
Abstract
The aim of this research was to develop a method for simultaneous quantification of proteins and main polyphenolic compounds extracted from oleaginous meal by aqueous media. Size exclusion chromatography with a Biosep column (exclusion range from 1 to 300 kDa) and acetonitrile/water/formic acid (10:89.9:0.1 v/v) eluent at 0.6 mL min−1 yielded the most efficient separation of sunflower proteins and chlorogenic acid monoisomers (3-caffeoylquinic acid, 5-caffeoylquinic acid, and 4-caffeoylquinic acid). After a study of the stability of the extract components, the incorporation of a stabilization buffer (0.5 mol L−1 tris(hydroxymethyl)aminomethane-hydrochloric acid/1.0 mol L−1 sodium chloride at pH 7) was proposed to avoid polyphenol-protein interactions and/or isomeric transformation. The use of 214 nm as the wavelength for protein quantification was also included to minimize the effect of interference from polyphenol-protein interactions on the quantification. Under the used experimental conditions, the protein and chlorogenic acid monoisomer signals remained stable during 300 min at 20 °C (95–125% of the starting value). The developed method was validated and parameters such as specificity, sensitivity, precision, and accuracy were determined. The results from size exclusion chromatography correlated well with the results of protein determination by the reference Kjeldahl method. The proposed method was successfully applied for rapeseed extract analysis making simultaneous quantification of proteins and major rapeseed polyphenols (sinapine and sinapic acid) possible.

Graphical abstract
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Sunflower (Helianthus annuus L.) is widely cultivated in Europe because of high content of edible oils in the seeds. After oil extraction process, a solid residue called meal is produced. This meal is a major source of proteins (30–50% on dry matter basis [1,2,3,4]) used in state for animal nutrition. Sunflower proteins have an attractive amino acid compositions and interesting functional properties such foaming, emulsifying, and gelling [1,2,3,4,5,6]. Therefore, they are considered very promising for human nutrition as protein isolates or concentrates.
The production of protein isolates and concentrates from oleoproteaginous meal is classically achieved in two main steps. The first one is a protein extraction from the meal using an aqueous liquid phase at pH and ionic strength ranging from 5 to 12.5 and from 0 to 2.1 mol L−1 [6,7,8,9,10], respectively. The second one is a purification step in which proteins are separated from the non-protein compounds extracted during the first step. This is usually achieved by protein precipitation or ultrafiltration [7,8,9,10].
However, sunflower meals also contain significant amounts of phenolic compounds (1–4% on dry matter basis), particularly chlorogenic acids (over 70% of all phenolic compounds) [2]. These polyphenols are solubilized with proteins during the protein extraction step. Chlorogenic acids are responsible of an unsuitable greenish color and off-flavors [2, 4]. Furthermore, they limit protein nutritional and functional properties [2, 4, 11]. Thus, these compounds need to be efficiently separated from proteins for the isolate production and food applications.
The process of extraction/purification revealed particularly challenging as chlorogenic acid interacts with proteins by non-covalent or covalent bonding. Non-covalent bonds may be hydrogen, ionic or hydrophobic interactions. They can be modulated by pH and ionic strength [11]. Covalent bonding occurs with protein amine functions when phenolic compounds are oxidized into quinones. The covalent interactions are also modulated by the pH since the chlorogenic acid oxidation mainly occurs under basic conditions [2, 4, 11]. The precise mechanism is not well understood to date though. Hence, quantification of both proteins and free polyphenols in the aqueous phases from extraction and purification steps is of major interest for either monitoring/optimizing the isolate production or to study phenol-protein interactions.
Although several methodologies have previously been proposed to analyze proteins and polyphenols in liquid samples, each technique has drawbacks and none allows for simultaneous quantification. The reference Kjeldahl procedures used in routine for protein measurement are highly fastidious, time-consuming, and harmful for both the researcher and the environment. In addition, rapid and simple colorimetric techniques (Bradford, Biuret, Lowry, and bicinchoninic acid assays) are not adapted for aqueous plant extract because of signal interferences in complex experimental matrix [3, 12,13,14]. On the other hand, the first choice for identification and quantification of phenolic compounds from plant material is reversed phase chromatography (RP-HPLC). Multiple methods have been reported using C18 columns with gradient elution of mobile phases composed of ultrapure water and polar acidified organic solvent [15,16,17]. However, most of those methods were aimed at analyzing polyphenols only. They were employed also for meal solvent extracts using methanol or ethanol and consequently contained very few interfering molecules.
The aim of this work was to develop a new chromatographic method suitable for simultaneous quantification of proteins and phenolic compounds co-extracted in sunflower aqueous phase. The strategy was divided into four main parts. First, optimal chromatographic parameters for satisfying separation of both proteins and polyphenols were established. Secondly, the stability of proteins and chlorogenic acids in extracts obtained in a range of typical extraction conditions (pH, ionic strength) was evaluated. This step was aimed at finding the more suitable stabilization conditions prior analysis. The developed assay was subsequently validated according to current recommendations of International Conference of Harmonization (ICH) [18] and protein quantification was compared with the reference Kjeldahl method. Finally, the performance of chromatographic method was tested for other oleaginous meal (rapeseed).
