
Abstract
The sea lamprey (Petromyzon marinus) is a destructive invasive species in the Great Lakes. Since the 1960s, tons of the lampricide 3-trifluoromethyl-4-nitrophenol (TFM) has been applied to selected tributaries each year to eliminate or reduce sea lamprey larval populations. Therefore, the environmental impact of TFM needs to be evaluated. However, the metabolism of TFM and its mechanism of selective toxicity in sea lamprey is not yet fully understood. Based upon our previous report on the identification, synthesis, and characterization of TFM metabolites observed in liver incubates from sea lamprey and non-target fishes, we now provide a robust assay for quantifying TFM and its metabolites in fish liver tissue. This method is important for assessing bioaccumulation of TFM in the ecosystems. The compounds purified in our previous report were used to develop and validate a quantitative ultra-high-performance liquid chromatography–tandem mass spectrometry (UHPLC-MS/MS) assay for TFM and TFM metabolites formed in vivo. Several sample preparation techniques were compared, and a protein precipitation method was selected. The unavailability of stable isotopic internal standards was overcome by using a matrix matching method. After a thorough validation, this method was applied to determine the concentrations of TFM and its metabolites in fish liver tissues from animals exposed to TFM, and in the comparison between dead animals and survivors. Seven of eight expected metabolites were observed, some for the first time in vivo. Our results indicate that in vivo nitroreduction, glucuronidation, sulfation, and glutathione conjugation are involved in TFM metabolism in sea lamprey.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The sea lamprey (Petromyzon marinus) is a jawless vertebrate and an invasive pest in the Laurentian Great Lakes. Parasitic phase sea lamprey feed upon economically and ecologically important fishes and contributed to the collapse of native fish populations in the 1900s [1]. After feeding upon fish in lakes, adult sea lampreys migrate into streams to spawn and their larvae remain buried in stream sediment for approximately 3–5 years. An intensive control program targets sea lamprey migratory adults using traps and barriers, and eliminates or reduces sea lamprey larvae using lamprey-specific pesticides (lampricides). Trifluoromethyl-4-nitrophenol (TFM) is the primary tactic used to treat infested streams and has reduced sea lamprey populations by up to 90% in some areas [2].
Although tons of TFM are applied to selected Great Lakes tributaries to control sea lamprey populations annually, its metabolism and toxicity to sea lamprey and other species (including humans) are not completely understood. As many xenobiotics, TFM undergoes phase I and II metabolisms. Previous research implicates phase II glucuronidation of the phenol ring of TFM as the major mechanism of TFM detoxification in non-target species, and sea lamprey is deficient in this mechanism [3,4,5]. In addition, sulfation in the phenol ring, and N-acetylation of the reduced TFM have also been reported [6]. Other pathways such as phase I metabolic reactions have also been identified in vitro using fish liver enzymes, including the reduction of the nitro group to an amino residue [7]. Under anaerobic conditions nitro aromatic can be reduced to aniline metabolites in fish liver extract through successive formation of reactive nitroso and hydroxylamine metabolites (Fig. 1). Indeed, TFM reduction occurs more frequently in sea lamprey compared to non-target species that are less affected by TFM (e.g., Oncorhynchus mykiss; rainbow trout), and at intermediate levels in species that are partially affected (e.g., lake sturgeon; Acipenser fulvescens) [8, 9]. In a previous study (part A), we found additional evidence for phase I metabolism of TFM and reduced TFM (TFMa) after incubation with liver fraction S9, known to contain phase I and phase II metabolic enzymes, from sea lamprey, rainbow trout, bluegill (Lepomis macrochirus), and lake sturgeon. We determined the metabolism of TFM and biomarkers associated with TFM metabolism [10]. TFMa was identified as a marker for TFM phase I reductive metabolism [9] while 3-trifluromethyl-4-amino-5-S-gluthationyl-phenol (TFMaGSH) was identified during the screening of TFMa oxidative metabolism. Phase II metabolites of TFM and TFMa with sulfate or glucuronate conjugates were also identified (Fig. 1). TFM-O-Glucuronide (TFMOGlu) and TFM-O-Sulfate (TFMOS) were identified as phase II metabolites from TFM conjugation and associated with TFM detoxification [10]. Other metabolites were all related to TFMa conjugation. These findings suggest that phase II metabolism contributes to selective detoxification in non-targeted fish while phase I metabolic transformations such as nitroreduction may be involved in selective bioactivation of TFM in lamprey [10].

