
Abstract
A selective and sensitive magnetic dispersive solid-phase microextraction (MDSPME) coupled with gas chromatography-mass spectrometry was developed for extraction and determination of organophosphorus pesticides (Sevin, Fenitrothion, Malathion, Parathion, and Diazinon) in fruit juice and real water samples. Zero valent Fe-reduced graphene oxide quantum dots (rGOQDs@ Fe) as a new and effective sorbent were prepared and applied for extraction of organophosphorus pesticides using MDSPME method. In order to study the performance of this new sorbent, the ability of rGOQDs@ Fe was compared with graphene oxide and magnetic graphene oxide nanocomposite by recovery experiments of the organophosphorus pesticides. Several affecting parameters in the microextraction procedure, including pH of donor phase, donor phase volume, stirring rate, extraction time, and desorption conditions such as the type and volume of solvents and desorption time were thoroughly investigated and optimized. Under the optimal conditions, the method showed a wide linear dynamic range with R-square between 0.9959 and 0.9991. The limit of detections, the intraday and interday relative standard deviations (n = 5) were less than 0.07 ngmL–1, 4.7, and 8.6%, respectively. The method was successfully applied for extraction and determination of organophosphorus pesticides in real water samples (well, river and tap water) and fruit juice samples (apple and grape juice). The obtained relative recoveries were in the range of 82.9%–113.2% with RSD percentages of less than 5.8% for all the real samples.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Organophosphorus pesticides (OPPs) compounds are widely used in agriculture to control pest insects by acting on the acetylcholinesterase enzyme. These compounds can affect the nervous system in insects, humans, and many other animals by irreversible inactivation of acetylcholinesterase enzyme [1, 2]. Therefore, these pesticides can lead to an acute cholinergic syndrome in human, increasing the acetylcholine neurotransmitter. Long-term exposure of humans to these compounds causes lower performance on standardized neuropsychological tests and neuropsychiatric symptoms reports [3]. Many pesticides may also leave residues in agricultural products for a long time, causing potential health risks in human and animal consumers [4]. Due to high consumption of these compounds in agriculture and their water solubility, they enter into the environment and lead to contamination of the environment and water sources. Therefore, techniques to determine these compounds with high sensitivity are essential because they are present in water and environmental samples at trace amounts [5, 6]. Moreover, due to matrix interferences in real samples, a sample preparation step is required prior to subsequent steps in the analytical processes. Microextraction methods are widely applied for sample preparation in real sample because analytes are concentrated and separated from the matrix of real samples in one step by these techniques [7].
Magnetic dispersive solid-phase microextraction (MDSPME) has recently attracted a great deal of attention as a new sample preparation technique [8,9,10,11]. In this procedure, magnetic SPME sorbents are ultrasonically dispersed into the sample solution and a cloudy suspension is formed, which provides a fast and efficient approach to extract and enrich the target analytes. After the extraction, the sorbent can be conveniently separated from the aqueous samples by a strong external magnet and the extracted analyte is then desorbed and analyzed using GC-MS. The method was widely used to extract a variety of analytes in real samples, including biological, food, real water, and etc. because this method has many advantages such as simplicity, low consumption of toxic organic solvents and SPME adsorbent, high preconcentration factor, and low extraction time. It also reduces the effects of interfering species in the sample matrix by extraction of analytes from the sample solution and therefore greatly affects the quality of analysis. One of the major problems to determine organophosphorus pesticides, especially at low concentrations in real samples, is the matrix effect [12,13,14]. Therefore, MDSPME was used to decrease these effects in the analysis of organophosphorus pesticides. The goal of MDSPME is to separate organophosphorus pesticides and preconcentrate from the real sample in one step. Thus, the choice of a sorbent in this procedure is the most important factor for extracting the analyte with high selectivity and preconcentration factor. Due to the unique features of nanomaterials such as large surface area and unique physical and suitable chemical properties, the use of these materials as SPME sorbent has greatly increased [15, 16].