Materials and methods
Plant materials and chemicals
Sunflower and rapeseed meals defatted by n-hexane were provided by Saipol (Bassens, France) and Olead (Pessac, France) respectively. Sodium chloride (NaCl, CAS 7647-14-201), sodium hydroxide pellets (NaOH, CAS 1310-73-2), and ammonium sulfate ((NH4)2SO4, CAS 7783-20-2) were obtained from VWR (Darmstadt, Germany). Hydrochloric acid (HCl, CAS 7647-01-0) was from Carlo Erba (Milan, Italy) and tris(hydroxymethyl)aminomethane (Tris, CAS 77-86-1) from Fisher Scientific (Hampton, USA). 5-O-caffeoylquinic acid (5-CQA, CAS 327-97-9), 4-O-caffeoylquinic acid (4-CQA, CAS 905-99-7), 3-O-caffeoylquinic acid (3-CQA, CAS 906-33-2), 3,4-O-dicaffeoylquinic acid (3,4-diCQA, CAS 14534-61-3), 3,5-O-dicaffeoylquinic acid (3,5-diCQA, CAS 2450-53-5), 4,5-O-dicaffeoylquinic acid (4,5-diCQA, CAS 57378-72-0), caffeic acid (CAS 331-39-5), quinic acid (CAS 77-95-2), trans-sinapic acid (CAS 7362-37-0), ferulic acid (CAS 1135-24-6), syringic acid (CAS 530-57-4), p-coumaric acid (CAS 501-98-4), and 4-hydroxybenzoic acid (CAS 99-96-7) were purchased from Sigma-Aldrich (St. Louis, USA). Caffeoylquinic acid names were established according to current IUPAC recommendations [19]. For chromatographic analysis acetonitrile (ACN), ultrapure water (H20) with resistivity ≥ 18.2 MΩ cm−1 and formic acid were used. All reagents were LC-MS grade supplied from Fisher Scientific (Hampton, USA).
Preparation of aqueous extracts and sunflower protein powder
Prior to extraction, oleaginous meal was milled and sieved through mesh size of 150 μm. The solid/liquid extraction was performed by stirring the meal with distilled water or 0.5 mol L−1 NaCl (1:9 w/w) during 30 min at room temperature. The pH of extraction mixture was adjusted to 5, 7, or 9 ± 0.05 using 1.0 mol L−1 NaOH or 1.0 mol L−1 HCl solutions. After centrifugation step (15,000×g for 30 min at 20 °C), supernatant was collected and filtered by 0.22-μm pore size membrane.
For calibration and validation purpose, further purification of proteins extracted at pH 7 and 0.5 mol L−1 from sunflower meal was performed using low-pressure chromatography Akta Pure system from GE Healthcare (IL, USA). Ammonium sulfate was added to aqueous extract up to 60% of saturation and stirred for 20 min at a room temperature. After this time and centrifugation step (15,000×g for 30 min at 20 °C), obtained pellet was dissolved in 60 mL of 0.25 mol L−1 Tris-HCl/0.5 mol L−1 NaCl buffer solution at pH 7. The elution was carried out at 60 mL min−1 with deionized water on Sephadex G-25 Fine silica gel (GE Healthcare, IL, USA). The peak corresponding to proteins recorded at 280 nm was collected and freeze-dried.
SE-HPLC analysis
The analyses were conducted on HPLC Shimadzu LC20 system equipped with a binary pump, a micro wave degasser, a thermostated autosampler and column compartment, a fraction collector (FRC-10A), a photodiode array (PDA) detector, and an electrospray quadrupole mass spectroscopy (MS) detector operated by LabSolutions software, all from Shimadzu Corporation (Kyoto, Japan). Five microliters of sample from autosampler set at 20 °C was injected into Biosep SEC s-2000 column (300 × 7.8 mm; 5 μm, molecular weight range 1–300 kDa) from Phenomenex (Torrance, USA) maintained at 35 °C. A mixture of a acetonitrile and water with 0.1% formic acid (ACN/H20/formic acid (10:89.9:0.1 (v/v)) was used as the eluent, at a flow of 0.6 mL min−1. The UV signal was acquired between 190 and 400 nm with maximal absorption wavelength set at 214 nm or 280 nm for protein detection. The absorbance of phenolic compounds was recorded at 325 nm (caffeoylquinic, di caffeoylquinic, sinapic, caffeic, and ferulic acid), 217 or 275 nm (syringic acid), 310 nm (p-coumaric acid), and 255 nm (4-hydroxybenzoic acid). Mass spectra for phenolic compounds were recorded in positive electrospray ionization mode using both total ion chromatogram (TIC) and selected ion monitoring (SIM) with the main signals for phenolic components being 355.3+ (caffeoylquinic acid), 517.5+ (dicaffeoylquinic acid), 181.2+ (caffeic acid), 193.2+ (quinic acid), 225.2+ (sinapic acid), 195.2+ (ferulic acid), 199.2+ (syringic acid), 165.1+ (p-coumaric acid), and 139.1+ (4-hydroxybenzoic acid).