Overview of TFM metabolism in fish. UDPGT, uridine diphosphatoglucuronosyltransferase; UDPGA, Uridine 5′-diphosphoglucuronic acid; PST phenol sulfate transferase; GST, glutathione S transferase; GSH, glutathione (reduced); GSSG, glutathione dimer (oxidized) and NAT N-acetyltransferase
We postulate that TFMa is associated with the formation of reactive metabolites (nitroso and hydroxylamine) and possible toxicity in vivo. The formation of reductive metabolites such as nitroso and hydroxylamine may contribute to toxic effects. For example, reductive metabolism of the nilutamide molecule (5,5-dimethyl-3-[3-trifluoromethyl-4-nitro-phenyl]imidazolidine-2,4-dione), that shows strong structural similarities with TFM (3-trifluoromethyl-4-nitrophenol) [8], can form nitroso and hydroxylamine metabolites [11] that inhibit the mitochondrial respiration chain activities [12]. Another known example is chloramphenicol, an antibacterial nitro aromatic, whose nitroso metabolites show inhibitory effects on mitochondrial oxidative phosphorylation [13]. Therefore, both detoxification reactions (phase II metabolism of TFM) and reductive and oxidative reactions (phase I metabolism of TFM) in lamprey and fish need to be deciphered in order to understand the mode of action and the selective toxicity of TFM. This can be achieved by monitoring the concentrations of TFM or TFMa phase II metabolites. Such metabolites have been identified in vitro through the reactions of glucuronidation, N-acetylation, glutathione addition and sulfation. The metabolites associated with these reactions can be measured and linked to TFMa detoxification. Full monitoring of TFM metabolic markers can help in understanding its mode of action through the detoxification and bio-activation. In addition, an assay capable of monitoring TFM metabolism can determine the fate of TFM in vivo and the rate of bioaccumulation in the ecosystems.
Here, we developed a robust assay for quantifying TFM and its metabolites in fish liver tissue. Protein precipitation, solid phase extraction and liquid/liquid extraction were compared for their sample preparation performance because these are the most commonly used extraction methods. A quantitative LC-MS/MS method was developed to allow determination of eight target metabolites at low levels. The method was validated with evaluation of process recovery, stability, and intra- and inter-day accuracy and precision. Finally, the method was used to quantify the TFM metabolites in sea lamprey tissues from fish that were exposed to TFM (both the dead and survivor animals) and also from fish that were not exposed to TFM (control animals). The comparison of the metabolite concentrations provides novel insight to the mechanisms of TFM bio-activation or detoxification in vivo. With slight modification, this method can be used to monitor bioaccumulation of TFM in the environment that may affect human health.
Material and methods
Chemicals
All reagents and HPLC-grade solvents were purchased from Sigma-Aldrich (Saint Louis, MO, USA). Note that ammonium carbonate, ammonium acetate, formic acid, ammonium hydroxide and tributylamine were ACS-reagent grade. Standards for TFM metabolites used in this study were synthesized, purified and characterized in part A of this study [10], which includes full characterizations of the synthesized metabolites. The identification methods included MSMS fragmentation, high-resolution mass spectrometry, and 13C, 1H and 19F NMR.
Method development
Sample preparation
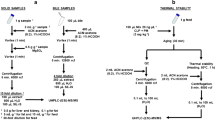
Liver tissues from adult sea lamprey were homogenized using a Potter-Elvehjem tissue grinder kept in a water/ice bath. Phosphate buffer (0.05 mol/L, pH 7.4) was added to the homogenate to reach the concentration of 0.5 g of wet tissue/mL. Sample preparation was optimized using 100 μL aliquots and five replicates per data point. Liquid/liquid extraction (LLE), protein precipitation (PPT) and solid phase extraction (SPE) were investigated for the extraction and clean-up of TFM metabolites. These methods were selected because they are the most common approaches for sample preparation upstream LC-MS experiments [14]. LLE and PPT conditions were standard methods (procedure described below) while SPE conditions followed the manufacturer’s instructions for the extraction of glucuronic acid conjugates. Five replicates were spiked with the targeted TFM metabolites before extraction to reach the final concentration of 100 nmol/L (volume of 1 mL). Absolute process recovery was calculated by the ratio of the signal area in samples spiked before the extraction process over the average signal area in standard samples.
Ethyl acetate (EtOAc) and methyl tert-butyl ether (MTBE) were used for liquid/liquid extraction. Homogenates were spiked with appropriate quantities of targeted analytes, vortexed, mixed with 1.2 mL of solvent and shaken for 20 min. Samples were then centrifuge (10 min; 9000g; 4 °C) and the aqueous layer (bottom layer) was frozen at − 18 °C to facilitate separation of the organic layer (top layer) for pipetting. The organic layer was transferred to a new tube and evaporated under reduced pressure. Residues were reconstituted in 1 mL of water: methanol (9:1) with tributylamine (10 mmol/L) and transferred to an auto-sampler vial.
