 15 mL of 50 mM ammonium bicarbonate and concentrated to 500 μL. The remained solution (500 μL) was transferred to a 4-mL glass vial. The filter was rinsed with 2 × 250 μL of 50 mM ammonium bicarbonate solution. The rinse fluid was combined with the remained solution and acidified using formic acid.
Digestion of isolated BChE with pepsin
The purified BChE was acidified with 2 × 15 µL of formic acid and digested with 8 mg of pepsin at 37 °C for 18 h. A DSC-18 cartridge (500 mg, 3 mL) was conditioned using 6 mL of 20 mM formic acid in acetonitrile followed by 6 mL of 20 mM formic acid in 95:5 (v/v) water-acetonitrile mixture. The digested solution was loaded on the SPE cartridge. The cartridge was washed with 3 mL of 20 mM formic acid solution in a 95:5 (v/v) water-acetonitrile mixture. Subsequently, the process of elution was carried out using 5 mL of 20 mM formic acid solution in a 10:90 (v/v) water-acetonitrile mixture. The eluent was concentrated to dryness under vacuum at 45 °C. The residue was re-dissolved in 100 µL of mobile phase A and a volume of 5 µL was injected into LC–MS/MS instrument.
As a reference standard, procured commercial lyophilized human serum BChE was dissolved in 1 mL of 50 mM NH4HCO3 solution, in its original vial, and 200 µL of this solution was treated by A234 and d1-A234 at a final concentration of 15 ng mL−1 for the agents. The resulting solution was incubated at 37 °C for 2 h. Protein digestion and SPE procedures were carried out using the same methods as those mentioned earlier.
Isolation and pronase E digestion of albumin adducts of metabolite
A 1 mL of treated plasma (see “Incubation of human plasma and urine samples with A234 (b)”) was added dropwise to 3 mL of acetone. Then, the mixture was centrifuged at 2000 g for 5 min. The supernatant was discarded and the pellet was washed with another 2 × 500 µL of acetone to separate the pellet from the intact agent and other interferents. Subsequently, the precipitate was dried under a mild nitrogen gas stream and re-dissolved in 1 mL of 50 mM NH4HCO3 solution. Then, 8 mg of pronase E enzyme was added to the solution of protein content and the solution was incubated at 37 °C while shaking for 6 h. After that, another portion containing 7 mg of pronase E enzyme was added and the solution was incubated at 37 °C while shaking for another 12 h. The reaction was quenched using 4 drops of formic acid, and the mixture was diluted with 2 mL aliquot of mobile phase A. The solution was centrifuged at 2000 g for 10 min. A 500-mg DSC-18 cartridge was conditioned using 6 mL of 20 mM formic acid in acetonitrile and 6 mL of 20 mM formic acid solution in 95:5 (v/v) water-acetonitrile mixture. The centrifuged sample solution was loaded onto the cartridge. The cartridge was washed with 3 mL of 20 mM formic acid in a 95:5 (v/v) water-acetonitrile mixture. The cartridge was eluted using 6 mL of 20 mM formic acid in a 70:30 (v/v) water-acetonitrile mixture. The solution was concentrated to dryness under vacuum at 45 °C. The residue was re-dissolved in 50 µL of mobile phase A, and a 15 µL aliquot of the resulting solution was injected into LC–MS/MS instrument for further analysis.
Isolation of human urine biomarker
An aliquot of 1 mL of the spiked urine sample (“Incubation of human plasma and urine samples with A234 (c)”) was introduced to an Oasis HLB cartridge (400 mg, 3 mL) which was preconditioned using 6 mL of acetonitrile and 6 mL of water. The cartridge was washed using 3 mL of water followed by 3 mL of 75:25 (v/v) water-acetonitrile mixture and then dried under a gentle stream of nitrogen gas. Next, the cartridge was eluted with 4 mL of 50:50 (v/v) water-acetonitrile mixture. The collected solution was concentrated to dryness under vacuum at 45 °C, and the residue was re-dissolved in 50 µL of mobile phase A, and 3 µL aliquot of this sample was injected into LC-ESI/MS/MS system for further analysis.
