
Abstract
Porcine reproductive and respiratory syndrome virus (PRRSV) has caused worldwide economic losses in the swine industry. Pigs infected with highly pathogenic (HP)-PRRSV display more severe symptoms than those infected with classical (C)-PRRSV. A rapid, sensitive, and reliable detection method to distinguish between HP-PRRSV and C-PRRSV is needed. In this study, we prepared a monoclonal antibody from a hybridoma that can distinguish HP-PRRSV(including TP, QJ, LQ, JN-HS, and TY strain) from C-PRRSV (CH-1A strain) using cell surface-fluorescence immunosorbent assays (CSFIA). Based on this monoclonal antibody (4D5), we developed a europium microsphere-based lateral flow immunochromatographic strip (EuNPs-LFICS) for the differential diagnostic detection of HP-PRRSV and C-PRRSV. Under optimized conditions, the method was rapid (15 min), sensitive (LOD: 2.57 ng mL−1, 606 TCID50/0.1 mL), selective for HP-PRRSV detection, and quantitative (DLR: 3.56–228 ng mL−1). In clinical samples, the EuNPs-LFICS assay was largely consistent with PCR results, indicating its practical clinical application.
Graphical abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Porcine reproductive and respiratory syndrome virus (PRRSV), an enveloped RNA virus that belongs to the family Arteriviridae in the order Nidovirales, is the most prevalent viral pathogen in pigs, arose nearly concurrently in the USA and Europe in the late 1980s and spread worldwide within a few years [1, 2], causing major economic losses to the pork industry worldwide [3, 4]. The most significant feature of PRRSV is its ability to rapidly evolve. This may be due to a lack of RNA-dependent RNA polymerase proofreading, as well as extensive viral recombination, which results in an extraordinarily mixed composition of isolates of varying pathogenicity [5].
PRRSV has two genotypes: the European genotype (PRRSV-1) and the North American genotype (PRRSV-2). The main type of transmission in China is PRRSV-2, including classic PRRSV (C-PRRSV) and highly pathogenic PRRSV (HP-PRRSV) [6,7,8]. However, compared to C-PRRSV, pigs infected with HP-PRRSV display more severe clinical symptoms such as diarrhea, conjunctivitis, and respiratory distress [9]. Furthermore, the vaccine currently under development does not have a good cross-protection effect on PRRSV [10], which leads to a rapid decline in pork production and a sharp rise in the market price of pork [11, 12]. The PRRSV strain in most parts of China is still HP-PRRSV [13]. Accordingly, the identification and control of HP-PRRSV infection is of vital importance to the Chinese pig industry [14].
The most distinguishing feature of HP-PRRSV is a 30-aa deletion in the nsp2-coding region compared to VR-2332 (a prototype strain), HB-1 (sh)/2002, and CH-1a (the earlier Chinese isolates). This unique deletion is considered to be a genetic marker of HP-PRRSV strains [15, 16]. The current method of HP-PRRSV detection is RT-PCR [8, 17, 18]. Although high-level amplification in PCR-based detection is accurate and reliable, the laboratory-based method relies on specialized professional equipment and sophisticated operation, which are time-consuming and inconvenient for rapid, on-site monitoring.
Many studies have shown that although PRRSV has great genetic variability, there are still some conserved amino acid sequences in the structural proteins and nonstructural proteins of HP-PRRSV, which are different from those of C-PRRSV. For example, the amino acid sequences of the N protein between HP-PRRSV and C-PRRSV were inconsistent at positions 15–21, 42–48, and 88–94 [19], and the N protein homology of HP-PRRSV, which is common in mainland China, reaches 97% [20]; GP3 protein of HP-PRRSV has 17 amino acid mutations compared with the GP3 proteins of C-PRRSV. Moreover, several studies showed that there were also differences in epitopes between C-PRRSV and HP-PRRSV. For example, 130KANATSMRFH139 has been identified as a linear B cell epitope on nonstructural protein 12 (NSP12), epitope T 59RQAAAEILE68 of dominant linear GP3, which were highly conserved in HP-PRRSV but do not contain similar sequences in C-PRRSV and PRRSV-1 [21,22,23]. And finally, as we all know, virus strains in the same branch have relatively high similarities in the conservative parts of gene sequences and protein epitopes [24, 25]. Therefore, immunological methods can be used to distinguish between HP-PRRSV and C-PRRSV [20, 26].
Recently, fluorescent signal-based immunochromatographic strips (F-ICS) have been used as a novel tool for point-of-care testing due to their portability, simplicity, speed, and high sensitivity [27,28,29]. In particular, europium microspheres (EuNPs) with a highly sensitive lanthanide label have been widely used in rapid quantitative immunoassays where they offer advantages including broad excitation spectrum, long fluorescence lifetime, sharp emission peaks, large stokes shift, high resolution, low background, and no photobleaching [30,31,32]. In order for EuNPs to be more effectively used as fluorescent probes for detection, the surface of EuNP microspheres is usually modified with specific functions to better connect to specific targets, which can greatly improve the stability and sensitivity of detection [33, 34]. Here, we generated a monoclonal antibody that can differentiate between HP-PRRSV and C-PRRSV, and a novel europium microsphere-based lateral flow immunochromatographic strip (EuNPs-LFICS) was developed to differentially diagnose HP-PRRSV and C-PRRSV.
Materials and methods
Cells and viruses
HP-PRRSV strains (TY, TP, QJ, LQ, and JN-HS) were kindly supplied by Yunnan Agricultural University, China. The PRRSV CH-1a strain was purchased from Shangdong Lvdu Bioscience & Technology Co., Ltd. SP2/0 myeloma cells and Marc-145 cells were from our laboratory.