Protein and polyphenolic compounds separation by SE-HPLC
The Biosep SEC s-2000 column was calibrated with different molar weight protein solutions (MWGF200 Kit from Sigma-Aldrich, St. Louis, USA) containing blue dextran (2000 kDa), β-amylase (200 kDa), alcohol dehydrogenase (150 kDa), bovine serum albumin (66 kDa), carbonic anhydrase (29 kDa), and cytochrome c (12.4 kDa). The standard solutions of proteins were prepared as recommended at concentration ranging from 2 to 10 g L−1 in 0.05 mol L−1 Tris-HCl buffer at pH 7.5 with 0.1 mol L−1 KCl and 5% of glycerol (v/v). Five microliters of sample was injected into chromatographic column under the suitable elution condition (ACN/H20/formic acid (10:89.9:0.1) (v/v)) and the signal was recorded at 280 and 214 nm. Three different runs were performed and calibration curve (Electronic Supplementary Material (ESM) Fig. S1) was expressed as log MW vs. Ve/V0, where Ve corresponds to elution volume of standard protein and V0 to dead volume determined using blue dextran.
To confirm the retention time of proteins, 5 μL of aqueous extract (pH 9/0 mol L−1 NaCl) was eluted under the same conditions and fractions were collected every 1 mL (about 1.67 min) at the UV-detector outlet. The UV signal was acquired from 190 to 400 nm with maximal absorption wavelength set at 280 or 214 nm for protein detection. The fractions were dried overnight through rotary evaporator using SpeedVap from Eppendorf (Hamburg, Germany) and then analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
The identification of phenolic compounds in aqueous extract was established by comparing the retention time of obtained peaks from sunflower and rapeseed extracts with those of commercially available standards using dual PDA/MS detection. The phenolic compound solutions were dissolved in ethanol or ethanol/water mixture at the concentrations ranged from 1 to 2 g L−1. The injection volume for all samples was set at 5 μL.
Sample stability
The stability of the UV signal of proteins and chlorogenic acid isomers in sunflower extract was studied with six extraction conditions including various pH (5, 7, and 9) and NaCl concentration (0 or 0.5 mol L−1). The protein stability was evaluated 300 min after extraction at 20 °C using two wavelengths, 280 and 214 nm. Additionally, the kinetics of signal modifications of chlorogenic acid isomers were carried out during 300 min at 325 nm after extraction at 20 °C with the time interval of 60 min. For stabilization assay, aqueous extract was combined with buffer solution (50:50 v/v) prior to analysis. The results were expressed as the percentage of UV surface peak in relation to initial value presumed to 100%. Each stability test was repeated in triplicate and the average values with standard deviation (% SD) were calculated.
Method validation
The performance of the developed method was evaluated by determination of validation parameters such as specificity, linearity, limit of detection and quantification, repeatability, intermediate precision, and accuracy.
Sunflower proteins, unlike chlorogenic acids, are not available commercially as a reference substance. The extraction and purification was carried out as described in the “Preparation of aqueous extracts and sunflower protein powder” section. The obtained sunflower protein powder with the purity determined by the reference Kjeldahl method (86.0% on dry matter basis) was used for validation procedures. Analytical grade standards of 3-CQA, 5-CQA, and 4-CQA with purity ≥ 99.0% were provided by Sigma-Aldrich (St. Louis, USA) and used in the validation.
Specificity was evaluated by comparing the retention times of sunflower proteins, 5-CQA, 3-CQA, and 4-CQA in individual and mixed solutions with those of sunflower extract components under addition of 0.25 mol L−1 Tris-HCl/0.5 mol L−1 NaCl at pH 7.
The calibration of protein content was based on sunflower proteins isolated in-house (86.0% on dry matter basis) in the range of concentration from 0.82 to 20.43 g L−1 in buffer solution of 0.25 mol L−1 Tris-HCl/0.5 mol L−1 NaCl at pH 7. The calibration curves of chlorogenic acid monoisomers were prepared by dissolving of commercially available 3-CQA, 5-CQA, and 4-CQA in 50% ethanol mixture (v/v) in the range of concentration from 0.05 to 5.00 g L−1 or from 0.01 to 1.00 g L−1 for 5-CQA or 3-CQA and 4-CQA, respectively. Each calibration plot represents an average of three independent repetitions for six concentration levels. Linear regression analysis was performed to determine curve slop and correlation coefficient (R2). Limit of detection (LOD) and limit of quantification (LOQ) defined as a ratio signal-to-noise ≥ 3 or ≥ 10, respectively, were also calculated.
The repeatability and intermediate precision were determined as measurement variability (% RSD) over the same day or 3 consecutive days, respectively. For this purpose, the standard solutions with three concentration levels of sunflower proteins (4.08, 8.17, 20.42 g L−1), 5-CQA (0.10, 0.50, 5.00 g L−1), and 3-CQA and 4-CQA (0.05, 0.20, 1.00 g L−1) were prepared and injected in triplicate.