Protein precipitation was also evaluated for the extraction of TFM metabolites. Homogenates were spiked with appropriate quantities of targeted analytes, vortexed, mixed with 1.2 mL of acetonitrile (or 0.1% formic acid in acetonitrile), incubated for one hour at 4 °C and shaken for 20 min. Samples were centrifuged (10 min; 9000×g; 4 °C) and the supernatants were transferred to new tubes and freeze-dried. Residues were then reconstituted in 1 mL of water: methanol (9:1) with tributylamine (10 mmol/L) and transferred to an auto-sampler vial.
SPE was carried out using two different types of sorbent [1]. Mixed-mode cation exchange cartridges (Waters MCX 3 cm3 60 mg) were activated with 3 mL of methanol and conditioned with 3 mL of water. Liver homogenate (diluted to 1 mL in 5% ammonium hydroxide solution) was loaded onto the cartridge and rinsed with 1 mL of 5% ammonium hydroxide solution. The cartridge was then eluted with 1 mL of methanol [2]. For the other mixed mode cartridges (Waters HLB PRiME 3 cm3 60 mg), liver tissue homogenate was diluted to 1 mL in ammonium carbonate buffer (10 mmol/L; pH 10) and directly loaded onto the cartridge. The cartridge was then rinsed with 1 mL of loading buffer and eluted with 1 mL of methanol. After evaporation under vacuum, residues were reconstituted in 1 mL of water/methanol (9:1) with tributylamine (10 mM) and transferred to an auto-sampler vial.
Ultra-high performance liquid chromatography tandem mass spectrometry (UHPLC-MS/MS) method
Chromatographic separation was achieved on a column with a C18 stationary phase (Acquity BEH C18 100 × 2.1 mm; particle size 1.7 μm; Waters Corporation) equipped with an in-line filter. A binary gradient between mobile phase A (10 mmol/L tributylamine and 10 mmol/L acetic acid in ultrapure water, pH 5) and B (HPLC-grade acetonitrile) at 200 μL/min was applied. The gradient was as follows (% of A; time in minutes): (80; 0.0), (50; 2.0), (10; 4.0), (1; 7.0), (1; 8.0), (80; 8.1) and (80; 10.0) with column temperature set at 35 °C. The flow through was diverted to waste from 0 to 2 min and from 7 to 10 min. The auto-sampler temperature was set at 4 °C and injection volume at 10 μL.
Quantitative tandem mass spectrometry method development was carried out on a Waters Xevo TQ-S UPLC-MS/MS triple quadrupole instrument. Both positive and negative modes of the electrospray source were investigated. The mode showing better sensitivity was selected for each target and the cone voltage parameter was optimized accordingly. Selected reaction monitoring (SRM) parameters (cone voltage and collision cell energy) were also individually optimized using QuanOptimized (Waters) to obtain three selective and sensitive MS/MS transitions. Finally, the best transition per target was selected among those three transitions by evaluating which one had the best signal-to-noise ratio. Ionization parameters were optimized as followed: negative mode, source voltage 2500 kV, temperature 120 °C, cone temperature 500 °C;, and desolvation gas flow rate (N2) 800 L/min.
NMR quantitation of the stock solution
Metabolites were determined using quantitative 1H NMR (1H qNMR) as described previously [10]. Synthesized compounds were dissolved in CD3OD and D2O respectively for phase I and phase II metabolites. NMR data were performed on an Agilent DirectDrive2 500 with 7600AS 96 with auto-sampler controlled by VnmrJ 3.2A software. 1H qNMR parameters were as followed: a pulse angle of 90°, a spectral width from − 0.5 to 9.5 ppm, and a recovery delay of 25 s for 32 scans. Aromatic 1H NMR signal area was used to estimate standard concentration based on external calibration.