Materials and reagents
Specific pathogen-free Balb/c mice were purchased from the experimental animal center of Southern Medical University. Adjuvant (Freund’s complete and incomplete), PEG2000, HAT, and HT were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO). RPMI-1640 medium was purchased from Gibco. Goat anti-mouse IgG conjugated with HRP was purchased from Abcam. N-(3-Dimethylaminopropyl)-N′-ethyl carbodiimide hydrochloride, ovalbumin (OVA), and bovine serum albumin (BSA) were purchased from Asegene (Guangzhou, China). Sheep anti-mouse IgG Fc fragment specific labeled with fluorescein isothiocyanate (FITC) was purchased from Jackson (West Grove, PA, USA). Oleyl-PEG4000-NHS-antigen TP was synthesized in our laboratory as described previously by Xiuqing-Li [35]. Europium microspheres (EuNPs), 1-ethyl-3-(3-dimethyl aminopropyl) carbodiimide hydrochloride (EDC), and sulfo-N-hydroxysuccinimide (sulfo-NHS) were purchased from Thermo (USA). Morpholine ethyl sulfonic acid (MES), bovine serum albumin (BSA), PEG4000, Tween-20, and sodium casein were purchased from Sigma-Aldrich (USA). Goat anti-mouse IgG antibody was purchased from Tianjin Sungene Biotech Co., Ltd (Tianjin, China). All oligonucleotides were synthesized from Sangon Biotech (Shanghai, China). Viral RNA/DNA extraction kit was purchased from Takara Bio (Dalian, China). DNA marker was purchased from Beyotime Biotech (Shanghai, China). PCR Mix was purchased from Tsingke Biotech Co., Ltd (Beijing, China). UltraSignal ECL Western Blotting Detection Reagent was purchased from 4A Biotech Co., Ltd (Beijing, China). Nitrocellulose (NC) membrane (CN140), conjugation pads, sample pad (Gl-b04), PVC plate, and absorbent pads (H2) were all purchased from JN-HS Bio Co., Ltd (Shanghai, China). All clinical samples were obtained from the Ministry of Agriculture Key Laboratory for the development of veterinary diagnostic products (Wuhan, China). Millipore Milli-Q water (> 18 MΩ cm) was used for all solutions. All chemicals used were of analytical grade or higher without further purification.
Apparatus
We used a Synergy H1 Hybrid Multi-Mode microplate reader (Bio-Tek) and ZEISS AXIOVERT A1 phase-contrast fluorescence microscope (Zeiss) with a 10× objective lens. A XYZ3060 series dispenser platform (Bio-Dot, China) and programmable strip cutter HGS201 (AUTOKUN, China), XW-80 Avortex Mixer (Shanghai, China), and 5810 R centrifuge (Eppendorf, Germany) were used to prepare EuNPs-LFICS. A JEOL 2100F transmission electron microscope (TEM, Japan), Carl Zeiss AG scanning electron microscopy (SEM, Germany), and Zetasizer Nano ZS (Malvern Panalytical Ltd.) were used. Additionally, our group designed a simple fluorescence camera.
Preparation and purification of monoclonal antibodies against PRRSV
Specific pathogen-free BALB/c mice were immunized 5 times with 200 μg HP-PRRSV per mouse. The serum antibody titer was detected by indirect ELISA. After intraperitoneal boost injection, the spleen cells of mice were harvested and fused with mouse myeloma cells SP2/0 using standard procedures [36]. Fused cells were cultured in RPMI-1640 medium containing 5% HAT (Sigma-Aldrich) and 10% FBS. After 5 days of fusion, cell surface-fluorescence immunosorbent assays (CSFIA) were used to screen for positive hybridoma cells, as previously reported [34]. Ascites monoclonal antibodies were purified using ammonium sulfate, desalting columns, and protein G affinity chromatography columns. Ten microliters of BSA solution with a concentration of 100 mg/mL was added.
ELISA
The antigens/antibodies were diluted to an appropriate concentration with the coating solution, and then an ELISA plate filled with 100 μL per well was incubated at 4 °C for 12 h. After that, the plate was washed 3 times with PBST buffer and then blocked with 200 μL 5% nonfat milk/well at 37 °C for 1 h. And then, after washing 3 times with PBST buffer, the plate was filled with 100 μL antibodies/antigens per well before incubation at 37 °C for 1 h. Next, the plate was washed 3 times with PBST buffer and incubated at 37 °C for 0.5 h after adding enzyme-labeled goat anti-mouse antibody with appropriate concentration. After washing 5 times with PBST buffer, the plate was added with 100 μL TMB and incubated at 37 °C for 0.2 h. Finally, the absorbance was read at 450 nm using a microplate reader after adding a stopping solution (50 μL, 2 M H2SO4).
Detection of antibody characteristics
The purified mAbs were identified by SDS-PAGE. The subtypes of mAbs were determined by a mouse monoclonal antibody isotyping ELISA kit according to the manufacturer’s protocol. The antibody titer and specificity were tested by indirect ELISA assay. The mAb pair for PRRSV detection was selected by sandwich ELISA.
Western blotting analysis
PRRSV was diluted in loading buffer and separated on SDS-PAGE gels. The protein samples were transferred to polyvinylidene fluoride (PVDF) membranes for 1 h at constant voltage (90 V) and then blocked with milk at 4 °C 12 h. The polyvinylidene fluoride (PVDF) membranes were washed three times with PBST and incubated for 1 h at an incubator (37 °C) with mAb 4D5. The PVDF membranes were washed three times with PBST and incubated for 1 h at an incubator (37 °C) with HRP-conjugated secondary antibodies. The result was visualized using an enhanced chemiluminescence system.
