
Abstract
Protein corona formed on nanomaterial surfaces play an important role in the bioavailability and cellular uptake of nanomaterials. Modification of surfaces with oligoethylene glycols (OEG) are a common way to improve the resistivity of nanomaterials to protein adsorption. Short-chain ethylene oxide (EO) oligomers have been shown to improve the protein resistance of planar Au surfaces. We describe the application of these EO oligomers for improved protein resistance of 30 nm spherical gold nanoparticles (AuNPs). Functionalized AuNPs were characterized using UV-Vis spectroscopy, dynamic light scattering (DLS), and zeta potential measurements. Capillary electrophoresis (CE) was used for separation and quantitation of AuNPs and AuNP-protein mixtures. Specifically, nonequilibrium capillary electrophoresis of equilibrium mixtures (NECEEM) was employed for the determination of equilibrium and rate constants for binding between citrate-stabilized AuNPs and two model proteins, lysozyme and fibrinogen. Semi-quantitative CE analysis was carried out for mixtures of EO-functionalized AuNPs and proteins, and results demonstrated a 2.5-fold to 10-fold increase in protein binding resistance to lysozyme depending on the AuNP surface functionalization and a 15-fold increase in protein binding resistance to fibrinogen for both EO oligomers examined in this study.

Using capillary electrophoresis, the addition of short-chained oligo(ethylene oxide) ligands to gold nanoparticles was shown to improve protein binding resistance up to 15-fold.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The unique features of gold nanoparticles (AuNPs), including ease of preparation [1], tunable optical properties [2], and facile surface functionalization [3], have attracted considerable interest from the scientific community over the past few decades. These properties have enabled AuNPs to be harnessed for applications in many fields, including biology and medicine, where they have been used for bioimaging, diagnostic assays, molecular delivery, and photothermal therapy [4,5,6,7]. On exposure to biological fluids, AuNPs interact with various native proteins, often resulting in the formation of an adsorbed protein coating on the particle surface, referred to as a “protein corona” [8,9,10,11,12]. The formation of a protein corona can significantly alter the apparent properties of the initial particle (e.g. size, shape, charge) and can adversely affect ligand conjugation and biodistribution, potentially leading to altered immune responses and potential toxicity [13,14,15,16,17,18]. As such, researchers have sought to develop methods to decrease protein binding to AuNP surfaces [19,20,21,22,23,24,25].
Surface modification with thiol compounds, which exploits the relatively strong Au-S bond to increase ligand stability, is the most common method of increasing the protein adsorption resistance of AuNPs [26]. While a variety of thiol compounds are available for AuNP surface functionalization, thiol-terminated polyethylene glycols (PEG-SHs) are amongst the most commonly used [22, 27,28,29]. PEG-SHs easily form monolayers on Au surfaces, are hydrophilic, have low toxicity, and reduce adsorption of proteins [22, 30]. Several studies have demonstrated the relationship between PEG molecular weight and surface coverage with the degree of protein resistance; in general, increasing the ligand surface coverage, especially with higher molecular weight (longer-chain) PEG ligands leads to increased resistance toward protein binding [31].
Planar Au surfaces modified with oligoethylene glycol (OEG), which are short-chain oligomers, have also been shown to resist protein binding at levels comparable to PEG [19, 20, 32,33,34]. In contrast to their PEG counterparts, OEG compounds have been shown to have maximum protein resistance with a surface coverage of approximately 55%, with increased surface coverage actually leading to reduced protein adsorption resistance [35,36,37,38]. This observation, along with findings that some PEG and OEG modified surfaces are not protein resistant, illustrates that the ethylene oxide (EO) motif common to both PEG and OEG compounds may not be the primary contributor to protein resistance [39, 40]. Unlike PEG compounds, OEG compounds can adopt well-defined chemical structures, making them better suited for understanding the mechanisms behind their protein resistive nature [41]. While the exact conditions have yet to be clarified, the molecular characteristics of OEG surfaces found to give rise to high protein resistance include uniform surface coverage, conformational mobility, relative systemic disorder, and high degree of hydration [40]. While the protein resistance of planar Au surfaces modified by short-chain OEG compounds has been intensely studied, the generation of protein resistance via surface functionalization of AuNPs by short-chain OEG compounds has received less attention [19, 20, 32,33,34, 42]. Understanding how the protein-binding resistance of short-chain OEGs on planar Au surfaces compares to spherical AuNP surfaces has implications for the design of AuNPs in biomedical applications since the curved surface can induce different conformations of the adsorbates and therefore different protein affinity constants [20, 33, 34].