For accuracy evaluation, the percent recovery method was used. Three standard solutions of sunflower proteins (0.82, 4.08, 8.24 g L−1), 5-CQA (0.10, 0.50, 5.00 g L−1), and 3-CQA and 4-CQA (0.05, 0.20, 1.00 g L−1) were spiked with sunflower aqueous extract. After triplicate analysis by SE-HPLC method, the average of recovery of nominal concentrations and the variability (% RSD) were calculated.
Protein quantification and characterization
Kjeldahl method
The procedure of the Kjeldahl assay was carried out as described in AOAC method 991.20 [20]. Assessment of total nitrogen content was performed on SpeedDigester K-439, Scrubber K-415, Kjel-Flex K-360, all from Butchi Labortechnik (Flawil, Swiss) coupled with Titrino Plus 877 from Metrohm (Herisau, Swiss). The mineralization of organic matter was achieved by heating samples with 96% sulfuric acid (v/v) and catalyzer at 450 °C during 150 min. Following distillation step, the nitrogen quantity was indirectly determined by titration against 0.01 mol L−1 HCl. The non-protein nitrogen in samples was measured in supernatant after protein precipitation using 50% trichloroacetic acid (w/v). A factor of N × 5.6 was applied to conversion of nitrogen to sunflower proteins. Each analysis was repeated in triplicate and averaged concentration was calculated.
SDS-PAGE analysis
SDS-PAGE characterization was carried out according to the Leammli method [21]. Dried samples were solubilized in 50 μL of Laemmli buffer containing 2% β-mercaptoethanol (v/v) and heated at 95 °C for 5 min. Ten microliters of samples and molecular weight markers (ranged from 250 to 10 kDa and from 26.6 to 6.5 kDa) were applied to 5 and 17% of polyacrylamide stacking and resolving SDS-PAGE gels, respectively. After migration step at 20 mA, the gels were stained with Coomassie Brilliant Blue and destained by incubation overnight in 10% acetic acid solution (v/v). All reagents and electrophoretic equipment were purchased from Bio-Rad (Hercules, USA).
Results and discussion
Protein and polyphenolic compounds separation by SE-HPLC
High-performance liquid chromatography (HPLC) is a powerful analytical technique that can offer efficient, rapid, and fully automated quantification of multicomponent mixtures. Among various chromatographic modes, the size exclusion chromatography (SEC) was taken into consideration for method development because of the significant difference in molecular weight between the target component, proteins (10–300 kDa), and phenolic compounds (0.1–0.6 kDa).
The representative chromatogram of analyzed sunflower aqueous extract using size exclusion high-performance liquid chromatography (SE-HPLC) method is shown in Fig. 1. The first peak is visible from 7 to 10.5 min of run time at the wavelength of 280 nm (Fig. 1a). From the column calibration (ESM Fig. S1), this peak with retention time at 8.6 min corresponds to molecular weight ranging from 12.4 to 200 kDa (expected size 72 kDa). Thus, the signal probably represents elution of the mix of sunflower globulins (300–350 kDa) and albumins (10–18 kDa). To verify the presence and identity of the proteins, the eluate from the column was fractionated from 0 to 60 min and evaluated by SDS-PAGE. The results revealed the presence of acidic and basic subunits of globulins (bands 20–50 kDa) and albumins (bands 10–18 kDa) [1] in the collected fractions 5 and 6 corresponding to elution times between 6.7 and 10.0 min. Hence, the elution of sunflower proteins was confirmed at the previously mentioned retention time.

SE-HPLC chromatogram of aqueous extract form sunflower meal (pH 9/0 mol L−1 NaCl) using mobile phase consisting of ACN/H20/formic acid (10:89.9:0.1 v/v). UV signal was recorded at 280 (a) and 325 nm (b). Internal figures represent SDS-PAGE gel of protein fractions (0–60 min) (a) and MS spectrum of sunflower extract and the mixture of 3-CQA, 5-CQA, and 4-CQA reference standards detected at m/z 355.3+ (b)
In addition, three distinct peaks were observed between 40 and 50 min (Fig. 1b). The recorded UV spectra of all peaks showing maximum absorption at 325 nm with a shoulder at 295 nm (ESM Fig. S2b). This indicated the detection of three different phenolic compounds [22]. The main phenolic compound is well known to be chlorogenic acid [15]. This molecule is an ester of quinic and cafeic acids. Depending on the substitution position of caffeoyl moiety, this molecule occurs under different isomeric forms. The most common chlorogenic acid isomers according to current IUPAC numbering are 5-caffeoylquinic acid (5-CQA), 3-caffeoylquinic acid (3-CQA or neochlorogenic acid), and 4-caffeoylquinic acid (4-CQA or cryptochlorogenic acid). As illustrated in Fig. 1b, elution of 3-CQA, 5-CQA, and 4-CQA standards was detected, respectively, at 41.9, 45.1, and 48.4 min at 325 nm or at 42.1, 45.1, and 48.5 using positive ionization mode (m/z 355.3+). Based on these results, the three phenolic peaks found in sunflower aqueous extract were unambiguously assigned to 3-CQA, 5-CQA, and 4-CQA. Interestingly, these compounds were eluted after total volume of column (about 15 min). This suggests secondary hydrophobic interactions between phenolic compounds and silica-bonded matrix. Usually, the interactions between stationary phase and analytes are undesirable in size exclusion chromatographic mode since they increase the elution duration. However, in this case, the secondary interactions, specific for the phenolic compounds, resulted in satisfactory resolution (Rs ≥ 2) between all target components. Hence, in the proposed method, mixed mechanism presents an important benefit for simultaneous quantification purpose.