Immunofluorescence staining
Marc-145 cells were infected with the TP strain for 2 h and cultivated for 48 h at 37 °C. Mock-infected Marc-145 cells served as a negative control. The cells were fixed with 4% (w/v) paraformaldehyde for 16 min and disposed of 0.2% Triton X-100 for 22 min. After three disposal of PBST buffer, the antibody 4D5 was added and disposed for 12 h at 4 °C. And then cells were incubated with secondary antibodies IgG labeled with FITC for 1 h in the dark before being disposed three times by PBST buffer. DAPI was used to dye nuclei for 5 min. The results were detected by confocal microscopy.
Preparation of EuNP-labeled anti-PRRSV monoclonal antibody (EuNPs-4D5)
EuNPs-4D5 was conjugated as follows: First, 10 μL of EuNPs were added to 990 μL MES (0.1 M, pH 6.0). Then, 10 μL EDC (1 mg mL−1) was added followed by 10 μL solfu-NHS (50 mg mL−1). The solution was mixed for 30 min to activate the carboxyl group on the EuNP surface. Thirty micrograms of 4D5 was then added to the mixture and incubated for 2 h. Next, 10 μL BSA solution with a concentration of 100 mg mL−1 was added and mixed for 2 h, followed by centrifugation at 15,000 rpm for 30 min. Finally, the supernatant was removed and the precipitate was suspended in 500 μL Gly-NaOH (0.1 M pH 8.5, containing 8% (w/v) sucrose, 1% (w/v) BSA, 0.5% (w/v) sodium casein, 2% (w/v) D-trehalose, and 0.02% (w/v) NaN3).
Preparation of EuNPs-LFICS for PRRSV detection
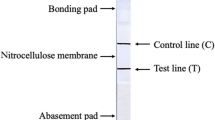
The sample pad was pre-processed using 6 mL of PBS. The EuNPs-4D5 solution was applied to the conjugate pad (5 μL/cm) using a dispensing platform and then dried at 37 °C. For the NC membrane, 1 mg mL−1 4D5 and 0.25 mg mL−1 goat anti-mouse IgG were separately distributed onto designated areas at a dispensing rate of 1 μL/cm as the test line (T-line) and control line (C-line), respectively. Finally, the sample pad, conjugate pad, NC membrane, and absorbent pad were arranged in order, assembled on the PVC plate with 2 mm overlap between each part, cut into 3.5 mm width, and placed in a plastic case for subsequent analysis.
Performance of the EuNPs-LFICS for HP-PRRSV detection
Determination of EuNPs-LFICS sensitivity
To accurately determine the sensitivity of EuNPs-LFICS, a series of different HP-PRRSV (TP strain) concentrations were diluted in 80 μL PBS and added to the sample pad. The solution flowed toward the absorbent pad by capillary action. Images were captured using a fluorescence camera and the fluorescence intensity data were detected using the fluorescence strip reader after 15 min.
Determination of EuNPs-LFICS specificity
EuNPs-LFICS was tested specificity using antigens from HP-PRRSV (including TP, QJ, LQ, JN-HS, and TY strains) and C-PRRSV (CH-1A strain) as well as pseudorabies virus (PRV), porcine epidemic diarrhea virus (PEDV), classical swine fever virus (CSFV), African swine fever virus (ASFV), porcine circovirus (PCV), and viral medium (medium). Each of these antigens was diluted to 100 ng mL−1 with PBS and individually applied to the EuNPs-LFICS. Results were detected using the fluorescence strip reader after 15 min.
EuNPs-LFICS application to clinical samples
Fresh swine tissue samples (such as tonsils, brain, heart, liver, spleen, lung, and kidney) were collected, weighed, and mixed with the PBS (15 mM, pH 7.4) at a ratio of 1:5 (w/v). Swine tissue homogenate samples were collected using a churning machine. After centrifugation at 4000×g for 5 min, the supernatant was collected and used as the sample solution. Fifty clinical samples from different swine tissue extracts were collected. After dilution, the samples were detected by EuNPs-LFICS and compared with RT-PCR results.
Results and discussion
Production of monoclonal antibodies
In order to quickly and conveniently obtain monoclonal antibodies, we used a new monoclonal antibody screening method [35]. In this method (Fig. 1a), antigens can anchor to the hybridoma cell surface with the help of Oleyl-PEG4000-NHS. Therefore, the antibodies secreted from the hybridoma cells could be captured by the antigens on the cell surface. Afterward, an anti-mouse IgG Fc fluorescent antibody was added to fluorescently stain the antigen-specific positive hybridoma cells. The positive hybridoma cells were observed under a fluorescence microscope. As shown in Fig. 1b, positive cells had a clear fluorescent signal, while the negative cells were not fluorescent. Ultimately, we obtained 36 clones of hybridoma cells that stably secrete anti-PRRSV antibodies. Then, we selected 6 clones of hybridoma cells that highly express and secrete anti-PRRSV antibodies, of which 4D5 monoclonal antibody can specifically recognize HP-PRRSV but not C-PRRSV. The following experiments will not be conducted on the remaining 30 strains.