Protein-binding resistance can be quantified through determination of equilibrium and rate constants. Heterogeneous binding assays, in which either the nanoparticle or protein is affixed to a solid substrate, or homogeneous binding assays, in which both the nanoparticle and protein are allowed to interact in free solution, may be employed. Previous studies using heterogeneous binding assays have been reported, such as immobilization of AuNPs on planar surfaces for quantitative analysis of protein corona by surface plasmon resonance (SPR) [43, 44]. However, to more fully understand the dynamics governing protein adsorption to AuNP surfaces, homogeneous binding assays are preferred to allow the species to interact in free solution without restricted movement. To this end, several homogeneous binding assays have been reported that utilize dynamic light scattering (DLS) [45], circular dichroism (CD) spectroscopy [46], and fluorescence-based assays [47, 48] for quantitative and semi-quantitative analysis of protein corona. The present study uses capillary electrophoresis (CE) for evaluation of protein-AuNP binding under homogeneous binding conditions.
CE is a high efficiency and rapid separation technique that relies on differences in the charge-to-size ratio of analytes as the basis for separation. It has been extensively used for the separation of biomolecules [49,50,51] and more recently for nanoparticles [52,53,54,55]. Kinetic capillary electrophoresis (KCE) can be used for the determination of equilibrium and rate constants and may be carried out in several modes [49]. One mode, affinity capillary electrophoresis (ACE) has been previously used to determine nanoparticle-protein dissociation constants, K d [23, 56]. Other modes, like the well-established nonequilibrium capillary electrophoresis of equilibrium mixtures (NECEEM) technique [57,58,59,60], allow for the determination of both equilibrium (K d) and rate (k off) constants in a single experimental run, assuming 1:1 binding stoichiometry (see Electronic Supplementary Material). The dissociation constant, K d, and unimolecular rate constant for protein-AuNP dissociation, k off, can be determined through peak integration and curve fitting of the electropherogram resulting from injection of a mixture containing unbound protein and unbound AuNPs in equilibrium with protein-bound AuNPs. NECEEM is typically employed for DNA- or small molecule-protein binding [61, 62], but here we demonstrate its application for evaluation of nanoparticle-protein binding.
Herein we describe the preparation and characterization of thiol-functionalized AuNPs, the application of NECEEM for the determination of protein affinity for citrate-stabilized AuNPs, and the evaluation of the improved protein-binding resistance of thiol-functionalized AuNPs.
Experimental1
Reagents
Hydroxythiol herein,Footnote 1HT, (IUPAC: 21-mercapto-3,6,9,12,15,18-hexaoxahenicosanol) and hydroxydithiol herein, HD, (IUPAC: 19-(3′-mercaptopropanyl)-22-mercapto-3,6,9,12,15,18-hexaoxado-cosan-1-ol) were previously synthesized and characterized (Fig. S1) [40]. Citrate-stabilized AuNPs (nanoXact, 30 nm nominal particle diameter, 50 mg/L Au in 2 mM citrate solution) were obtained from nanoComposix (San Diego, CA) and are herein referred to as cit-AuNPs. Ultrapure deionized water (H2O, 18.2 MΩ*cm) was produced by a Millipore (Darmstadt, Germany) Milli-Q UV Plus Type I water purification system. Tris (hydroxymethyl) amino methane (Tris, ≥ 99.9%), glycine (Gly, ≥ 99.0%), fibrinogen (Fb, 65% -85% protein, from bovine plasma) and lysozyme (Lz, ≥ 90%, from chicken egg white) were obtained from Sigma-Aldrich (St. Louis, MO). All chemicals were used as received without further purification.