The presence of minor dimeric forms of chlorogenic acids (3,4-dicaffeoylquinic acid, 3,5-dicaffeoylquinic acid, 4,5-dicaffeoylquinic acid) in sunflower kernels and shells has been reported previously in methanol or ethanol extracts of meal [15, 16]. These compounds were not extracted in water phase. This may be related to poor polarity of dimeric chlorogenic acids that should be far less soluble in aqueous extracts.
Sample stability
It is widely known that phenol-protein covalent bonding occur under basic conditions. Such interactions after sampling would result in a bias in chlorogenic acid or protein quantification. Figure 2 shows the SE-HPLC chromatogram at 325 nm of alkaline sunflower extract (pH 9) just after sampling and 300 min later. Under this condition, the signal of 5-CQA clearly decreased in favor of 3-CQA and 4-CQA. Meanwhile, a slight increase of the signal at the elution time of proteins was also noted. Hence, two types of changes were observed in 300-min storage time under basic conditions. The first one was the expected covalent bonding of chlorogenic acid to sunflower proteins. This might result in inaccurate determination of overall chlorogenic acid and protein contents. The second one was isomerization of 5-CQA leading to bias in chlorogenic isomer compositions in the sample. Noteworthy, 5-CQA was not degraded into other molecules.

SE-HPLC chromatogram of aqueous extract form sunflower meal (pH 9/0 mol L−1 NaCl) recorded at 325 nm at initial time (dashed black line) and 300 min after extraction (continuous red line). Temperature was kept at 20 °C
Thus, the stability of chlorogenic isomers and protein signals in the classical pH and ionic strength range of protein extraction or purification processes was investigated. The stability test was carried out during 300 min, which would correspond to the average storage time from sample collections to analysis. This study was performed in order to find conditions in which reduced interaction/bias occurs for stabilizing samples.
Chlorogenic acid stability
The graphs in Fig. 3a–c show the kinetics of 3-CQA, 5-CQA, and 4-CQA isomerization during 300 min at pH (5, 7, and 9) with or without NaCl (0.5 mol L−1). This figure clearly demonstrated that the stability of chlorogenic acid isomers is mainly pH-dependent. Changes were only observed under alkaline conditions where 5-CQA signal was gradually decreased to reach the ultimate value close to 50% after 300 min of analysis. Simultaneously, the increase in 3-CQA signal (about 250% of initial value) and 4-CQA (about 150% of initial value) was noticed over test time. In contrast, in neutral and acid pH range, the content of chlorogenic acid isomers remains relatively stable. The ionic strength has a secondary importance on stability of phenolic compounds in aqueous solution. This observation is in line with recent researches, where 3-CQA, 5-CQA, and 4-CQA have been denoted as strong reactive molecules able to positional acyl migration in basic pH values. Consequently, 5-CQA concentration reduces, whereas 3-CQA and 4-CQA signal increases. Moreover, it has also been confirmed that 3-CQA is the most stable chlorogenic monoisomers [23,24,25]. These previous data could explain the rapid increase of 3-CQA in comparison to the transformation rate of other isoforms. Besides these observations, no new degradation product was identified after 300 min after extraction.

Kinetic of phenolic compound isomerization as a function of pH and salt concentration of sunflower extract during 300 min without stabilization: 3-CQA (a), 5-CQA (b), 4-CQA (c), and after addition of stabilization buffer (0.5 mol L−1 Tris-HCl/1.0 mol L−1 NaCl buffer solution at pH 7): 3-CQA (a’), 5-CQA (b’), 4-CQA (c’)
In light of these results, various buffer systems (citric, phosphate, bicarbonate, Tris-HCl), concentrations (0.1–1.0 mol L−1), and pH (3–9) were tried to stabilize the sample whatever its pH or ionic strength conditions. Figure 3a’–c’ presents the modification of 3-CQA, 5-CQA, and 4-CQA signals after stabilization with the most suitable buffer (0.5 mol L−1 Tris-HCl/1.0 mol L−1 NaCl at pH 7 (50:50 v/v)). This figure shows an acceptable stability of phenolic peaks (between 95 and 125% of initial signal of 3-CQA, 5-CQA, and 4-CQA) over the storage time. Indeed, addition of this buffer solution allows maintaining a neutral pH of extract, thereby reducing the transformation of chlorogenic acid isomers. Furthermore, due to high ionic strength of buffer no protein precipitation occurs in stabilized extract. Thus, the buffer efficiently limited the bias associated to the extraction and/or purification conditions.