Production of monoclonal antibodies. a The monoclonal antibody screening methods. b Fluorescent images and bright-field images of hybridoma cell (4D5, 3E9, 7B8, 11C11, 4B4, 5F10)
Characterization of mAbs
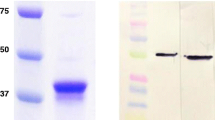
To further characterize the 6 mAb strains, we generated large-scale hybridoma cultures and intraperitoneally injected them into mice. The mAbs were then harvested, purified, and characterized. The purified mAbs gave two bands in the SDS-PAGE with molecular masses of approximately 55 kDa and 25 kDa (Supplementary Information (ESM) Fig. S1), showing that the antibodies were successfully purified. We diluted the purified antibodies to 1 mg/mL and determined their titers. The titers of purified antibodies 4B4, 5F10, 3E9, 4D5, 11C11, and 7B8 were 1:64,000, 1:8000, 1:8000, 1:16,000, 1:16,000, and 1:32,000, respectively (Fig. S2, Table S1). All of the mAbs were IgG1 isotype (Table S1). Subsequently, we used ELISA to determine the specificity of the 6 mAbs to different viruses. The results showed that the 6 mAbs did not cross-react with other common porcine viruses, such as classical swine fever virus (CSFV), porcine epidemic diarrhea virus (PEDV), and pseudorabies virus (PRV) (Fig. S3). And ELISA experiments proved that monoclonal antibody 4D5 can distinguish 5 strains of HP-PRRSV (TP, QJ, LQ, TY, JN) and from C-PRRSV (CH-1a strain) (Fig. S4). According to literature reports, the antibodies produced by PRRSV virus immunization are mainly directed against PRRSV structural proteins [37]. Therefore, in this experiment, it is hoped to understand the relationship between antibody 4D5 and various structural proteins through docking, so as to predict the protein targeted by monoclonal antibody 4D5. At first, the 3D model structure of 4D5 antibodies (Fig. S5a) was built with Discovery Studio. And then, we obtained the docking scores of antibody 4D5 and structural proteins GP2, GP3, GP4, M, and N. The docking conformation score of antibody 4D5 and N protein is higher, indicating that antibody 4D5 may be directed against N protein (Fig. S5b). To verify the results of the antigen–antibody molecular docking, we performed an indirect ELISA reaction between the monoclonal antibody 4D5 and the purified N protein. The positive control comes from the serum of mice immunized with QJ virus for 5 weeks, which contains a variety of antibodies against different viral structural proteins. The results showed that mAb 4D5 can react with the N protein. The epitope targeted by mAb4D5 is located on the N protein (Fig. S5c). And then, the docking model of mAb4D5 and N protein further shows that mAb4D5 can interact well with the N protein of PRRSV (Fig. S5d).
In 1995, two strains of porcine reproductive and respiratory syndrome virus (PRRSV), named CH-1a and BJ-4, were isolated from China for the first time. The highly pathogenic PRRSV (HP-PRRSV) was first discovered in mainland China in 2006. Subsequent studies confirmed that HP-PRRSV originated from C-PRRSV (a CH-1a-like strain) [38]. To determine whether the mAbs that we generated could distinguish HP-PRRSV (QJ, TP, LQ, JN-HS-HS, TY) from C-PRRSV (CH-1a), we further identified the 6 mAbs using ELISA, including antibodies 3E9, 4B4, 5F10, 4D5, 11C11, and 7B8. Our results showed that antibody 4D5 could distinguish HP-PRRSV from C-PRRSV (Fig. 2a), which was further confirmed by western blot (Fig. 2b). An immunofluorescence assay (IFA) was then used to further characterize antibody 4D5. Our results showed that 4D5 reacted well with Marc-145 cells infected with PRRSV, but no reaction was found on the Marc-145 cells not infected with PRRSV (Fig. 2c). This demonstrated that 4D5 specifically recognized PRRSV. To obtain the best PRRSV mAb pairs, a double-antibody sandwich ELISA was used. Our results showed that 4D5-4D5 was the best mAb pair (Table S2).

Characterization of mAbs. a ELISA analysis of antibody 4D5 distinguish HP-PRRSV (QJ, TP, LQ, JN-HS, TY) from C-PRRSV (CH-1a). b Western blot analysis of mAbs distinguishes HP-PRRSV (QJ, TP, LQ, JN-HS, TY) from C-PRRSV (CH-1a). c Marc-145 cells infected with TP strain were analyzed by confocal microscopy, using antibody 4D5. Uninfected MARC-145 cells are used as control. Nuclei are stained with DAPI
EuNPs-LFICS fabrication and function
EuNPs-LFICS were manufactured using a sample pad (for the applied test sample), a conjugation pad (for loading EuNPs-4D5), an NC membrane (for loading 4D5 and goat anti-mouse IgG), an absorption pad (for liquid absorption), and a PVC plate (for support material) (Fig. 3). In the absence of samples containing PRRSV, EuNPs-4D5 was not captured on the T-line, so only the C-line can watch the fluorescent signal (Fig. 3a). In contrast, when PRRSV was present in the sample, a double-antibody sandwich form was formed on the T-line and both the T-line and C-line can watch the fluorescent signal (Fig. 3b).

a, b The fluorescence lateral flow immunoassays for PRRSV detection. T-line and C-line can watch the fluorescent signal with positive samples while negative samples had no fluorescent signal at the T-line
Characterization of EuNPs and EuNPs-4D5
TEM analysis showed that the average sizes of the EuNPs and EuNPs-4D5 were about 280 nm (Fig. 4a) and 300 nm (Fig. 4b), respectively. We next tested the hydrodynamic diameter and surface charge of the EuNPs-4D5. The EuNPs-4D5 diameter was increased slightly compared to EuNPs (Fig. 4c). When the EuNPs were activated, their surface charge showed little change; however, the negative surface charge was significantly reduced after immobilization of the antibody on the EuNPs (Fig. 4d). These data demonstrated that the antibody was successfully conjugated with EuNPs.