Graphene and graphene oxide as unique sorbents have found wide application for extraction or removal of organic pollutants. Due to special structure, graphene oxide has various superior properties, including excellent thermal conductivity, mechanical properties, specific magnetism, and large surface area [17, 18]. The aim of this study is to develop a magnetic dispersive solid-phase microextraction coupled with GC-MS for preconcentration and determination of five organophosphorus pesticides (sevin, fenitrothion, malathion, parathion, and diazinon) in real water samples. Zero valent Fe- reduced graphene oxide quantum dots were prepared as a new and efficient SPME sorbent and used to extract organophosphorus pesticides. This microextraction procedure has several advantages, such as simplicity, rapidity, solvent-free, and cost effective. The analytical performance of the proposed method was compared with other microextraction techniques and demonstrated the suitability of the proposed method to analyze the target compounds in real water samples.
Experimental
Chemicals and materials
Organophosphorus pesticides (sevin, fenitrothion, malathion, parathion, and diazinon) as pure compounds were purchased from National Institute of Metrology (Beijing, China). Potassium permanganate, sulfuric acid, graphite powder (purity 99%, mesh 325), sodium nitrate, hydrogen peroxide, hydrochloric acid, and other chemicals were used to synthesise the zero valent Fe- reduced graphene oxide quantum dots and were obtained from Merck (Darmstadt, Germany).
Apparatus
The analysis of organophosphorus pesticides were performed by Shimadzu GCMS-QP2010/PLUS system (Shimadzu, Kyoto, Japan), operated at 70 eV electron impact. Helium carrier gas was maintained at a constant flow of 1.04 mL min−1. A fused silica capillary column RTX-5MS Shimadzu (30 m × 0.25 mm i.d., 0.25 μm film thickness) was used. The GC temperature was programmed as follows: start temperature 60 °C (hold for 1 min), increased to 180 °C at 20 °C min −1 and held for 5 min and finally to 250 °C at 20 °C min −1 and held for 9 min. The temperature of the interface and the ion source was at 230 °C. The retention time and the fragment ions were identified by injecting the multi-compound standard in full-scan mode (m/z 46–500). The retention time and selected ions for each OPPs is presented in Electronic Supplementary Material (ESM) Table S1.
Synthesized of zero valent Fe-reduced graphene oxide quantum dots
Graphene nanosheets were prepared by synthesis of graphene oxide and reduced to graphene with NaBH4 [17, 19]. In a typical procedure, a mixture of graphite flakes (0.5 g) and NaNO3 (0.5 g) was cooled to 0 °C in an ice bath followed by dropwise addition of 23 mL of concentrated H2SO4; 3.0 g of KMnO4 was gradually added to the mixture and the temperature of the suspension was kept below 20 oC. The suspension was ultrasonically treated for 20 min at room temperature and diluted with 40 mL of deionized water. In order to remove any excess of KMnO4, a mixture of 3 mL H2O2 (30%) and 100 mL deionized water was then added drop-wise to the suspension until the color of the suspension changed from dark brown to yellow. The resulting product was washed several times with 5% HCl aqueous solution and distilled water to remove any metal ion impurities, sulfate ions, and excess of acid, respectively. After filtration, graphene oxide was dried under vacuum at 80 oC for 24 h.
Hydrothermal cutting method was used to synthesize graphene oxide quantum dots (GOQDs) [20]. One hundred mL of dimethylformamide (DMF) was added to graphene oxide (0.5 g) and ultrasonically treated for 2 h. The resulting mixture was transferred into an autoclave and heated at 250 oC for 8 h. After treating, the resulting mixture was filtered through a nylon membrane (0.2 μm) and heated to vaporize the excesses of DMF.
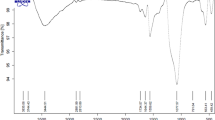
In order to prepare zero valent Fe- reduced graphene oxide quantum dots (rGOQDs@ Fe), 0.5 g of GOQDs in 150 mL distilled water was ultrasonically dispersed for 1 h. A solution containing 0.8 g of FeCl3 in 20 mL of deionized water was added drop-wise to GOQDs under nitrogen gas and was ultrasonically treated for 1 h at room temperature. The mixture was heated up to 90 oC, and 40 mL of NaBH4 solution (8.5 % w/v) was added drop-wise until the color of the suspension changed from dark brown to black. The mixture was treated for another 4 h at 90 oC, under stirring. In order to remove any oxygen gas from the solution, the overall procedure was performed under constant bubbling of nitrogen gas. The resulting product was filtered, and washed with deionized water and ethanol several times, respectively. Zero valent Fe-reduced graphene oxide quantum dots (rGOQDs@ Fe) were finally dried in vacuum at 80 oC for 48 h [19]. SEM images of magnetic graphene oxide (MGO) and rGOQDs@ Fe and FTIR spectra for rGOQDs@ Fe, MGO, and GO are shown in Figs. 1 and 2, respectively. According to Fig. 1, the particle size of rGOQDs@ Fe is much smaller and their dispersion is much more uniform than MGO.