Protein stability
The histogram of Fig. 4 represents the protein signal modification (protein peak area at 280 nm) after 300 min from sampling. The protein signal remained stable under acidic or neutral conditions with salt addition (100–105% of initial value after 300 min). In contrast, the protein peak area slightly increased in alkaline conditions regardless of ionic strength (> 120% of starting value after 300 min). Nevertheless, no degradation products or shift in retention time of proteins were observed. This observation is probably the consequence of the phenolic covalent bonding occurring under these conditions. Indeed, such interactions probably added a phenolic contribution in absorbance at 280 nm to the proteins peak (Fig. 2). This leads to a somewhat overestimation of protein content in samples at pH 9 as chlorogenic acids absorb in the range of protein absorption as well.

Stability of sunflower protein surface peak as a function of extracting conditions (pH and salt concentration) measured after 300 min by SE-HPLC method. The results for 280 and 214 nm of analytical wavelengths were compared
Interestingly, the extinction coefficient of proteins is more than ten times higher at 214 nm comparing to 280 nm [26], while absorption of chlorogenic acid does not differ much at those two wavelengths (ESM S2). Hence, protein quantification at 214 nm would probably minimize the interferences of the protein signals originating from the signal of complexed phenolic compounds. To verify this hypothesis, the stability of protein signal was also studied at 214 nm. As it is shown in Fig. 4, the significant improvement in signal stability measured at 214 nm in comparison with 280 nm was noticed. All values are within the 95–105%, including alkaline extraction conditions. Thus, the application of the wavelength at 214 nm for protein analysis reduces the observed bias of phenolic contribution on protein quantification.
Performance of SE-HPLC method for simultaneous quantification
Specificity, linearity, limit of detection and quantification, precision, and accuracy
The developed SE-HPLC method was validated according to the internationally recommendations described in ICH Guideline Q2 (R1) [18]. The performance for simultaneous quantification was examined in terms of specificity, limit of detection (LOD), limit of quantification (LOQ), repeatability, intermediate precision, and accuracy. Table 1 summarizes in details the results of these validation procedures.
Specificity was assessed by controlling the ability of SE-HPLC method to distinguish between sunflower proteins and chlorogenic acid isomers present in sunflower extract. The chromatograms of sunflower proteins, 5-CQA, 3-CQA, and 4-CQA in individual and mixed solutions were compared to that of the sunflower extracts under the addition of 0.25 mol L−1 Tris-HCl/0.5 mol L−1 NaCl at pH 7 (ESM Fig. S3). In all above combinations, the retention times of sunflower proteins, 3-CQA, 5-CQA, and 4-CQA were found at around 8, 42, 45, and 48 min. The absence of signal interference confirms unambiguous elution of target compounds.
The calibration curves were plotted as the peak surface against the known concentration of analytes (ESM Fig. S4). For this purpose, a set of solutions consisting of six concentration levels of sunflower proteins powder (0.82–20.43 g L−1), 3-CQA (0.01–1.00 g L−1), 5-CQA (0.05–5.00 g L−1), and 4-CQA (0.01–1.00 g L−1) were analyzed by SE-HPLC method. The regression equations, intercepts, and regression coefficients (R2) are displayed in Table 1. The excellent R2 close to 1 determined for all calibration curves reflects the linear relationship between the signal intensity and analyte content in the sample. Moreover, these calibration ranges cover completely the classical concentrations found in aqueous extracts of sunflower meal. Under chromatographic conditions, the LOD and the LOQ were calculated as a ratio signal-to-noise ≥ 3 or ≥ 10. The low values of LOD and LOQ determined for sunflower proteins (17.6 mg L−1 and 58.7 mg L−1), 3-CQA (0.7 mg L−1 and 2.2 mg∙L−1), 5-CQA (0.9 mg∙L−1 and 3.0 mg L−1), and 4-CQA (1.5 mg L−1 and 5.0 mg L−1) guarantee a high sensitivity of the developed SE-HPLC method.
Precision of SE-HPLC method was measured using the samples of sunflower proteins and chlorogenic acid monoisomers with three different concentration levels. The intra-day precision (repeatability) was obtained by repeating three times quantification of prepared samples by SE-HPLC within the same day. The same samples were analyzed subsequently over 3 consecutive days in order to determine the inter-day precision (intermediate precision). The coefficients of variation (RSD %) of repeatability and intermediate precision measured for all compounds of interest were found to be within acceptable limits: RSD < 3% (repeatability) and RSD < 5% (intermediate precision), respectively.
Accuracy was evaluated by determining the recovery of analytes spiked with sunflower aqueous extract. For this purpose, three different concentration levels of sunflower proteins (0.82, 4.08, 8.24 g L−1), 5-CQA (0.10, 0.50, 5.00 g L−1), and 3-CQA and 4-CQA (0.05, 0.20, 1.00 g L−1) were added. The average accuracy calculated on the basis of triplicate analysis of each sample ranged from 99.4 to 100.6% with the maximal RSD ≤ 2.5%. Above results clearly demonstrate a good performance of the developed analytical method for simultaneous quantification of all target compounds.
Comparison protein quantification by SE-HPLC and reference Kjeldahl method
In addition to statistical validation tests, the performance of SE-HPLC method for quantification of proteins in aqueous extract of sunflower meal was examined in relation to the reference Kjeldahl method.