Method validation
This method was validated using 100 μL aliquots of sea lamprey liver homogenate (0.5 g wet tissue/mL). This corresponds to 50 mg of liver tissue, which is a typical mass of a larval sea lamprey liver. Given the unavailability of stable isotope internal standard for TFM, the absolute quantification of TFM and its metabolites was achieved by matrix matching. The calibration curve samples were spiked in liver matrix aliquots (50 mg tissue/100 μL aliquot, with a spike volume of 10 μL) before extraction of the matrix. The matrix match approach compensates for the bias introduced by both sample preparation (protein precipitation) and matrix effect (during LC-MS). However, matrix matching cannot compensate for the unique response of each analyte toward electrospray analysis. This can only be eliminated by the use of standard material. Standard materials were synthesized, purified and characterized to serve in the quantitative method development and validation. The calibration curve data points were 0, 0.5, 1, 2.5, 5, 10, 25, 50, 100 and 250 nmol/L. The calibration curve samples and validation samples were extracted by precipitation of proteins with 1 mL of ice-cold acetonitrile (see detailed procedure in section 2.2.1). The limits of detection and quantification (LOD and LOQ, respectively) were defined by a signal-to-noise ratio (S/N) of greater than or equal to three and ten, respectively.
The validation of the absolute process recovery was carried out using 50 μL of sea lamprey liver homogenate (1 g wet tissue/mL) mixed with 250 μL of water. Process recovery was determined by the ratio of the signal area in matrix spiked before extraction over the signal area in standard solution. Five replicates of each data point were acquired and used to calculate the standard deviation. Stability was assessed by comparing the signal area normalized to the highest area over 3 days. Five replicates were analyzed per day.
The validation criteria in this report were based on the FDA Guidelines for Bioanalytical Method Validation [15]. Accuracy and precision parameters were evaluated at three concentrations. Concentrations of low, medium and high quality control (LQC, MQC and HQC) were set at 1, 100 and 250 nmol/L respectively, except for TFM, TFMa and TFMaNAc with respective LQCs of 10, 5 and 2 nmol/L. Precision was determined by the standard deviation within replicates and accuracy was calculated with the ratio of the determined concentration over the spiked concentration. The goal was to achieve the guideline that the accuracy should not exceed (100 ± 20) % for LQC and (100 ± 15) % for MQC and HQC, and the precision (standard deviation) should be lower or equal to 20% for LQC and 15% for MQC and HQC. Intraday parameters were determined within 15 replicates analyzed in the same day, whereas interday parameters were calculated using 15 replicates distributed across 3 consecutive days.
Exposure of sea lamprey to TFM
Sea lamprey larvae were exposed to TFM at the concentration of 1.31 ± 0.02 mg/L (mean ± standard deviation). This concentration corresponds to a theoretical LC50 of TFM for sea lamprey based on water chemistry and previous LC50 determination at The US Geological Survey, Great Lakes Science Center, Hammond Bay Biological Station. Concentrations used for field application generally ranged from 3 to 10 mg/L. Animals were held in 9 tanks with 10 animals/tank and exposed to TFM for 24 h. Control animals (n = 20) were not exposed to TFM and were kept in a separate tank. Water alkalinity and hardness were 79 mg/L and 95 mg/L, respectively. Water chemistry was monitored and found to be constant over the 24-h experimental period among the tanks. The pH was monitored at 1, 3, 5, 6 and 24 h in the tanks and measured at 8.11 ± 0.18 (mean ± standard deviation; n = 50). The temperature was monitored at 1, 3, 5, 6 and 24 h in the tanks and measured at 11.99 ± 0.64 °C (mean ± standard deviation; n = 50). Dissolved oxygen (D.O.) was monitored at 1, 5, and 24 h in the tanks and measured at 10.43 ± 0.66 mg/L (mean ± standard deviation; n = 30). Fish were euthanized and liver tissues were snap frozen in liquid nitrogen and stored at − 80 °C until use. For LC-MS/MS determination, whole liver was homogenized and extracted.
Results and discussion
Quantitative method development
Stock solution determination
LC-MS techniques are reliable for the detection of small organic molecules. However, LC-MS is often unreliable for assessing purity. In fact, LC-MS methods often fail to quantify impurities such as inorganic salts or residual solvents. In order to use the synthesized compounds as standards for LC-MS/MS quantitative method development, absolute concentrations of each standard need to be determined. Therefore, stock solution concentrations were authenticated by 1H qNMR. Indeed, contrary to other techniques, qNMR allowed determination of the absolute concentration of targeted molecules [16] regardless of the nature or ratio of the counter ion, or the presence of impurities such as salts or water. In addition, NMR is a non-destructive method and data can be acquired even with limited amount of materials, which makes qNMR suitable for this study. Three aromatic 1H resonances of each molecule (except TFMaGSH showing a single 1H NMR aromatic signal) were used to determine the mean concentration. For TFMaGSH, two 1H aromatic signals were used. Precision on the determination of the stock solution was acceptable with standard deviation within three replicates ranged from 0.1 to 1.53%. Accuracy was evaluated using a TFM sample (commercially available standard). TFM concentration (prepared at 150 mmol/L in CD3OD) was measured at 151.9 mmol/L, giving an estimated error of 1.25%. Therefore, qNMR (external calibration) serves as a relevant tool for the authentication of stock solution concentrations in order to obtain absolute and quantitative reference for each targeted metabolite.