SEM images of MGO (a) and rGOQDs@ Fe (b)

FTIR spectra for GO (a), M-GO (b), and rGOQDs@ Fe (c)
Magnetic dispersive solid-phase microextraction
In order to carry out the extractions procedure, 10.0 mL each of five 15 ng mL–1 OPPs standard solutions were placed in a 20 mL conical test tube. The pH of the solutions was adjusted at 7.0 by adding sodium dihydrogen phosphate/ disodium hydrogen phosphate buffer; 15 mg of rGOQDs@ Fe as sorbent was then added and ultrasonically treated for 5 min. The SPME sorbent was dispersed to fine particles and a cloudy mixture of sorbent was formed in the bulk of aqueous sample. After extraction of analyte, the sorbent was rapidly separated from the solution under a strong external magnetic field and the supernatant solution was discarded. The adsorbed analytes onto the sorbents were eluted with 0.4 mL of acetone under sonication for 3.5 min and then 10 μL of the supernatant acetone phase was injected through a split ratio five injector into the GC-MS system to analyze the five OPPs.
Results and discussion
Optimization of the MDSPME procedure
In order to obtain maximal extraction efficiency, several factors affecting the extraction and desorption steps, such as amount of sorbent, volume of donor phase, pH, extraction time, desorption time, and the type of desorption solvent were selected and optimized. Each experiment was repeated three times and the mean peak area was applied as the response.
Amount of sorbent
It is essential that the analyte must be interacted with sorbent surface in order to be extracted from the sample solution. Therefore, the extraction efficiency increases with increase in the amount of sorbent and number of active positions on its surface. However, the extraction efficiency becomes approximately constant with further increase of sorbent amount because the extraction procedure is an equilibrium process for distributed analyte between the liquid and solid phase (sample solution and sorbent surface) [21]. The amount of analyte extracted onto the sorbent is directly related to the sorbent volume, distribution coefficiency between SPME sorbent and sample solution, and concentration of the analyte in the sample matrix [21, 22]. Therefore, the effect of sorbent amount on the extraction efficiency was investigated in the range of 10–30 mg at constant experimental conditions. As shown in Fig. 3, the extraction efficiency was increased with the amount of sorbent up to 15 mg and remained nearly constant afterwards. According to the above results, 15 mg of sorbent was selected for subsequent experiments.

Effect of amount of sorbent in extraction of OPPs
pH of sample solution
A factor that plays an important role for adsorption of the analytes onto the sorbents is the pH of the sample solution. A suitable interaction between the analyte and sorbent is created to adsorb the analyte when the polarity of functional groups on the sorbent surface and analyte is the same, and this polarity can be controlled by adjusting the pH of the sample solution. In order to achieve the best interaction between the analyte and sorbent, the effect of pH on the extraction efficiency of OPPs was investigated. For this propose, the pH of solutions was varied in the range of 4.0–9.0 using suitable buffer solutions, and the extracted process was performed for about 8 min. The results show that the peak areas were dramatically increased in the pH range of 4 –7 and decreased at higher pH (Fig. 4). It seems that at pH 7.0, the functional groups on the organophosphates and sorbent mainly exist as neutral groups and a strong interaction occurs between them. Therefore, the extraction efficiency of OPPs is highest at pH7.0 and was chosen for further studies.

Effect of feed solution pH in extraction of OPPs
Donor phase volume
An important parameter in microextraction methods is preconcentration factor defined as the ratio of donor phase volume to desorption solvent volume. Therefore, the preconcentration factor has a direct and inverse relationship with the donor phase volume and desorption solvent volume, respectively. Owing to probable decrease of interaction between the analyte and sorbent surface using large volumes of sample solution, the preconcentration factor and extraction efficiency are reduced. Thus, the effect of sample volume was studied in the range 5.0–15.0 mL (Fig. 5). The results show that the peak areas increased for up to 10.0 mL of sample solution and levelled off at higher volumes. Thus, a sample volume of 10.0 mL was selected for further studies.