Characterization of EuNPs and EuNPs-4D5. a TEM images of EuNPs. b TEM images of EuNPs-4D5. c Changes in EuNP particle size before and after antibody labeling. d Changes in EuNP particle zeta potential before and after antibody labeling
Distinguishing the HP-PRRSV strain from the C-PRRSV strain
To further determine the selectivity of HP-PRRSV and C-PRRSV, we had to verify that the HP-PRRSV (TP strain) had strong positive signals, but the C-PRRSV (CH-1A strain) was not detected as a false positive. The antigens (TP and CH-1A strain) were diluted to 50 ng mL−1 in PBS buffer solution and added to the EuNPs-LFICS. Fifteen minutes later, the results were detected by the fluorescence strip reader. As shown in Fig. 5, in the presence of HP-PRRSV, a strong fluorescence signal existed on the T-line of the EuNPs-LFICS, while no signal was detected in the presence of C-PRRSV (Fig. 5a, b). To determine whether EuNPs-4D5 was captured on the T-line, we measured SEM images of HP-PRRSV and C-PRRSV. In the presence of C-PRRSV, no immune complex was formed in the test area (Fig. 5c). In contrast, immune complexes were observed on the NC membrane of the HP-PRRSV-positive specimens (Fig. 5d). These results suggested that EuNPs-LFICS had good performance in the differential diagnosis of HP-PRRSV and C-PRRSV.

Differential diagnosis between HP-PRRSV and C-PRRSV. a Fluorescent images were recorded by the fluorescence camera. b Fluorescence intensity of H-PRRSV and C-PRRSV on the T-line. Each value represents the mean of three replicates (n = 3). SEM images of the EuNPs-LFICS on T-line in the presence of C-PRRSV (c) and HP-PRRSV(d)
Optimal EuNPs-LFICS conditions and feasibility
We dispensed a series of EuNPs-4D5 concentrations onto the conjugation pad. 200 ng mL−1 PRRSV was added to the EuNPs-LFICS and the fluorescence intensity on the T-line was analyzed. The results showed that the fluorescence of the T-line increased with fluorescent probe concentration. The correlation coefficient was R2 = 0.995, showing a strong, positive correlation between fluorescence intensity and EuNPs-4D5 concentration (Fig. 6a). Besides, the fluorescence signal intensity of the T-lines reached the platform maximum after 15 min, suggesting that the optimal reaction time was 15 min (Fig. 6b). To improve the EuNPs-LFICS performance, we optimized different concentrations of 4D5 on the T-line as well as the amount of 4D5 labeled EuNPs. After the addition of 200 ng mL−1 PRRSV, the ratio of the positive and negative sample fluorescence intensity was the highest when the T-line concentration was 1.0 μg cm−1 (Fig. 6c) and 30 μg 4D5 were used to bind EuNPs (Fig. 6d). Therefore, we used these optimized conditions for future experiments.

Optimal EuNPs-LFICS conditions and feasibility. a Correlation between EuNPs-4D5 and fluorescence intensity. b The reaction time and corresponding fluorescence intensity. Optimization of 4D5 concentration on the T-line (c) and the number of labeled 4D5 on EuNPs (d). Each value represents the mean of three replicates (n = 3)
EuNPs-LFICS PRRSV detection performance
EuNPs-LFICS analytical sensitivity
The EuNPs-LFICS reliably indicated the presence of HP-PRRSV. To analyze the performance range of the EuNPs-LFICS, serial HP-PRRSV dilutions (TP strain, 1.78, 3.56, 7.13, 14.25, 28.5, 57, 114, 228, 456, and 912 ng/mL diluted in PBS) were applied to the sample pad under the optimal conditions. We used a fluorescence camera to capture images of the EuNPs-LFICS (Fig. 7a). To quantify the detection performance of EuNPs-LFICS, fluorescence intensity data were collected using a professional fluorescence strip reader. Fluorescence peaks indicated that fluorescence intensity on the T-line was significantly correlated with HP-PRRSV concentration (Fig. 7b). Furthermore, we used HP-PRRSV concentration as the independent variable and the peak integral area of the data on the T-line as the ordinate variable to establish a standard curve. The linear range of detection (DLR) was 3.56–228 ng/mL, while the limit of detection (LOD) was 2.57 ng/mL (606 TCID50/0.1 mL) (Fig. 7c). These results show that EuNPs-LFICS was a method with good suitability and sensitivity for the detection of PRRSV.

Determination of sensitivity of EuNPs-LFICS for the detection of PRRSV. a Fluorescent images were recorded by the fluorescence camera. b Fluorescence peaks on the T- or C-lines of each test read-out using a fluorescent strip reader. c The standard curve is used for PRRSV detection. Each value represents the mean of three replicates (n = 3)
EuNPs-LFICS analytical specificity
To analyze the specificity of the EuNPs-LFICS, we added 100 ng mL−1 of the following virus antigens to the strips: HP-PRRSV (including TP, QJ, LQ, JN-HS, and TY strains), C-PRRSV(CH-1A strain)as well as PRV, PEDV, CSFV, ASFV, PCV, PBS, and medium. After reacting for 15 min, the T-line fluorescence signal was measured using a fluorescence strip reader. The corresponding PRRSV concentration (ePRRSV) was calculated by interpolating the ePRRSV value into the standard curve (Fig. 7c), and the corresponding cross-reaction rate was expressed as (ePRRSV)/100 × 100%. The cross-reactivity of the other viruses was almost imperceptible, indicating that the EuNPs-LFICS had inherently high specificity (Fig. 8).