Quantitative liquid chromatography tandem mass spectrometry development
The lowest limits of detection were achieved using negative-mode electrospray ionization for all targets. Table 1 summarizes the detection parameters and detection performances after optimization of the mass spectrometry parameters. The MS/MS transitions selected using the QuanOptimize software from Waters were identical to the fragmentations observed with high resolution mass spectrometry. The use of high resolution mass spectrometry allowed the identification of the neutral losses. TFM showed the neutral loss of NO (30 Da) while TFMa and TFMaNAc showed the loss of HF molecules. TFMa showed two peaks associated with the neutral loss of one or two HF while TFMaNAc showed only the neutral loss of three HF molecules. Phase II metabolites (glucuronic acid and sulfonate conjugates) showed the loss of anhydroglucuronic acid (TFMOGlu and TFMaOGlu) or SO3 (TFMOS and TFMaOS). Finally, the glutathione conjugate TFMaGSH was monitored with the transition (481 > 272) corresponding to the cleavage of the CH2-S bond.
The separation of the target compounds was achieved by reverse phase UHPLC. Several solvent conditions and different stationary phases were investigated including five aqueous buffers: 10 mmol/L ammonium acetate, ammonium acetate and formic acid (10 mmol/L each), 10 mmol/L triethylamine (no acid), 10 mmol/L formic acid, and tributylamine with acetic acid (10 mmol/L each). Most conditions achieved good separation of the analytes. However, glucuronide conjugates eluted early in the chromatographic separation and often co-eluted with matrix interferences. The glucuronide conjugate was retained only in the presence of the ion-pairing agent TBA combined with acetic acid. Thus, TBA with acetic acid (10 mmol/L each) was selected as the aqueous mobile phase (solvent A) and acetonitrile as the organic mobile phase (solvent B). Several stationary phases were also investigated to reach the optimal chromatographic separation of the TFM metabolites. CSH Fluorophenyl and HSS, CSH, BEH C18 columns from Waters were compared and BEH C18 (100 × 2.1 mm, particle size 1.7 μm) was selected for further method development and validation based on peak shape and higher retention times. Figure 2 shows the extracted ion chromatograms for TFM and its seven identified metabolites. A baseline separation and sharp peaks minimized interferences between the targets.

Extracted ion chromatograms of TFM metabolites added to extract from sea lamprey liver at 100 nM (MQC). From top to bottom: TFM, TFMa, TFMaNAc, TFMOS, TFMOGlu, TFMaOS, TFMaOGlu and TFMaGSH
Sample preparation optimization
Sample preparation was optimized after testing techniques for sample concentration and clean-up upstream of LC-MS/MS experiments. These techniques included liquid/liquid extraction (LLE), solid phase extraction (SPE), and protein precipitation (PPT).
Liquid/liquid extraction (LLE) was tested using a ratio of 1:3 (sample homogenate: solvent). Two solvents, MTBE and ethyl acetate, were evaluated. Samples extracted with MTBE appeared to have high fat content after solvent evaporation, making it inappropriate for LC-MS/MS experiments. Ethyl acetate extracts showed minute amount of phase II metabolites (glucuronic acid, sulfate and glutathione conjugates) compared to other extraction techniques. This was attributed to the high polarity and charge state of these metabolites in ethyl acetate solution that resulted in significant loss, and was therefore not further investigated.
Solid phase extraction (SPE) was investigated with two types of sorbent; mixed-mode (Waters HLB PRiME 60 mg) and mixed-mode cation exchange (Waters MCX 60 mg). For the extraction of TFMaGSH, MCX and HLB cartridges showed similar performances. However, all other targets showed lower recoveries with MCX SPE sample preparation (< 30%). Thus, MCX cartridges were not further considered.
Two experimental conditions were evaluated for protein precipitation (acetonitrile and 0.1% formic acid in acetonitrile). Protein precipitation with ice-cold acetonitrile was preferred over ice-cold acetonitrile with 0.1% formic acid for higher recovery yields. In fact, the recovery of all analytes with acidic protein precipitation was lower or equal to recoveries obtained with ice-cold acetonitrile (Fig. 3). Similarities in process recoveries showed that HLB PRiME SPE of liver homogenate was competitive with PPT (Fig. 3). Absolute process recoveries using PPT with ice-cold acetonitrile ranged from 28.5 to 99.3% (Table 2). PPT was therefore selected for further method validation experiments and application to biological sample analysis for the ease of implementation and the low cost.