Effect of donor phase volume in extraction of OPPs
Extraction time
Microextraction is an equilibrium- and time-dependent process. During the microextraction procedure, the analyte is distributed between the two phases, the donor phase and SPME sorbent, to reach equilibrium [23]. Therefore, it is necessary to select an extraction time during which the analyte reaches its equilibrium between the two phases for an appropriate extraction efficiency. The effect of extraction time was investigated in the range of 2–10 min at constant experimental conditions. According to ESM Fig. S1, an extraction time of 5 min was selected to obtain high extraction efficiency.
Effect of desorption condition
Desorption conditions are important factors in extraction procedure because the adsorbed analyte should be desorbed ultrasonically from the surface of SPME sorbent prior to injecting into the GC/MS. Therefore, the type and volume of desorption solvent and desorption time have great influence on the extraction efficiency and should be optimized. Several organic solvents (methanol, acetonitrile, acetone, and dichloromethane) were selected and studied as desorption solvents for desorbing the OPPs. The results show acetone gave the highest desorption efficiency and was chosen as the desorption solvent (ESM Fig. S2). The effect of volume of desorption solvent on the extraction efficiency was studied in the range of 0.3–2.0 mL, and 0.4 mL acetone was used as the optimum value in all subsequent experiments. Desorption time was also investigated in the range of 2–5 min. The results indicate that the highest extraction efficiency was obtained at 3.5 min for all analytes. It is logical that peak areas are reduced because desorption equilibrium could not be completed at a shorter times and the desorbed analytes are diffused to the pores of hollow fiber for longer times [24].
According to the results, the optimal experimental conditions are: pH 7.0; donor phase volume, 10.0 (mL); extraction time,5 min; desorption time, 3.5 min; sorbent amount, 15 mg; acceptor phase volume, 0.4 mL; type of acceptor phase, acetone.
Comparison of the MD-SPME efficiency of the SPME sorbent
In order to investigate the ability of new sorbent in the extraction of OPPs, magnetic graphene oxide (M-GO) and graphene oxide (GO) were prepared according to previous articles and used as sorbents [25, 26]. The results are shown in Table 1 and indicate that the new sorbent compared with two other sorbents was quite effective for extraction of OPPs. Owing to nonpolar structure and small numbers of polar functional groups in these analytes, especially for malathion, the extraction efficiency was increased by decreasing and removing polar function groups such as carboxyl, ether, and hydroxyl groups on the sorbent surface. Therefore, rGOQDs@ Fe as a sorbent showed the highest extraction efficiency because the number of polar function groups in its surface are few. The FTIR spectra of sorbents show that the number of polar function groups on the surface of rGOQDs@ Fe and M-GO were low and high, respectively (Fig. 2). The stretching vibrations of the hydroxyl group located at 3365, 3121, and 3419 for GO and M-GO shifted to shorter wave numbers for rGOQDs@ Fe. In addition, the reduction in the peak width and its intensity in rGOQDs@ Fe indicate that the strength of hydrogen bonds has declined. Moreover, the presence of the stretching vibrations of C=O at 1722 and 1717 cm–1 for GO and M-GO were absent in the FTIR spectrum of rGOQDs@ Fe. Therefore, the analyte extraction efficiency is increased by reduction of strength of hydrogen bond for hydroxyl groups and removal of carbonyl groups in the rGOQDs@ Fe surface.
Method validation
The performance of the proposed method for determination of OPPs was evaluated under the optimum conditions by several important parameters such as the linear range, relative standard deviation (RSD), limit of detection (LOD), and enrichment factor. The results obtained are summarized in Table 2. LOD was the lowest analyte concentration that could be detected and was calculated as the ratio of three times standard deviation of blank readings to the slope of the calibration curve after preconcentration. The limit of quantitation (LOQ) was the level above which quantitative results may be obtained with a specified degree of confidence [27]. LOQ of the assay was evaluated as the concentration for which S/N was equal to 10. The LODs and LOQs were less than 0.07 and 0.21 ngmL-1 for all analytes, respectively. The results also indicated that the lowest and the highest LODs and LOQs were related to malathion and parathion, respectively. The proposed method showed a wide linear dynamic range for analysis of OPPs with R-square greater than 0.9959. Intra-day precision of the method was evaluated for five replicate experiments for the spiked samples at 10 ng mL–1 of each analyte on the same day. Inter-day precision of the method was tested for 3 d at the same concentration levels (10 ng mL–1of each analyte). The relative standard deviations (%RSD) of intraday precision and interday precision are less than 4.7% and 8.6% for all the analytes, respectively, and show good reproducibility and precision for the proposed method (Table 2). The preconcentration factor (PF) was calculated as the ratio of the analyte concentration in acetone as acceptor phase and aqueous sample. According to Table 2, the preconcentration factor was more than 112 for extraction of OPPs. Comparing the results for the analysis of these compounds show that the proposed method has the lowest relative standard deviations and LODs, and highest preconcentration factor for malathion.