Results and discussion
Enzyme activity assay
Although Ellman’s assay method is widely used for screening purposes, it is known that this method is not suitable for the identification of NAs. In Ellman’s method, usually, the BTC substrate is hydrolyzed by active BChE in blank plasma. When the serine active site of BChE is inhibited by an NA such as A234, according to Scheme S3 (represented in Supplementary Information), BTC would not be hydrolyzed. In our study, it was observed that when the concentration level of A234 in a plasma sample was approximately 5 ng mL−1, the ChE activity was decreased by 85% from 3.99 for a blank sample to 0.59 units mL−1 (described in Supplementary Information).
Biomarker identification
In plasma sample analysis, there are two analytical approaches for the identification of the utilized agent [25]. In the first approach, the target proteins are isolated and enzymatically cleaved into peptides, and the sample would be investigated looking for phosphonylated or phosphorylated peptides such as Tyr- and BChE-nonapeptide adducts using LC–MS/MS. In the second approach, the agent is regenerated from protein adducts and would be subsequently analyzed using GC–MS/MS or LC–MS/MS techniques. The second procedure does not require the analysis of large protein molecules or their peptides. Both of these approaches were used in the present work to unambiguously verify the presence of A234 in plasma samples based on OPCW criteria. According to these criteria, “at least two biomarkers are required for unambiguous identification of the CWC-related chemicals, where each biomarker is identified by one analytical method giving consistent results with respect to the spiking chemical; alternatively, in exceptional circumstances where only one biomarker is detectable, the biomarker may be reported by two (or three) analytical techniques giving consistent results” [26] (described more in Supplementary Information).
Fluoride regenerated chemical
Through this method, added fluoride ions react with protein adducts, and as a result, phosphoryl moiety of A234 would be released from plasma proteins, generating the original agent again which can be directly analyzed using GC–MS/MS. Due to the high resistance of this agent against hydrolysis [27, 28] (hydrolysis rates of some agents are represented in Table S3 in Supplementary Information) and considerable proton affinity of their amidine nitrogen atoms, unlike other NAs, A234 could be also analyzed using LC–MS/MS without any derivatization [29].
It is important to note that results from fluoride regeneration, in general, produce a fluoro-analog of the agent. Therefore, only A234-like substance can be identified as any other leaving group remains unknown.
The product ion spectra of protonated A234 were recorded by GC-CI/MS/MS and LC-ESI/MS/MS instruments using different collision energies, which are shown in Figs. S3 and S4 (in Supplementary Information). A pair of precursor-product ions was considered for quantitative measurements, and the other transitions were used to confirm the identity of the biomarkers. Table 1 demonstrates the selected transitions and corresponding optimized collision energies. The proposed fragmentation pathways of protonated A234 are illustrated in Scheme S4.
To remove free or unbound A234 from plasma samples, two different methods were used in this work. In both methods, the regeneration process for spiked samples at 50 ng mL−1 concentration level of A234 in the absence of fluoride ions was investigated to ensure complete separation to minimize the risk of false-positive detection and to show that fluoride is the active reagent in regeneration. As shown in Fig. S5 (in Supplementary Information), the peak of A234 was not observed in the absence of fluoride ions. These results indicate that free A234 has been eliminated.
Figure 1 a depicts the overlaid GC-CI/MS/MS chromatograms of a plasma sample treated by A234 at 10 ng mL−1 concentration level, with two MRM transitions. In order to confirm the peak identification, A234 standard solution was added to the sample and the recorded chromatograms were compared with those of the sample. The area ratio of two MRM transitions was also considered an extra identification criterion, which could be compared with the area ratio of the same quantity of a reference chemical.

Analyses of the regenerated A234 from a plasma sample which was treated by the agent at 10 ng mL−1 concentration level after acetone precipitation. a GC-CI/MS/MS in MRM mode chromatograms (at m/z 225 → 124 and 225 → 197). b LC-ESI/MS/MS in MRM mode chromatograms (at m/z 225 → 74 and 225 → 197); overlaid with the chromatograms of the same samples after A234 standard addition
Figure S6 illustrates retention times (rt) and peak areas, which were used to calculate relative intensities. The obtained results were within the acceptable limits for the reliable identification [26] (|Δrt|≤ 0.1 min for GC-CI/MS/MS in MRM mode, |Δrt|≤ 0.2 min for LC-ESI/MS/MS analysis, and the relative mass fragmentation tolerance between 20 and 50%). Table 2 represents the corresponding calculated data.