Comparison of sample preparation techniques for the extraction of TFM, TFM metabolites and internal standards. The techniques investigated include protein precipitation (PPT) and SPE (HLB and MCX). Absolute process recovery is represented by the ratio of the signal area in matrix spiked before extraction over the signal area in standard solution. Error bars represent standard deviations within replicates
Method validation
The limits of quantification in standard solution ranged from 0.01 to 0.5 nmol/L. However, due to background noise, the linearity of the method was limited to 0.5 to 250 nmol/L except for TFM, TFMa and TFMaNAc, which gave linear responses over 5–250, 2–250 and 1–250 nmol/L, respectively. Concentrations selected for the lowest validation point of TFM, TFMa and TFMaNAc were therefore set at 10, 5 and 2 nmol/L, respectively.
Extract stability was monitored for 3 days and shown in Fig. 4. Extracts were stored either at 4 °C (in the dark inside a UHPLC auto-sampler) or in a − 20 °C freezer between validation days. Normalized signal areas were all within 80 to 100% range, highlighting metabolite stability over the validation period of 3 days. These data indicate the stability of TFM and its metabolites in the experimental conditions described here (sample preparation & storage).

Stability assay of TFM and its metabolites over 3 days in spiked tissue extracts. Signal area was normalized to the highest signal area measured. Each bar represents the average of five replicates. Error bars represent the standard deviation from five replicates
Accuracy parameters (Tables 3 and 4) were within the acceptable range set by FDA guidance for bioanalytical method development (80–120% for LQC and 85–115% for MQC and HQC) except for the interday accuracy of TFMaNAc at HQC (83.5%). Precision parameters (Tables 3 and 4) were also within the values set by FDA guidance (≤ 20% for LQC and ≤ 15% for MQC and HQC) except for TFM interday precision at LQC and TFMa and TFMaNAc. TFMa showed precision ranging from 19 to 29% for intraday and 14.7 to 20.5% for interday, whereas TFMaNAc showed intraday precision ranging from 16.9 to 21%. The trend of higher variation within replicates of TFMa and TFMaNAc is due to their relative instability toward oxidation. Oxidized phenol aniline and phenol amide tend to spontaneously oxidized and polymerized in solution [17]. The successful validation of accuracy and precision parameters highlighted the efficiency of using the matrix match approach to quantify TFM and its metabolites in fish liver homogenates.
Determination of TFM metabolites in biological samples
Livers from sea lamprey larvae exposed to TFM were extracted and determined as described above. The experiment resulted in the death of 27 fish (63 survivors). Survivor and control animals were euthanized with 0.025% MS-222. There was no significant difference in the mass and length (ANOVA; p > 0.05) of survivor, control, and dead animals. Control fish weighed 89.7 ± 12.9 mg and were 98.8 ± 47.7 mm long. Survivors weighed 89.7 ± 16.5 mg and were 105.8 ± 48.2 mm long. Dead fish weighed 80.6 ± 5.3 mg and were 61.2 ± 12.3 mm long. The mass measurements of liver tissues were not significantly different among groups (ANOVA; p > 0.05). Tissue mass measurements were 18.3 ± 7.6 mg for control animals (n = 10), 18.6 ± 5.9 mg for survivors (n = 20), and 14.8 ± 5.2 mg for dead animals (n = 20).