Real samples analysis
The described method was employed to analyze five OPPs in three kinds of real water samples (well, river, and tap water) and two kinds of fruit juice samples (apple and grape). The tap and well water samples were obtained from the outskirts of Mashhad (Iran). Primary and secondary river water samples were collected from Golestan River (Torghabeh, Iran) in April (1 wk after spraying) and May 2017, respectively. The real fruit juice samples (Sunich brand, Alifard Co.) were purchased from a supermarket in Mashhad (Iran). All real samples were spiked with 20 and 50 ng mL−1 of OPPs and centrifuged at 6000 rpm for 5 min and then filtrated through a 0.45 mm filter membrane to remove all solid impurities in the samples prior to extraction and analysis. Each sample was analyzed in triplicate and the results are presented in Table 3. The results indicate that none of OPPs were detectable by the proposed method in all the real samples, except for malathion in the river samples. The amount of malathion was more than the allowed limit in the primary river water sample but its concentration decreased dramatically in the secondary river water sample. This may be due to seasonal and daily rainfall, the high consumption of malathion, and its consuming time [28]. In all cases, the obtained relative recoveries are between 82.9% and 113.2% with RSD percentages of less than 5.8%. According to the results in Table 3, all the analytes can be quantitatively recovered from the real samples by the proposed method, showing the ability of the method to extract and determine OPPs in real water and fruit juice samples. A representative chromatogram of spiked well water samples at the outskirts of Mashhad (Iran) is shown in Fig. 6.

Chromatogram of a spiked well water samples at 20 ng mL1– for determination of OPPs under optimum conditions. (1. sevin, 2. diazinon, 3. fenitrothion, 4. malathion, 5. parathion)
Comparison to other determination methods
Preconcentration and determination of OPPs by the proposed method was compared with previous methods and the results are summarized in Table 4. The investigation revealed that no studies for simultaneous microextraction and determination of these analytes have been carried out so far. The present technique provides wide linear dynamic range and suitable detection limits in comparison with other methods. Moreover, extraction and desorption time for preconcentration of OPPs with this method is less than with the other methods (data is not in Table 4). The results show that LODs for the analyte determination is lower than in the previous works except for fenitronthion and diazinon by using air-assisted liquid–liquid microextraction/ GC-FPD [32] and for diazinon by using solid phase microextraction method/GC-Ms [12]. The linear dynamic range of more than two orders was also obtained by the proposed method, which is equal to or greater than by the other methods. Other advantages of the proposed method over the previous methods are relatively high enrichment factor and low sorbent consumption. However, one disadvantage of the method is higher relative standard deviations compared with most previous approaches, particularly solid-phase extraction coupled to GC methods (Table 4).
Conclusion
A new and efficient dispersive solid phase microextraction was developed as sample preparation for extraction of five organophosphorus pesticide compounds in real water samples prior to analysis by GC-MS. Effective factors in microextraction approach were thoroughly optimized. The new SPME sorbent, Zero valent Fe-reduced graphene oxide quantum dots, was synthesized and applied for extraction of OPPs. The performance of new sorbent was compared with magnetic graphene oxide and graphene oxide. The results indicate that the new sorbent is very suitable for extraction of these analytes. Advantages of this method are low LOD and short extraction time, simple operation, low sample solution and sorbent consumption, high preconcentration factor, and organic solvent free. In order to investigate the performance of the proposed method, several spiked real water and fruit juice samples were analyzed and the relative recoveries were obtained.
References
Yang Y, Asiri AM, Du D, Lin Y. Acetylcholinesterase biosensor based on a gold nanoparticle–polypyrrole–reduced graphene oxide nanocomposite modified electrode for the amperometric detection of organophosphorus pesticides. Analyst. 2014;139(12):3055–60.