Identification of the regenerated A234 using LC-ESI/MS/MS was considered the second analytical technique. Figure 1 b demonstrates the overlaid LC-ESI/MS/MS chromatograms of the sample in the MRM mode after the A234 regeneration process, in which the retention times and ion ratios are comparable with those of the A234 reference chemical.
By applying the second preparation method, the probable free agent was removed through filtration of plasma samples using a 10-kD MWCO filter, and then, A234 was regenerated by fluoride ion. The chromatograms and calculated data are presented in Supplementary Information (Fig. S7 for GC-CI/MS/MS in MRM mode, Fig. S8 and Table S4 for LC-ESI/MS/MS in MRM mode). No significant differences were observed between the analytical data obtained from acetone precipitated samples and the filtered sample.
BChE adduct
The hydroxyl group of a serine residue, which is the active site of BChE and AChE, is the main biochemical target of organophosphorus nerve agents (OPNAs) [30, 31]. However, BChE adducts are subjected to dealkylation through a process named aging, which makes it difficult, or impossible, to accurately diagnose the chemical structure of the used agent. In plasma sample analysis, due to the low concentration of BChE in human plasma (approximately 5 mg L−1) [32], the isolation of BChE adducts from albumin and the other abundant plasma proteins would be required. In this study, two-dimensional chromatography using anion exchange and procainamide-based affinity chromatography was used to enhance the recovery of the purification process.
After BChE isolation, pepsin digestion and cleanup using DSC-18 cartridge were applied to separate A234-nonapeptide (FGES * AGAAS) from the other peptides, and LC-ESI/MS/MS under optimized gradient elution program (Table S2) was employed to monitor non-aged A234-nonapeptide at m/z 1000.4 and two other probable ions corresponding to the aging process: the product of the loss of N,N-diethylacetamidine group at m/z 904.3 and the product of the loss of N,N-diethylacetamide and ethyl groups from non-aged A234-nonapeptide at m/z 876.3 (Scheme S5 in Supplementary Information). The product ion scan using a collision energy of 40 eV was performed on both A234 and d1-A234 nonapeptide adducts in order to select the appropriate daughter ions for MRM analyses (Fig. S9 in Supplementary Information). The proposed fragmentation pattern of the protonated A234-nonapeptide is illustrated in Scheme S6 (in Supplementary Information).
Three MRM transitions at m/z 1000.4 → 778 (quantitation transition), 1000.4 → 673, and 1000.4 → 602 (confirmation transitions) were selected for MRM experiments. Corresponding transitions were also selected for d1-A234-nonapeptide (at m/z 1001.4 → 778, 1001.4 → 673, and 1001.4 → 602).
In order to achieve the best sensitivity through monitoring each transition, three fragmentor voltages including 100, 120, and 135 V were applied in MRM mode. For all transitions, the best results were obtained when the fragmentation voltage was set at 135 V. In a similar manner, three collision energies including 20, 30, and 40 eV were applied for each transition. The largest peak areas were obtained using the collision energy of 30 eV for the transition of m/z 1000.4 → 778 and 40 eV for both of the other transitions. The optimization experiments were carried out on a plasma sample which was spiked with A234 at a concentration level of 10 ng mL−1 and LC-ESI/MS/MS in MRM mode was employed for the analysis. The results of these experiments are shown in Fig. 2. Identification of A234- and d1-A234-nonapeptide in human plasma sample was performed through comparison of the ratios of the peak areas related to different transitions, and LC retention times with those of reference chemicals (Fig. S10 in Supplementary Information for d1-A234-nonapeptide). Table S5 (in Supplementary Information) summarizes the results of LC-ESI/MS/MS analyses in MRM mode, which were used for identification of A234-nonapeptide.


LC-ESI/MS/MS in MRM mode chromatograms (at m/z 1000.4 → 602, 1000.4 → 673, and 1000.4 → 778) of blank plasma sample (a, b, and c), and A234-nonapeptide derived after pepsin digestion of BChE isolated from plasma exposed to A234 at 10 ng mL−1 concentration level (d, e, and f). Chromatograms of g, h, and i correspond to analysis of standard solution of BChE exposed to A234 at 15 ng mL−1
Optimized LC-ESI/MS/MS methodology in MRM mode was also employed to investigate the aging products of the biomarker (at m/z 904.3 and 876.3 as precursor ions); however, no product was observed. Therefore, unlike the other OPNA-BChE adducts [33], at least 1 week after spiking the plasma with A234, the aging process was not taken place for BChE-A234 adduct at a detectable level.