All targets were detected in the liver tissues except TFMaOGlu (Fig. 5). TFM, TFMa and TFMaGSH were observed in higher concentrations and samples had to be diluted. To determine the significance of the differences in metabolite concentrations among groups, a two-way ANOVA was performed. Significant differences were observed among groups for all observed metabolites except for TFMaNAc. Student t tests were performed to investigate significant differences between groups. The control group had negligible levels of TFM and its metabolites. For TFM, the control group was significantly different from the survivors and the dead group (P < 0.0001) while the dead and the survivors were similar (P > 0.42). Fish were captured in the wild and held in house until TFM exposure experiment. It is therefore possible that previous exposure while in streams led to the bioaccumulation of the most hydrophobic metabolites (TFM and TFMNAc). There is no consequence of differential uptake or detoxification between the dead and the survivors in the concentration of TFM in vivo. Although no significant difference was observed between the survivor and the dead groups in TFMaGSH (P < 0.31) and TFMaOS (P < 0.12) levels, these two targets were in significantly higher concentrations in TFM treated animals than in control animals (P < 0.01). This is the first evidence that TFMaGSH and TFMaOS exist in sea lamprey larvae. Formation of GSH conjugates can be used as a proxy for the formation of quinoid metabolites that can undergo protein arylation [18] and redox cycling [19], therefore presenting two mechanisms of toxicity. The levels of TFMa, TFMOS and TFMOGlu differed from each other (P < 0.05). TFMa was significantly higher in survivors group compared to dead animals. This may be attributed to the reactive nature of p-aminophenol metabolite such as TFMa that may be metabolized or undergo spontaneous binding to surrounding biomolecules. In this case, the nitro reduction leading to the production of TFMa may stop at the death of the animal, and the spontaneous degradation or reactivity toward biomolecules results in the decrease of TFMa. TFMOS and TFMOGlu are TFM conjugated metabolites contributing to the detoxification of TFM in vivo. TFMOGlu has been identified and widely discussed [20]. However, this is the first evidence of TFM sulfation in sea lamprey larvae. The low levels of the conjugates are associated with the limited capability of sea lamprey to detoxify TFM through glucuronidation and sulfation. Although TFMOS and TFMOGlu are considered stable, their hydrophilicity can lead to excretion and result in low in vivo concentration.

Concentrations of TFM and TFM metabolites in sea lamprey larval liver tissues in control animals and animals exposed to TFM for 24 h at 1.31 mg/L. Bars and error bars represent the means and associated standard deviations
Conclusion
In summary, we developed a sensitive and robust UHPLC-MS/MS method to quantify TFM and its metabolites. After screening, identifying, and synthesizing metabolites of TFM in sea lamprey and non-target species, a method was needed to quantify metabolites that are likely important in TFM detoxification or bio-activation. We developed, optimized, and validated a method for quantifying TFM metabolites in sea lamprey liver homogenates. The method was applied to biological samples and led to the determination of seven of eight targets in the liver of sea lamprey larvae exposed to TFM for 24 h. Consistent with previous reports, we detected TFM and TFMOGlu in vivo. However, this is the first time that TFMa, TFMaNAc, TFMaGSH and TFMOS have been identified in vivo. Glucuronidated and sulfated TFM were measured in significantly higher concentrations in the survivors compared to dead animals. These results confirmed the limited capability of sea lamprey to detoxify TFM through glucuronidation and sulfation and showed the first evidence of in vivo nitro reduction resulting in the formation of reactive metabolites, reflected in part by the substantial conversion to TFMa and its glutathione conjugate. Clear evidence is provided for both detoxification (sulfation and glucuronic acid conjugations) and bio-activation (nitroreduction and dehydrogenation) metabolism. This method not only implicates a new mechanism of TFM metabolism, but also provides a sensitive tool for future investigations of TFM as an important pesticide for invasive sea lamprey. The quantification of TFM metabolites could contribute to the optimization of TFM application, or the development of the next generation of lampricides by identifying key chemical functions in TFM selective bioactivation and/or detoxification. The optimization of TFM application can be supported by the evaluation of the impact of treatment conditions (e.g. water pH and hardness), and inhibitory or activation effects of other pesticides (e.g. niclosamide) on TFM intake and metabolism. The quantitative determination of TFM could have broader impacts, in particular in the field of human health. Although, this would require partial method validation for the analysis of new matrices, a broader outcome of this study is to make the analysis of TFM available for the monitoring the exposure of humans to this pesticide.
References
Smith BR, Tibbles JJ. Sea lamprey (Petromyzon marinus) in Lakes Huron, Michigan, and Superior: history of invasion and control, 1936–78. Can J Fish Aquat Sci. 1980;37(11):1780–801.
Mullett K, Sullivan P. Sea lamprey control in the Great Lakes 2016 in Great Lakes fishery commission. 2016. http://www.glfc.org/pubs/slcp/annual_reports/ANNUAL_REPORT_2016.pdf:. Accessed 20 Feb 2017.
Lech JJ, Costrini NV. In vitro and in vivo metabolism of 3-trifluoromethyl-4-nitrophenol (TFM) in rainbow trout. Comp Gen Pharmacol. 1972;3(10):160–6.
Kawatski JA, Bittner MA. Uptake, elimination, and biotransformation of the lampricide 3-trifluoromethyl-4-nitrophenol (TFM) by larvae of the aquatic midge chironomus tentans. Toxicology. 1975;4(2):183–94.
Lech JJ, Statham CN. Role of glucuronide formation in the selective toxicity of 3-trifluoromethyl-4-nitrophenol (TFM) for the sea lamprey: comparative aspects of TFM uptake and conjugation in sea lamprey and rainbow trout. Toxicol Appl Pharmacol. 1975;31(1):150–8.