Berijani S, Sadigh M, Pournamdari E. Homogeneous liquid–liquid microextraction for determination of organophosphorus pesticides in environmental water samples prior to gas chromatography-flame photometric detection. J Chromatogr Sci. 2016;54:1061–7.
Fulco CE, Liverman CT, Sox HC, Effects of long-term exposure to organophosphate pesticides in humans.National Academies Press (US); 2000.
Deng X, Guo Q, Chen X, Xue T, Wang H, Yao P. Rapid and effective sample clean-up based on magnetic multiwalled carbon nanotubes for the determination of pesticide residues in tea by gas chromatography–mass spectrometry. Food Chem. 2014;145:853–8.
Zhang W, Asiri AM, Liu D, Du D, Lin Y. Nanomaterial-based biosensors for environmental and biological monitoring of organophosphorus pesticides and nerve agents. Trends Anal Chem. 2014;54:1–10.
Meng X, Wei J, Ren X, Ren J, Tang F. A simple and sensitive fluorescence biosensor for detection of organophosphorus pesticides using H 2 O 2-sensitive quantum dots/bi-enzyme. Biosens Bioelectron. 2013;47:402–7.
Ghorbani M, Chamsaz M, Rounaghi GH. Glycine functionalized multiwall carbon nanotubes as a novel hollow fiber solid-phase microextraction sorbent for pre-concentration of venlafaxine and o-desmethylvenlafaxine in biological and water samples prior to determination by high-performance liquid chromatography. Anal Bioanal Chem. 2016;408(16):4247–56.
Ghorbani M, Chamsaz M, Rounaghi GH. Ultrasound-assisted magnetic dispersive solid-phase microextraction: a novel approach for the rapid and efficient microextraction of naproxen and ibuprofen employing experimental design with high-performance liquid chromatography. J Sep Sci. 2016;39(6):1082–9.
Mahpishanian S, Sereshti H. Graphene oxide-based dispersive micro-solid phase extraction for separation and preconcentration of nicotine from biological and environmental water samples followed by gas chromatography-flame ionization detection. Talanta. 2014;130:71–7.
Wen Y, Niu Z, Ma Y, Ma J, Chen L. Graphene oxide-based microspheres for the dispersive solid-phase extraction of nonsteroidal estrogens from water samples. J Chromatogr A. 2014;1368:18–25.
Chen D, Zhao Y, Miao H, Wu Y. A novel dispersive micro solid phase extraction using PCX as the sorbent for the determination of melamine and cyromazine in milk and milk powder by UHPLC-HRMS/MS. Talanta. 2015;134:144–52.
Zhu F, Ruan W, He M, Zeng F, Luan T, Tong Y, et al. Application of solid-phase microextraction for the determination of organophosphorus pesticides in textiles by gas chromatography with mass spectrometry. Anal Chim Acta. 2009;650(2):202–6.
Xie J, Liu T, Song G, Hu Y, Deng C. Simultaneous analysis of organophosphorus pesticides in water by magnetic solid-phase extraction coupled with GC-MS. Chromatographia. 2013;76(9/10):535–40.
Fenik J, Tankiewicz M, Biziuk M. Properties and determination of pesticides in fruits and vegetables. Trends Anal Chem. 2011;30(6):814–26.
Mehdinia A, Aziz-Zanjani MO. Recent advances in nanomaterials utilized in fiber coatings for solid-phase microextraction. Trends Anal Chem. 2013;42:205–15.
Tian J, Xu J, Zhu F, Lu T, Su C, Ouyang G. Application of nanomaterials in sample preparation. J Chromatogr A. 2013;1300:2–16.
Esmaeili A, Entezari MH. Facile and fast synthesis of graphene oxide nanosheets via bath ultrasonic irradiation. J Colloid Interface Sci. 2014;432:19–25.
Liu Y, Deng R, Wang Z, Liu H. Carboxyl-functionalized graphene oxide–polyaniline composite as a promising supercapacitor material. J Mater Chem. 2012;22(27):13619–24.
Karamani AA, Douvalis AP, Stalikas CD. Zero-valent iron/iron oxide-oxyhydroxide/graphene as a magnetic sorbent for the enrichment of polychlorinated biphenyls, polyaromatic hydrocarbons and phthalates prior to gas chromatography-mass spectrometry. J Chromatogr A. 2013;1271(1):1–9.