Albumin adduct
Another protein biomarker of NAs is the albumin adduct. In comparison with the less abundant and aging-prone ChE adduct, the adduct of Tyr-411 of plasma albumin is more stable, and easier to extract. In most of the cases, during analysis, the purification of albumin adduct would not be required. Nevertheless, the mechanism and the kinetics of the reaction of OPNAs with albumin are unique and the corresponding adducts are formed in dissimilar amounts [16].
In the present study, various incubation times including 24, 48, and 72 h were applied for the formation of A234-Tyr adduct. The incubation time of 48 h demonstrated a peak area for A234-Tyr in LC–MS/MS experiment, which was significantly more than that of 24 h, but not less than that of 72 h. Regarding the incubation times, it was found that the reactivity of A-series for albumin would be less than that for the other NAs such as VX. The low reactivity of A234 to Tyr might be related to the nature of the isomerized structure. Under basic and neutral conditions, A234 would be first isomerized to keto-enol form (Scheme S7), making the agentless electrophilic.
LC-ESI/MS/MS was employed using an optimized gradient elution program (Table S2). MS/MS experiments were performed in product ion scan mode using the collision energy of 15 eV and fragmentor voltage of 120 V to select appropriate MRM transitions. Proposed fragmentation pathways of protonated A234-Tyr and d1-A234-Tyr are illustrated in Scheme S8 (in Supplementary Information).
As is apparent from Fig. 3, in LC-ESI/MS/MS spectrum of A234-Tyr, a signal at m/z 244 was observed, while in the spectrum of d1-A234-Tyr, two peaks with almost the same intensities at m/z 245 and 244 were observed. To explain such spectral features, different fragmentation pathways leading to the formation of non-identical ions at m/z 244 were proposed, which are represented in Scheme S8 (in Supplementary Information).

Product ion spectra of the protonated A234-Tyr at m/z 386.2 (top), and d1-A234-Tyr at m/z 387.3 (bottom) obtained after pronase E digestion of albumin
For identification and quantification of this biomarker at trace levels, two MRM transitions including m/z 386.2 → 244.3 and 386.2 → 285.3 were selected. The fragmentation voltage and collision energy for each transition were optimized. Five different collision energies (5, 10, 15, 20, and 25 eV) and three fragmentor voltages (80, 100, and 120 V) for each selected transition were considered. It was revealed that for both transitions, the best results were obtained when the fragmentor voltage and collision energy were set at 120 V and 15 eV, respectively.
A234-Tyr in human plasma (spiked with 100 ng mL−1 of A234) was identified by comparison of the relative peak areas of the transitions and the retention time of the suspected chemical in the sample with those of the A234-Tyr reference chemical, as mentioned for A234-nonapeptide. The method was successfully employed to investigate a plasma sample spiked at 15 ng mL−1 concentration level; the related chromatograms and identification data are represented in Fig. 4 and Table S6 (in Supplementary Information), respectively.


LC-ESI/MS/MS in MRM mode analyses (at m/z 386.2 → 244.3 and 386.2 → 285.3) of a pronase E digestion of albumin from plasma samples exposed to blank solvent (a and b) and A234 at 15 ng mL−1 concentration level (c and d), looking for A234-Tyr. Chromatograms of e and f correspond to the A234-Tyr reference chemical at 100 ng mL−1 concentration level
Urinary biomarker
Since most OPNAs are rapidly metabolized in vivo and are excreted via urine [34], the identification of intact agents in biomedical fluids would be almost impossible. Unlike the other OPNAs, due to the high resistance of A234 against hydrolysis, it would be possible to detect and identify this agent in its original form in urine samples, and subsequently, the isolated A234 agents from urine samples could be directly analyzed using GC–MS/MS and LC–MS/MS techniques. In the present work, A234 was extracted and cleaned up from urine samples using SPE method and was subsequently detected using LC-ESI/MS/MS technique in MRM mode. An optimized method was developed for monitoring three transitions including m/z 225 → 74, 124, and 197, as mentioned in “Fluoride regenerated chemical.” Fig. 5 illustrates LC-ESI/MS/MS (in MRM mode) chromatograms of the isolated A234 from urine sample treated with A234 at 1 ng mL−1 concentration level, overlaid with the chromatogram of the same sample after A234 standard addition. The observed retention time and ion intensity ratios were within the acceptable limits of the OPCW identification criteria (Fig. S11 and Table S7 in Supplementary Information).