Hubert TD. Environmental fate and effects of the lampricide TFM: a review. J Geat Lakes Res. 2003;29(1):456–74.
Buhler DR, Rasmusson ME. Reduction of p-nitrobenzoic acid by fishes. Arch Biochem Biophys. 1968;124:582–95.
Bussy U, Chung-Davidson YW, Li K, Li W. Phase I and phase II reductive metabolism simulation of nitro aromatic xenobiotics with electrochemistry coupled with high resolution mass spectrometry. Anal Bioanal Chem. 2014;406(28):7253–60.
Bussy U, Chung-Davidson YW, Li K, Li W. A quantitative assay for reductive metabolism of a pesticide in fish using electrochemistry coupled with liquid chromatography tandem mass spectrometry. Environ Sci Technol. 2015;49(7):4450–7.
Bussy U, Chung-Davidson Y-W, Buchinger TJ, Li K, Smith SA, Jones DA, et al. Metabolism of a sea lamprey pesticide by fish liver enzymes. Part A. Identification and synthesis of TFM metabolites. ABC-01582-2017.
Ask K, Dijols S, Giroud C, Casse L, Frapart YM, Sari MA, et al. Reduction of nilutamide by NO synthases: implications for the adverse effects of this nitroaromatic antiandrogen drug. Chem Res Toxicol. 2003;16(12):1547–54.
Berson A, Schmets L, Fisch C, Fau D, Wolf C, Fromenty B, et al. Inhibition by nilutamide of the mitochondrial respiratory chain and ATP formation. Possible contribution to the adverse effects of this antiandrogen. J Pharmacol Exp Ther. 1994;270(1):167–76.
Abou-Khalil S, Abou-Khalil WH, Yunis AA. Differential effects of chloramphenicol and its nitrosoanalogue on protein synthesis and oxidative phosphorylation in rat liver mitochondria. Biochem Pharmacol. 1980;29(19):2605–9.
Li W, Zhang J, Tse FLST. Handbook of LC-MS bioanalysis: best practices, experimental protocols, and regulations. Wiley; 2013. p. 704.
Food and Drug Administration. Guidance for industry, bioanalytical method development. Food and Drug Administration. 2013. https://www.fda.gov/downloads/Drugs/Guidances/ucm368107.pdf. Accessed 15 Jan 2017.
Giraudeau P. Challenges and perspectives in quantitative NMR. Magn Reson Chem. 2017;55(1):61–9.
Bussy U, Giraudeau P, Tea I, Boujtita M. Understanding the degradation of electrochemically-generated reactive drug metabolites by quantitative NMR. Talanta. 2013;116:554–8.
Cohen SD, Khairallah EA. Selective protein arylation and acetaminophen-induced hepatotoxicity. Drug Metab Rev. 2010;29(1–2):59–77.
Yin H, Vergeade A, Shi Q, Zackert WE, Gruenberg KC, Bokiej M, et al. Acetaminophen inhibits cytochrome c redox cycling induced lipid peroxidation. Biochem Biophys Res Commun. 2012;423(2):224–8.
Hubert TD, Bernardy JA, Vue C, Dawson VK, Boogaard MA, Schreier TM, et al. Residues of the lampricides 3-trifluoromethyl-4-nitrophenol and niclosamide in muscle tissue of rainbow trout. J Agric Food Chem. 2005;53(13):5342–6.
Acknowledgements
This research was supported by the Great Lakes Fishery Commission. A.D.J. was supported by the USDA National Institute of Food and Agriculture Hatch project MICL-02143. The authors also thank Andrew Paul Buchinger (local angler) and Professor Kim Scribner (MSU Dept. of Fisheries & Wildlife) for the gifts of fresh blue gill livers and fresh lake sturgeon livers, respectively.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
We declare no conflict of interests. Animal tissues used in analyses were collected by staff of USGS Lake Huron Biological Station, Millersburg, MI 49759, USA. Experimental protocols involving the handling of fishes were carried out in accordance with United States federal guidelines for care and use of animals and were approved by the American Fisheries Society through the “Use of Fishes in Research Committee, 2014”.
Rights and permissions
About this article

Cite this article
Bussy, U., Chung-Davidson, YW., Buchinger, T. et al. Metabolism of a sea lamprey pesticide by fish liver enzymes part B: method development and application in quantification of TFM metabolites formed in vivo. Anal Bioanal Chem 410, 1763–1774 (2018). https://doi.org/10.1007/s00216-017-0831-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-017-0831-7