Stock solutions of HT and HD were prepared to final concentrations of 2 mM in water. Lyophilized proteins were prepared to a stock concentration of 1 mg/mL in water. Tris-Gly buffer was prepared to a final concentration of 5 mM Tris and 500 mM Gly with an unadjusted pH of 7.6.
Gold nanoparticle sample preparation
Functionalization and wash of AuNPs
AuNP solutions were combined with water, HT solution, or HD solution in a 1:1 volumetric ratio (1.4 mL total volume) and the resultant mixtures were vortexed for 30 s and allowed to incubate for a minimum of 40 h. After incubation, the AuNP solutions were centrifuged at 3900 g for 12 min using a MiniSpin Plus microcentrifuge (Eppendorf, Hamburg, Germany). The supernatant was removed and the AuNPs were reconstituted in H2O and vortexed for 30 s. The AuNPs were centrifuged a second time (at 3900 g for 12 min), the supernatant was removed, and the AuNPs were suspended in either H2O or Tris-Gly buffer to half of the original solution volume (0.7 mL) so that the final concentration of AuNPs was ≈ 50 mg/L (≈ 0.3 nM). The final solution was resuspended by vortexing for 30 s. AuNPs suspended in water are herein referred to as cit-AuNPs, while AuNPs functionalized with HT or HD are herein referred to as HT-AuNPs and HD-AuNPs, respectively.
Pre-wash of AuNPs
To test if the initial citrate concentration hindered functionalization of the AuNPs with HT and HD, a pre-wash step was performed prior to incubation with the thiol ligands. In this step, 0.7 mL AuNPs were centrifuged at 3900 g for 12 min and the supernatant was discarded. Then, H2O or Tris-Gly buffer was added to a final volume of 0.7 mL and the AuNPs were resuspended by vortexing for 30 s. Pre-washed AuNPs resuspended in H2O were then subjected to the functionalization and wash procedure described above. Pre-washed AuNPs resuspended in Tris-Gly buffer were used as control samples.
Gold nanoparticle characterization
Instrumentation and methodology
The pH of AuNP solutions was recorded with an Orion™ 2-Star pH Meter (Thermo Scientific, Waltham, MA) using an InLab® Semi-Micro Combination pH Electrode (Mettler Toledo, Columbus, OH). pH values are reported as the mean of four measurements ± one standard deviation (SD).
Dynamic Light Scattering (DLS) measurements were collected on a Zetasizer Nano ZS (Malvern Instruments, Westborough, MA) operated in 173° backscatter mode with a laser wavelength of 633 nm.
Zeta Potential (ZP) measurements were obtained on the same instrument using a palladium dip cell and applying the Smoluchowski equation; sample conductivity was measured during the ZP runs. The DLS measurement procedure followed NIST-NCL protocol PCC-1 [63] with the z-average diameter (d z), polydispersity index (PDI), ZP and suspension conductivity values reported as the mean of five measurements plus or minus one standard deviation.
UV-Vis absorbance measurements were collected in UV-transparent disposable plastic semimicro cuvettes (Brandtech, Essex, CT) with a 1 cm path length using a Lambda 750 spectrophotometer (Perkin-Elmer, Waltham, MA). The spectrometer was a split-beam style equipped with an 8 + 8 cell changer and water-jacketed temperature control. All measurements were recorded at ≈ 23 °C.