Identification of the isolated intact A234 from urine sample treated with A234 at 1 ng mL−1 concentration level after HLB SPE cleanup, using LC-ESI/MS/MS in MRM mode with three transitions at m/z 225 → 74, 225 → 124, and 225 → 197; overlaid with the chromatograms of the same sample after A234 standard addition
Quantitative analytical performance
The performance of the developed method was evaluated under optimized conditions, and the obtained results are represented in Table 3. The minimum concentration of the analyte that could produce a peak-to-peak S/N ratio ≥ 5:1 for quantitation transitions in MRM mode was defined as the LOD of the method. The LOQ was determined as the lowest concentration that could be quantified accurately and precisely with a peak-to-peak S/N ratio ≥ 10:1 for quantitation transitions in MRM mode. LOD and linear range for the intact agent in the urine sample were measured as 0.1 ng mL−1, and 1–100 ng mL−1, respectively. In blood samples, analysis of two different protein adduct biomarkers, as well as determination of regenerated A234, was considered. LOD for BChE adduct, the regenerated A234, and A234-Tyr adduct was 1, 1, and 10 ng mL−1, while the related calibration curves were linear within the range of 2–100, 2–100, and 15–100 ng mL−1, respectively.
A typical calibration curve for the determination of the isolated intact A234 from the urine sample is depicted in Fig. S12. Repeatability (within day RSDs, n = 5 sample, at 50 ng mL−1 concentration level of A234) and reproducibility (between day RSDs, n = 3 days, at 50 ng mL−1 concentration level of A234) of the analytical methods are also demonstrated in Table 3. Within day RSDs and between day RSDs for the identification of albumin adduct, fluoride regenerated chemical and urinary biomarker were less than 4 and 5%, 6 and 7.5, 4.5, and 5, respectively.
Conclusions
In this paper, for the first time, several approaches were presented for the detection, identification, and measurement of A234 different biomarkers in plasma and urine samples. To this end, various sample preparation methods were introduced, allowing accurate analysis of intact A234, A234-Tyr, and BChE A234-nonapeptide as different A234 biomarkers in blood and urine samples using GC–MS/MS and LC–MS/MS.
Considering all developed analytical methods, the obtained LOD and LOQ values for A234-Tyr were higher than those of other studied analytes, and due to the hydrolysis resistance of A234, this compound can be directly monitored in urine samples without any derivatization using GC–MS/MS and LC–MS/MS techniques.
It was expected that these experimental approaches could be also applicable for unambiguous identification of A234 for verification purposes, as well as OPCW Biomedical Proficiency Tests.
References
Kloske M, Witkiewicz Z. Novichoks–the A group of organophosphorus chemical warfare agents. Chemosphere. 2019;221:672–82. https://doi.org/10.1016/j.chemosphere.2019.01.054.
Costanzi S, Machado JH, Mitchell M. Nerve agents: what they are, how they work, how to counter them. ACS Chem Neurosci. 2018;9:873–85. https://doi.org/10.1021/acschemneuro.8b00148.
Hatfill SJ. Chemical warfare: nerve agents. J Am Physicians Surg. 2019;24:19–23.
Costanzi S, Koblentz GD. Updating the CWC: how we got here and what is next. Arms Control Today. 2020;50:16–20.
Nepovimova E, Kuca K. Chemical warfare agent Novichok-mini-review of available data. Food Chem Toxicol. 2018;121:343–50. https://doi.org/10.1016/j.fct.2018.09.015.
Mirzayanov VS. State secrets: an insider’s chronicle of the Russian chemical weapons program. Parker: Outskirts Press, Inc; 2009. (ISBN 1432725661).
Hoenig SL. Compendium of chemical warfare agents. Berlin: Springer; 2007. (ISBN 9780387692609).
Ellison DH. Handbook of chemical and biological warfare agents. Hoboken: CRC Press; 2008. (ISBN 9780849314346).