Specificity analysis among different viruses of EuNPs-LFICS
EuNPs-LFICS analytical stability
We further evaluated the stability of EuNPs-LFICS in PRRSV detection. The same concentrations of PRRSV were assessed for the intra-batch and inter-batch variability following similar procedures. The coefficients of variance (CVs) generated from the same batch and repeated batches were both below 15% (Tables S3 and S4). Besides, we compared different time points using the same EuNPs-LFICS at 37 °C. The results remained basically unchanged at 37 °C for at least 4 weeks, corresponding to 4 °C storage for about 1 year (Table S5). This result demonstrated the stability of EuNPs-LFICS and their feasibility for use in PRRSV detection.
EuNPs-LFICS application to swine clinical samples
To estimate the applicability of our EuNPs-LFICS in clinical samples, we used EuNPs-LFICS to detect PRRSV in 50 clinical tissue extracts and compared the results with an RT-PCR kit. As shown in Table 1, the total coincidence rate was 98%. This indicated that the strip assay sensitivity was slightly lower than the RT-PCR kit. Table S6 shows a comparison of different HP-PRRSV detection methods. Importantly, EuNPs-LFICS were integrated with a 3D-printed read-out platform designed by our group that can be read by the naked eyes [39], thus improving the practical application in rapid monitoring during potential PRRSV outbreaks (Fig. S7). In short, EuNPs-LFICS could be used for rapid clinical detection of PRRSV.
Conclusions
In this study, we prepared a monoclonal antibody (4D5) and used it to develop a novel EuNPs-LFICS for HP-PRRSV detection. We first produced hybridoma strain 4D5, which can distinguish HP-PRRSV (including TP, QJ, LQ, JN-HS, and TY strains) from C-PRRSV (CH-1A strain) by CSFIA with high specificity and affinity, and then fabricated a rapid, sensitive, specific, and stable EuNPs-LFICS for the differential diagnostic detection of PRRSV. When applied to swine clinical samples, the EuNPs-LFICS results were largely consistent with RT-PCR results. Thus, this novel EuNPs-LFICS has the potential for on-site PRRSV detection and can help to prevent its spread.
Abbreviations
- PRRSV:
-
porcine reproductive and respiratory syndrome virus
- HP-PRRSV:
-
highly pathogenic porcine reproductive and respiratory syndrome virus
- CSFIA:
-
cell surface-fluorescence immunosorbent assays
- EuNPs-LFICS:
-
europium microsphere-based lateral flow immunochromatographic strip
- C-PRRSV:
-
classic porcine reproductive and respiratory syndrome virus
- NSP 12:
-
nonstructural protein 12
- F-ICS:
-
fluorescent signal-based immunochromatographic strips
- EuNPs:
-
europium microspheres
- OVA:
-
ovalbumin
- BSA:
-
bovine serum albumin
- FITC:
-
fluorescein isothiocyanate
- sulfo-NHS:
-
sulfo-N-hydroxysuccinimide
- MES:
-
morpholine ethyl sulfonic acid
- NC:
-
nitrocellulose
- PVDF:
-
polyvinylidene fluoride
- T-line:
-
test line
- C-line:
-
control line
- PRV:
-
pseudorabies virus
- PEDV:
-
porcine epidemic diarrhea virus
- CSFV:
-
classical swine fever virus
- ASFV:
-
African swine fever virus
- PCV:
-
porcine circovirus
- IFA:
-
immunofluorescence assay
- DLE:
-
range of detection
- LOD:
-
limit of detection
- CV:
-
coefficients of variance
References
Wensvoort G, Terpstra C, Pol JM, ter Laak EA, Bloemraad M, de Kluyver EP, et al. Mystery swine disease in The Netherlands: the isolation of Lelystad virus. Vet Q. 1991;13:121–30. https://doi.org/10.1080/01652176.1991.9694296.
Lee S-C, Choi H-W, Nam E, Noh Y-H, Lee S, Lee YJ, et al. Pathogenicity and genetic characteristics associated with cell adaptation of a virulent porcine reproductive and respiratory syndrome virus nsp2 DEL strain CA-2. Vet Microbiol. 2016;186:174–88. https://doi.org/10.1016/j.vetmic.2016.03.002.
Holtkamp DJ, Kliebenstein JB, Neumann EJ, Zimmerman JJ, Rotto HF, Yoder TK, et al. Assessment of the economic impact of porcine reproductive and respiratory syndrome virus on United States pork producers. J Swine Health Prod. 2013;21:72–84.
Neumann EJ, Kliebenstein JB, Johnson CD, Mabry JW, Bush EJ, Seitzinger AH, et al. Assessment of the economic impact of porcine reproductive and respiratory syndrome on swine production in the United States. J Am Vet Med Assoc. 2005;227:385–92. https://doi.org/10.2460/javma.2005.227.385.
Kappes MA, Faaberg KS. PRRSV structure, replication and recombination: origin of phenotype and genotype diversity. Virology. 2015;479:475–86. https://doi.org/10.1016/j.virol.2015.02.012.
Yin B, Qi S, Sha W, Qin H, Liu L, Yun J, et al. Molecular characterization of the Nsp2 and ORF5 (ORF5a) genes of PRRSV strains in nine provinces of China during 2016-2018. Front Vet Sci. 2021;8:605832. https://doi.org/10.3389/fvets.2021.605832.