In the graph of Fig. 5, the protein contents as determined by SE-HPLC method for ten different extraction conditions of pH (5, 6, 7, 8, 9) and NaCl concentrations (0 or 0.5 mol L−1) are plotted against the results obtained by the Kjeldahl procedure. As previously described, the calibration of chromatographic methods was carried out using the homemade sunflower protein powder (purity of 86.0% on dry matter basis). The aqueous extracts were stabilized by adding of buffer (50:50 v/v) containing 0.25 mol L−1 Tris-HCl/0.5 mol L−1 NaCl at pH 7 prior to SE-HPLC analysis. For the Kjeldahl method, the non-protein nitrogen was deduced after protein precipitation by TCA solution (40% w/v). As illustrated in the graphs of Fig. 5, acceptable agreement (R2 = 0.952) was achieved between concentrations of proteins measured by SE-HPLC and the reference Kjeldahl method. These results demonstrate that the proposed SE-HPLC method represents a reliable and powerful alternative assay.

Result comparison for protein quantification in sunflower extracts obtained by Kjeldahl method SE-HPLC method
Simultaneous quantification of sunflower extract components
The data in Table 2 display the results of simultaneous measurement of proteins and chlorogenic acid isomers obtained by SE-HPLC method. The compounds were extracted from sunflower meal in different pH (5, 6, 7, 8, 9) and NaCl concentration (0 or 0.5 mol L−1). Prior to SE-HPLC analysis, aqueous extracts were stabilized by adding of buffer (50:50 v/v) containing 0.25 mol∙L−1 Tris-HCl/0.5 mol L−1 NaCl at pH 7. As described above, chromatographic method was calibrated using the homemade sunflower protein powder (purity of 86.0% on dry matter basis) and commercially available 3-CQA, 5-CQA, and 4-CQA.
Regarding the protein content, the concentration increases mainly with pH to reach maximal value (14–15 g L−1) under alkaline conditions. The effect of salt addition was especially significant for acidic and neutral extraction (pH 5–7). This finding fits well with the general trend of protein extractability from sunflower meal described before by Pickardt et al. [3] and Ivanova et al. [27]. Similar conclusion has been drawn for phenolic quantification. The present results indicate 5-CQA as the predominant isomer found in sunflower meal. This observation is in good accordance with the majority of previous data [2, 15, 16, 28]. On the other hand, the amounts of 3-CQA and 4-CQA rise for high pH values due to 5-CQA degradation into those isoforms [23,24,25]. However, comparison the obtained content of phenolic compounds with already reported studies is difficult, because of lack of methods for polyphenol quantification directly in aqueous sunflower extract.
Application of SE-HPLC method for rapeseed extract
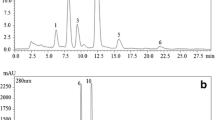
The performance of the proposed SE-HPLC method for simultaneous quantification of proteins and phenolic compounds was also evaluated for rapeseed aqueous extract. The same mobile phase composition used for sunflower meal (ACN/H20/formic acid (10:89.9:0.1 v/v)) was applied for rapeseed extract. A chromatogram obtained for a rapeseed extract at pH 9 (Fig. 6) shows similarities to the sunflower extract. An initial signal at 8.7 min at 214 nm is probably due to protein present in the sample. To verify this, as was done for sunflower samples, fractions were collected during elution. SDS-PAGE shows the presence of rapeseed globulin (20–30 kDa) and albumin (5–10 kDa) subunits bands [8] in the collected fractions 5 and 6. Simultaneously, the elution of two well-defined peaks of phenolic derivatives was recorded at 325 nm at 11–45 min of run time. To identify the observed phenolic derivatives, the retention times were compared to those of reference standards. As a result, elution of the major phenolic compounds in rapeseed sinapine and sinapic acid was confirmed at 11.0 and 39.5 min, respectively. Hence, the developed SE-HPLC method could be also successfully applied to the quantification of the principal extract components from this oleaginous source.

SE-HPLC chromatogram of aqueous extract form rapeseed meal (pH 9/0 mol L−1 NaCl) recorded at 214 and 325 nm. Internal figure represents reducing SDS-PAGE gel of collected protein fractions (0–15 min)
Conclusion
Quantification of proteins and phenolic compounds in aqueous extract is challenging because of complex protein/phenolic behaviors during extraction/purification process. The present work showed a reliable and consistent SE-HPLC method for simultaneous quantification of proteins and main chlorogenic acid isomers from sunflower aqueous extract. For the first time, a complete and efficient separation between target analytes has been achieved in one simple analysis. Furthermore, the impact of protein-polyphenol interactions and/or phenolic transisomerization on signal stability in sample was significantly minimized using suitable experimental conditions (buffer addition and 214 nm for protein measurement). The developed method was also validated in terms of specificity, sensitivity, precision, and accuracy according to the international recognized ICH procedures. Acceptable correlation for protein concentrations in comparison with the reference Kjeldahl method confirmed the performance of the proposed assay. Moreover, the applicability of SE-HPLC method was demonstrated for proteins and phenolic compounds (sinapine and sinapic acid) from rapeseed aqueous extract. Thus, the developed chromatographic method represents an advanced and alternative bioanalytical tool allowing understanding of protein-phenol system in liquid phase and optimizing of protein extraction and purification process.