Franca TCC, Kitagawa DAS, de A. Cavalcante SF, da Silva JAV, Nepovimova E, Kuca K. Novichoks: the dangerous fourth generation of chemical weapons. Int J Mol Sci. 2019;20:1222. https://doi.org/10.3390/ijms20051222.
Costanzi S, Koblentz GD. Controlling Novichoks after Salisbury: revising the chemical weapons convention schedules. Nonproliferation Rev. 2019;26:599–612. https://doi.org/10.1080/10736700.2019.1662618.
OPCW. Technical Secretariat, Consolidated text of adopted changes to Schedule 1 of the Annex on chemicals to the Chemical Weapons Convention. 2019; S/1820/2019.
Costanzi S, Slavick CK, Hutcheson BO, Koblentz GD, Cupitt RT. Lists of chemical warfare agents and precursors from international nonproliferation frameworks: structural annotation and chemical fingerprint analysis. J Che Inf Mode. 2020;60:4804–16. https://doi.org/10.1021/acs.jcim.0c00896.
OPCW. Technical Secretariat, Summary of the report on activities carried out in support of a request for technical assistance by Germany. 2020; S/1906/2020.
Koryagina N, Savel’eva E, Karakashev G, Babakov V, Dubrovskii YA, Ukolova E, Khlebnikova N, Murashko E, Koneva VY, Ukolov A. Determination of protein adducts of organophosphorus nerve agents in blood plasma. J Anal Chem. 2016;71:849–59. https://doi.org/10.1134/S1061934816080086.
Wang L, Du D, Lu D, Lin CT, Smith JN, Timchalk C, Liu F, Wang J, Lin Y. Enzyme-linked immunosorbent assay for detection of organophosphorylated butyrylcholinesterase: a biomarker of exposure to organophosphate agents. Anal Chim Acta. 2011;693:1–6. https://doi.org/10.1016/j.aca.2011.03.013.
Crow BS, Pantazides BG, Quiñones-González J, Garton JW, Carter MD, Perez JW, Watson CM, Tomcik DJ, Crenshaw MD, Brewer BN. Simultaneous measurement of tabun, sarin, soman, cyclosarin, VR, VX, and VM adducts to tyrosine in blood products by isotope dilution UHPLC-MS/MS. Anal Chem. 2014;86:10397–405. https://doi.org/10.1021/ac502886c.
Lee JY, Lim KC, Kim HS. Characterization and study on fragmentation pathways of a novel nerve agent, ‘Novichok (A234)’, in aqueous solution by liquid chromatography–tandem mass spectrometry. Molecules. 2021;26:1059. https://doi.org/10.3390/molecules26041059.
Noort D, Fidder A, van der Riet-van OD, Busker R, van der Schans MJ. Verification of exposure to Novichok nerve agents utilizing a semitargeted human butyrylcholinesterase nonapeptide assay. Chem Res Toxicol. 2021;34:1926–32. https://doi.org/10.1021/acs.chemrestox.1c00198.
Jeong WH, Lee JY, Lim KC, Kim HS. Identification and study of biomarkers from Novichok-inhibited butyrylcholinesterase in human plasma. Molecules. 2021;26:3810. https://doi.org/10.3390/molecules26133810.
Kranawetvogl A, Küppers J, Siegert M, Gütschow M, Worek F, Thiermann H, Elsinghorst PW, John H. Bioanalytical verification of V-type nerve agent exposure: simultaneous detection of phosphonylated tyrosines and cysteine-containing disulfide-adducts derived from human albumin. Anal Bioanal Chem. 2018;410:1463–74. https://doi.org/10.1007/s00216-017-0787-7.
Ellman GL, Courtney KD, Andres V Jr, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. https://doi.org/10.1016/0006-2952(61)90145-9.
Dubrovskii Y, Murashko E, Chuprina O, Beltyukov P, Radilov A, Solovyev N, Babakov V. Mass spectrometry based proteomic approach for the screening of butyrylcholinesterase adduct formation with organophosphates. Talanta. 2019;197:374–82. https://doi.org/10.1016/j.talanta.2019.01.059.