Shi Y, Pramanik A, Tchounwou C, Pedraza F, Crouch RA, Chavva SR, et al. Multifunctional biocompatible graphene oxide quantum dots decorated magnetic nanoplatform for efficient capture and two-photon imaging of rare tumor cells. ACS Appl Mater Interfaces. 2015;7(20):10935–43.
Pawliszyn J, Handbook of solid phase microextraction. 1ed. Elsevier; 2012.
Wercinski SA , Solid phase microextraction: a practical guide. CRC Press:Taylor & Francis Group; 1999.
Es’haghi Z, Khalili M, Khazaeifar A, Rounaghi GH. Simultaneous extraction and determination of lead, cadmium, and copper in rice samples by a new preconcentration technique: hollow fiber solid phase microextraction combined with differential pulse anodic stripping voltammetry. Electrochim Acta. 2011;56(9):3139–46.
Song X-Y, Shi Y-P, Chen J. A novel extraction technique based on carbon nanotubes reinforced hollow fiber solid/liquid microextraction for the measurement of piroxicam and diclofenac combined with high performance liquid chromatography. Talanta. 2012;100:153–61.
Sun J, Liang Q, Han Q, Zhang X, Ding M. One-step synthesis of magnetic graphene oxide nanocomposite and its application in magnetic solid phase extraction of heavy metal ions from biological samples. Talanta. 2015;132:557–63.
Wang C, Feng C, Gao Y, Ma X, Wu Q, Wang Z. Preparation of a graphene-based magnetic nanocomposite for the removal of an organic dye from aqueous solution. Chem Eng J. 2011;173(1):92–7.
Armbruster DA, Pry T. Limit of blank, limit of detection, and limit of quantitation. Clin Biochem Rev. 2008;29(Suppl 1):S49–52.
Fadaei A, Dehghani MH, Nasseri S, Mahvi AH, Rastkari N, Shayeghi M. Organophosphorous pesticides in surface water of Iran. Bull Environ Contam Toxicol. 2012;88(6):867–9.
Ballesteros E, Parrado M. Continuous solid-phase extraction and gas chromatographic determination of organophosphorus pesticides in natural and drinking waters. J Chromatogr A. 2004;1029(1):267–73.
Ravelo-Pérez LM, Hernández-Borges J, Ángel Rodríguez-Delgado M. Multiwalled carbon nanotubes as solid-phase extraction materials for the gas chromatographic determination of organophosphorus pesticides in waters. J Sep Sci. 2008;31(20):3612–9.
Su R, Xu X, Wang X, Li D, Li X, Zhang H, et al. Determination of organophosphorus pesticides in peanut oil by dispersive solid phase extraction gas chromatography-mass spectrometry. J Chromatogr B. 2011;879(30):3423–8.
You X, Xing Z, Liu F, Jiang N. Air-assisted liquid–liquid microextraction used for the rapid determination of organophosphorus pesticides in juice samples. J Chromatogr A. 2013;1311:41–7.
Wang Q, Yin J, Pan H, Xu F, Yang Y. A novel method based on combining ultrasonic-assisted dispersive solid-phase extraction and low-density solvent dispersive liquid–liquid microextraction (UA-DSPE-LDS-DLLME) for the determination of organophosphorus pesticides in soil. Anal Methods. 2014;6(18):7335–42.
Acknowledgements
The authors gratefully acknowledge the support of this research by the Ferdowsi University of Mashhad, Mashhad, Iran (No. 3/32788 dated 12/04/2014) and Central Lab Rural Water and Waste Water Company, Khorasan Razavi, Iran.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This article does not contain any studies with animal or human subjects.
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
ESM 1
(PDF 153 KB)
Rights and permissions
About this article

Cite this article
Akbarzade, S., Chamsaz, M., Rounaghi, G.H. et al. Zero valent Fe-reduced graphene oxide quantum dots as a novel magnetic dispersive solid phase microextraction sorbent for extraction of organophosphorus pesticides in real water and fruit juice samples prior to analysis by gas chromatography-mass spectrometry. Anal Bioanal Chem 410, 429–439 (2018). https://doi.org/10.1007/s00216-017-0732-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-017-0732-9