Xie S, Liang W, Wang X, Chen H, Fan J, Song W, et al. Epidemiological and genetic characteristics of porcine reproduction and respiratory syndrome virus 2 in mainland China, 2017-2018. Arch Virol. 2020;165:1621–32. https://doi.org/10.1007/s00705-020-04661-z.
Qiu W, Meng K, Liu Y, Zhang Y, Wang Z, Chen Z, et al. Simultaneous detection of classical PRRSV, highly pathogenic PRRSV and NADC30-like PRRSV by TaqMan probe real-time PCR. J Virol Methods. 2019;282:113774. https://doi.org/10.1016/j.jviromet.2019.113774.
Xiao S, Chen Y, Wang L, Gao J, Mo D, He Z, et al. Simultaneous detection and differentiation of highly virulent and classical Chinese-type isolation of PRRSV by real-time RT-PCR. J Immunol Res. 2014;2014:1–7. https://doi.org/10.1155/2014/809656.
Huang Y, Li Z, Li J, Yibo K, Yang L, Mah CK, et al. Efficacy evaluation of three modified-live PRRS vaccines against a local strain of highly pathogenic porcine reproductive and respiratory syndrome virus. Vet Microbiol. 2019;229:117–23. https://doi.org/10.1016/j.vetmic.2018.12.016.
Han J, Zhou L, Ge X, Guo X, Yang H. Pathogenesis and control of the Chinese highly pathogenic porcine reproductive and respiratory syndrome virus. Vet Microbiol. 2017;209:30–47. https://doi.org/10.1016/j.vetmic.2017.02.020.
Wei ZY, Liu F, Li Y, Wang HL, Zhang ZD, Chen ZZ, et al. Aspartic acid at residue 185 modulates the capacity of HP-PRRSV nsp4 to antagonize IFN-I expression. Virology. 2020;546:79–87. https://doi.org/10.1016/j.virol.2020.04.007.
Xiao XL, Wu H, Yu YG, et al. Rapid detection of a highly virulent Chinese-type isolate of Porcine Reproductive and Respiratory Syndrome Virus by real-time reverse transcriptase PCR. J Virol Methods. 2008;149:49–55. https://doi.org/10.1016/j.jviromet.2008.01.009.
Ge M, Li RC, Gong W, Tu C. Determination of antibody induction by highly pathogenic porcine reproductive and respiratory syndrome virus (HP-PRRSV) vaccine: a comparison of two ELISA kits. J Vet Med Sci. 2019;81:1173–6. https://doi.org/10.1292/jvms.18-0482.
Zhou L, Yang H. Porcine reproductive and respiratory syndrome in China. Virus Res. 2010;154:31–7. https://doi.org/10.1016/j.virusres.2010.07.016.
Wang G, Yu Y, Tu Y, Li Y, Tong J, Zhang C, et al. Characterizing the thymic lesions in piglets infected with attenuated strains of highly pathogenic porcine reproductive and respiratory syndrome virus. Vet Immunol Immunopathol. 2015;168:258–61. https://doi.org/10.1016/j.vetimm.2015.10.007.
Yang K, Li Y, Duan Z, Guo R, Liu Z, Zhou D, et al. A one-step RT-PCR assay to detect and discriminate porcine reproductive and respiratory syndrome viruses in clinical specimens. Gene. 2013;531:199–204. https://doi.org/10.1016/j.gene.2013.09.017.
Liang W, Zhao T, Peng Z, Sun Y, Stratton CW, Zhou D, et al. Epidemiological and genetic characteristics of porcine reproductive and respiratory syndrome virus circulating in central and South China in 2016. Acta Trop. 2019;190:83–91. https://doi.org/10.1016/j.actatropica.2018.11.004.
Fan B, Liu X, Bai J, Li Y, Zhang Q, Jiang P. The 15N and 46R residues of highly pathogenic porcine reproductive and respiratory syndrome virus nucleocapsid protein enhance regulatory T lymphocytes proliferation. PLoS One. 2015;10:e0138772. https://doi.org/10.1371/journal.pone.0138772.
Yan JJ, Yang QL, Lin YJ, Shi LH, Wang H, Tang Y. Codon optimization expression of PRRSV NSP7 protein and immunological activity identification. Chin J Microbiol Immunol. 2019;35:829–33. https://doi.org/10.3969/j.issn.1000-484X.2019.07.012.
Song Y, Zhou Y, Li Y, Wang X, Bai J, Cao J, et al. Identification of B-cell epitopes in the NSP1 protein of porcine reproductive and respiratory syndrome virus. Vet Microbiol. 2012;155:220–9. https://doi.org/10.1016/j.vetmic.2011.09.018.
Bi C, Shao Z, Zhang Y, Hu L, Li J, Huang L, et al. Identification of a linear B-cell epitope on non-structural protein 12 of porcine reproductive and respiratory syndrome virus, using a monoclonal antibody. Arch Virol. 2017;162:2239–46. https://doi.org/10.1007/s00705-017-3355-8.
Chen JZ, Wang Q, Bai Y, Wang B, Zhao HY, Peng JM, et al. Identification of two dominant linear epitopes on the GP3 protein of highly pathogenic porcine reproductive and respiratory syndrome virus (HP-PRRSV). Res Vet Sci. 2014;97:23843. https://doi.org/10.1016/j.rvsc.2014.07.011.
Feng X, Z CM, Wang HW. Genome-wide identification of ZmEXO70s gene family in Zea mays L. and association analysis of natural variations in ZmEXO70s genes with thermotolerance in seedlings. J South Agric. 2021;52:1–17.
Mao YJ, Gao BL. Comparative genomic analysis of the distribution and evolution of quorum sensing pathways in the Vibrio genus. J Trop Oceanogr. 2021;1–15.