Pantazides BG, Watson CM, Carter MD, Crow BS, Perez JW, Blake TA, Thomas JD, Johnson RC. An enhanced butyrylcholinesterase method to measure organophosphorus nerve agent exposure in humans. Anal Bioanal Chem. 2014;406:5187–94. https://doi.org/10.1007/s00216-014-7718-7.
Ralston JS, Main A, Kilpatrick BF, Chasson A. Use of procainamide gels in the purification of human and horse serum cholinesterases. Biochem J. 1983;211:243–50. https://doi.org/10.1042/bj2110243.
Mirbabaei F, Mohammad-Khah A, Babri M, Naseri MT. Verification of exposure to sarin nerve agent through the chemical analysis of red blood cell samples. Microchem J. 2020;158: 105174. https://doi.org/10.1016/j.microc.2020.105174.
OPCW. Work instruction for the reporting of the results of the OPCW Biomedical Proficiency Tests. 2020; QDOC/LAB/WI/BioPT04.
Harvey SP, McMahon LR, Berg FJ. Hydrolysis and enzymatic degradation of Novichok nerve agents. Heliyon. 2020;6: e03153. https://doi.org/10.1016/j.heliyon.2019.e03153.
Imrit YA, Bhakhoa H, Sergeieva T, Danés S, Savoo N, Elzagheid MI, Rhyman L, Andrada DM, Ramasami P. A theoretical study of the hydrolysis mechanism of A-234; the suspected novichok agent in the Skripal attack. RSC Adv. 2020;10:27884–93. https://doi.org/10.1039/D0RA05086E.
Blanca M, Shifrovitch A, Madmon M, Elgarisi M, Dachir S, Lazar S, Baranes S, Egoz I, Avraham M, Jaoui HD. Retrospective determination of regenerated nerve agent sarin in human blood by liquid chromatography–mass spectrometry and in vivo implementation in rabbit. Arch Toxicol. 2020;94:103–11. https://doi.org/10.1007/s00204-019-02622-3.
Golime R, Chandra B, Palit M, Dubey D. Adductomics: a promising tool for the verification of chemical warfare agents’ exposures in biological samples. Arch Toxicol. 2019;93:1473–84. https://doi.org/10.1007/s00204-019-02435-4.
Aryal UK, Lin CT, Kim JS, Heibeck TH, Wang J, Qian WJ, Lin Y. Identification of phosphorylated butyrylcholinesterase in human plasma using immunoaffinity purification and mass spectrometry. Anal Chim Acta. 2012;723:68–75. https://doi.org/10.1016/j.aca.2012.02.023.
Onder S, Schopfer LM, Cashman JR, Tacal O, Johnson RC, Blake TA, Lockridge O. Use of hupresin to capture red blood cell acetylcholinesterase for detection of soman exposure. Anal Chem. 2018;90:974–9. https://doi.org/10.1021/acs.analchem.7b04160.
Aurbek N, Thiermann H, Szinicz L, Eyer P, Worek F. Analysis of inhibition, reactivation and aging kinetics of highly toxic organophosphorus compounds with human and pig acetylcholinesterase. Toxicology. 2006;224:91–9. https://doi.org/10.1016/j.tox.2006.04.030.
Kim H, Lee BS, Lee Y, Lee JK, Choi IS. Solid-phase extraction of nerve agent degradation products using poly [(2-(methacryloyloxy) ethyl) trimethylammonium chloride] thin films. Talanta. 2019;197:500–8. https://doi.org/10.1016/j.talanta.2019.01.048.
Acknowledgements
This project was supported by the Defense Chemical Research Laboratory of Iran.
Author information
Authors and Affiliations
Contributions
All authors contributed to the design; conceived and planned the experiments; and verified the analytical methods, implementation of the research, the analysis of the results, and the writing of the manuscript. All authors discussed the results and contributed to the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Human plasma and urine samples were provided by volunteers from the DCRL laboratory, based on general ethical guidelines on conducting scientific research which was approved and disseminated by the Iran Ministry of health and medical education, 2013. All volunteers completed and signed the respected forms.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mirbabaei, F., Mohammad-Khah, A., Naseri, M.T. et al. Unambiguous identification and determination of A234-Novichok nerve agent biomarkers in biological fluids using GC–MS/MS and LC–MS/MS. Anal Bioanal Chem 414, 3429–3442 (2022). https://doi.org/10.1007/s00216-022-03964-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-022-03964-1