References
Gonzalez-Perez S. Sunflower proteins: overview of their physicochemical, structural and functional properties. J Sci Food Agric. 2007;87:2173–91.
Wildermuth S. Chlorogenic acid oxidation and its reaction with sunflower proteins to form green-colored complexes. Compr Rev Food Sci Food Saf. 2016;15:829–43.
Pickardt C. Optimisation of mild-acidic protein extraction from defatted sunflower (Helianthus annuus L.) meal. Food Hydrocoll. 2009;23:1966–73.
Bongartz V. Evidence for the formation of benzacridine derivatives in alkaline-treated sunflower meal and model solutions. Molecules. 2016;21:91.
Ivanova P. Functional properties of proteins isolated from industrially produced sunflower meal. Int J Food Sci. 2014;3:203–12.
Pickardt C. Pilot plant preparation of light-coloured protein isolates from de-oiled sunflower (Helianthus annuus L.) press cake by mild-acidic protein extraction and polyphenol adsorption. Food Hydrocoll. 2015;44:208–19.
Gueguen J. Plant proteins: context and potentialities for human food. Cahiers de nutrition et de dietetique. 2016;51:177–85.
Aider M. Canola proteins: composition, extraction, functional properties, bioactivity, applications as a food ingredient and allergenicity - a practical and critical review. Trends Food Sci Technol. 2011;22:21–39.
Tan SH. Canola proteins for human consumption: extraction, profile, and functional properties. J Food Sci. 2011;76:16–28.
Gonzalez-Perez S. Isolation and characterization of undenatured chlorogenic acid free sunflower (Helianthus annuus) proteins. J Agric Food Chem. 2002;50:1713–9.
Ozdal T. A review on protein-phenolic interactions and associated changes. Food Res Int. 2013;51:954–70.
Sapan CV. Colorimetric protein assay techniques. Anal Biochem. 1999;29:99–108.
Reichelt WN. Bioprocess monitoring: minimizing sample matrix effects for total protein quantification with bicinchoninic acid assay. Anal Biochem. 2016;43:1271–80.
Winters AL. Modification of the Lowry assay to measure proteins and phenols in covalently bound complexes. Anal Biochem. 2005;346:43–8.
Weisz GM. Identification and quantification of phenolic compounds from sunflower kernels and shells by HPLC-DAD/ESI-MSn. LWT Food Chem. 2009;115:758–65.
Pedrosa M. Determination of caffeic and chlorogenic acids and their derivatives in different sunflower seed. J Sci Food Agric. 2000;80:459–64.
Li J. Identification and quantification of phenolic compounds in rapeseed originated lecithin and antioxidant activity evaluation. LWT Food Sci Technol. 2016;73:397–405.
ICH I. Q2 (R1): Validation of analytical procedures: text and methodology. In: International Conference on Harmonization, Geneva. 2005.
Clifford MN. Chlorogenic acids and other cinnamates - nature, occurrence, dietary burden, absorption and metabolism. J Sci Food Agric. 2000;80:1033–43.
AOCS International. Official Method 991.20. Nitrogen (total) in milk. Official methods of analysis, 19th edn. Gaithersburg: AOCS International; 1995.
Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5.
Friedman M. Effect of pH on the stability of plant phenolic compounds. J Agric Food Chem. 2000;48:2101–10.
Xue M. Stability and degradation of caffeoylquinic acids under different storage conditions studied by high-performance liquid chromatography with photo diode array detection and electrospray ionization mass spectrometry. Molecules. 2016;21:948.
Yusaku N. Degradation kinetics of chlorogenic acid at various pH values and effects of ascorbic acid and epigallocatechin gallate on its stability under alkaline conditions. J Agric Food Chem. 2013;61:966–72.
Deshpande S. Investigation of acyl migration in mono- and di-caffeoylquinic acids under aqueous basic, aqueous acidic, and dry roasting conditions. J Agric Food Chem. 2014;62:9160–70.
Hong P. A review size exclusion chromatography for the analysis of protein biotherapeutics and their aggregates. J Liq Chromatogr Relat Technol. 2012;35:2923–50.
Ivanova P. Optimization of protein extraction from sunflower meal produced in Bulgaria. Bulg J Agric Sci. 2012;18:153–60.
Weisz GM. Sustainable sunflower processing - II. Recovery of phenolic compounds as a by-product of sunflower protein extraction. Innov Food Sci Emerg Technol. 2013;17:169–79.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Human/animal rights
This article does not contain any studies with human or animal subjects performed by the any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 350 kb)
Rights and permissions
About this article

Cite this article
Albe Slabi, S., Mathé, C., Framboisier, X. et al. A new SE-HPLC method for simultaneous quantification of proteins and main phenolic compounds from sunflower meal aqueous extracts. Anal Bioanal Chem 411, 2089–2099 (2019). https://doi.org/10.1007/s00216-019-01635-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-019-01635-2