Wang G, Yu Y, Zhang C, et al. Immune responses to modified live virus vaccines developed from classical or highly pathogenic PRRSV following challenge with a highly pathogenic PRRSV strain. Dev Comp Immunol. 2016;62:1–7. https://doi.org/10.1016/j.dci.2016.04.019.
Gong X, Cai J, Zhang B, Zhao Q, Piao J, Peng W, et al. A review of fluorescent signal-based lateral flow immunochromatographic strips. J Mater Chem B. 2017;5:5079–91. https://doi.org/10.1039/c7tb01049d.
Shen H, Chen H, Cheng Z, Ma L, Huang L, Xiao M, et al. A novel fluorescent immunochromatographic strip combined with pocket fluorescence observation instrument for rapid detection of PRV. Anal Bioanal Chem. 2018;410:7655–61. https://doi.org/10.1007/s00216-018-1379-x.
Xie K, Chen H, Peng B, Jin Z, Xiao W, Zhang Z, et al. On-site determination of classical swine fever virus (CSFV) by a fluorescent microsphere-based lateral flow immunoassay strip (FM-LFIAs) based on monoclonal antibodies. Anal Lett. 2020:1–16. https://doi.org/10.1080/00032719.2020.1860998.
Song X, Knotts M. Time-resolved luminescent lateral flow assay technology. Anal Chim Acta. 2008;626:186–92. https://doi.org/10.1016/j.aca.2008.08.006.
Qiao B, Li Y, Hu P, Sun Y, Si Z, Lu S, et al. EuNPs-MAb fluorescent probe based immunochromatographic strip for rapid and sensitive detection of fluorene. Sensors Actuators B Chem. 2018;262:221–7. https://doi.org/10.1016/j.snb.2018.01.231.
Chen H, Zhang X, Jin Z, Huang L, Dan H, Xiao W, et al. Differential diagnosis of PRV-infected versus vaccinated pigs using a novel EuNPs-virus antigen probe-based blocking fluorescent lateral flow immunoassay. Biosens Bioelectron. 2020;155:112101. https://doi.org/10.1016/j.bios.2020.112101.
Roy D, Park JW. Spatially nanoscale-controlled functional surfaces toward efficient bioactive platforms. J Mater Chem B. 2015;3(26):5135–49. https://doi.org/10.1039/c5tb00529a.
Xu F, Zhen G, Yu F, Kuennemann E, Textor M, Knoll W. Combined affinity and catalytic biosensor: in situ enzymatic activity monitoring of surface-bound enzymes. J Am Chem Soc. 2005;127(38):13084–5. https://doi.org/10.1021/ja050818q.
Li X, Bian H, Yu S, Xiao W, Shen J, Lan C, et al. A rapid method for antigen-specific hybridoma clone isolation. Anal Chem. 2018;90:2224–9. https://doi.org/10.1021/acs.analchem.7b04595.
Galfre G, Milstein C. Preparation of monoclonal antibodies: strategies and procedures. Methods Enzymol. 1981;73:3–46.
Van Breedam W, Costers S, Vanhee M, Gagnon CA, Rodriguez-Gomez IM, Geldhof M, et al. Porcine reproductive and respiratory syndrome virus (PRRSV)-specific mAbs: supporting diagnostics and providing new insights into the antigenic properties of the virus. Vet Immunol Immunopathol. 2011;141:246–57. https://doi.org/10.1016/j.vetimm.2011.03.008.
Wu Q, Li Z, Zhang G, Niu J, Zeng X, Sun B, et al. Genetic diversity and phylogenetic analysis of porcine reproductive and respiratory syndrome virus in southern China from 2007 to 2014. J Vet Sci. 2017;18:317–26. https://doi.org/10.4142/jvs.2017.18.3.317.
Xiao M, Huang L, Dong X, Xie K, Shen H, Huang C, et al. Integration of a 3D-printed read-out platform with a quantum dot-based immunoassay for detection of the avian influenza A (H7N9) virus. Analyst. 2019;144:2594–603. https://doi.org/10.1039/c8an02336k.
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2016YFD0500600).
Author information
Authors and Affiliations
Contributions
Junjie Yan: validation, formal analysis, visualization, investigation, resources
Bin Peng: validation, formal analysis, visualization, investigation, resources
Hui Chen: conceptualization, methodology, investigation, writing—original draft
Zhiyuan Jin: validation, visualization, investigation
Dongni Cao: investigation, supervision, validation
Qifang Song: investigation, writing—review and editing
Jianhua Ye: supervision, writing—review and editing
Hong Wang: supervision, writing—review and editing
Yong Tang: conceptualization, methodology, resources, investigation, funding acquisition, writing—review and editing
Corresponding authors
Ethics declarations
Ethics approval
All animal experiments in the present study were approved by the Jinan University Experimental Animal Welfare Ethics Committee.
Statement on animal welfare
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Conflict of interest
The authors declare no competing interests.
Source of biological material
HP-PRRSV strains (TY, TP, QJ, LQ, and JN-HS) were provided by Professor Gefen Yin and her team from the College of Veterinary Medicine, Yunnan Agricultural University.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(DOCX 1724 kb)
Rights and permissions
About this article

Cite this article
Yan, J., Peng, B., Chen, H. et al. On-site differential diagnostic detection of HP-PRRSV and C-PRRSV using EuNPs-mAb fluorescent probe-based immunoassay. Anal Bioanal Chem 413, 5799–5810 (2021). https://doi.org/10.1007/s00216-021-03558-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-021-03558-3