Transmission electron microscope (TEM) images were taken on a (FEI, Hillsboro, OR) Titan 80–300 analytical TEM operated at an accelerating voltage of 300 kV in bright-field mode using an Orius (Gatan, Pleasanton, CA) digital camera. Samples were prepared for TEM imaging by drop-casting the respective dispersions onto carbon-coated copper TEM grids (Ted Pella Inc., Redding, CA). Particle size analysis was manually performed using ImageJ and counting 210 particles for each sample.
Capillary electrophoresis
CE studies were performed on a Beckman Coulter P/ACE MDQ CE System with UV detection at 214 nm. UV detection of AuNPs may also be performed at 520 nm (where they exhibit a strong SPR band, however, detection at 214 nm provided increased analyte signal relative to 520 nm, with no added background signal. An uncoated, fused-silica capillary (Polymicro Technologies, Phoenix, AZ) was used for all CE experiments, with a length and inner diameter of 60.2 cm (50.0 cm from inlet to detector) and 75 μm, respectively. A 20 kV separation voltage was applied and samples were injected at 4 psi (≈ 27,500 Pa) for 10 s (≈ 355 nL injection volume).
All samples were diluted to final concentration in Tris-Gly buffer. To prepare AuNP-protein mixtures, working stock solutions of protein were prepared at various concentrations in Tris-Gly buffer, so that the final volume of protein added to AuNP solutions would be constant and therefore, the concentration of AuNPs would be the same. Then, protein and AuNPs were combined, the resulting mixtures were vortexed for 10 s, and incubated at 4 °C for a minimum of 1 h. AuNP-protein mixtures were vortexed for 10 s just prior to CE analysis.
32 KARAT software was used for CE control and data analysis, including manual peak integration and determination of analyte migration times. These data were subsequently processed using Microsoft Excel to determine K d , k off , and k on according to NECEEM theory (see ESM).
Results and discussion
Gold nanoparticle characterization
Excess citrate in AuNP suspensions can affect separation efficiency by CE. Thus, preliminary experiments focused on optimizing the number of wash cycles required to minimize the citrate concentration while maintaining the integrity of the AuNP suspension. The stock AuNP solutions were subjected to one, two, or three wash cycles. Conductivity measurements were used to estimate the concentration of citrate remaining in the AuNP suspension after successive wash cycles (Fig. S2). The suspension conductivity of the stock AuNPs was 0.5778 mS/cm ± 0.0040 mS/cm, which decreased to 0.0089 mS/cm ± 0.0006 mS/cm, 0.0021 mS/cm ± 0.0002 mS/cm, and 0.0016 mS/cm ± 0.0001 mS/cm after each wash cycle. While the first wash cycle removed the vast majority of the citrate, it was clear that the second wash cycle also removed a significant amount of citrate. The differences between the second and third wash cycles were small, with the absolute values of the measurements approaching that of pure water at ambient levels of CO2 (≈ 0.001 mS/cm) [64]. The stock (unwashed) AuNPs had an absorption peak centered at 520 nm (Fig. S3), characteristic of the SPR band of ~30 nm AuNPs, and the hydrodynamic diameter (d z) was 31.0 nm ± 0.6 nm, with a monomodal distribution (PDI = 0.10 ± 0.02; Table S1). After one wash cycle, the integrity of the AuNP suspension was essentially the same as the stock suspension, with the magnitudes of the SPR band (qualitative estimate of AuNP concentration) [65] and DLS size distributions nearly identical (Fig. S3). The second wash cycle resulted in slight aggregation of AuNPs (leading to a slight reduction in Au concentration, as seen from the reduced absorbance), and after the third wash cycle the AuNP suspension was fully compromised (low absorbance, high PDI; Table S1). These observations show that the third wash cycle was ineffective at removing additional citrate and caused significant aggregation and reduction in absorbance. Ultimately, two wash cycles were selected as optimal in the present study in order to remove the maximum amount of citrate without inducing significant AuNP aggregation.
Next, studies were performed to test if the Tris-Gly buffer used for NECEEM would influence the physicochemical properties of the AuNPs. Stock AuNPs were subjected to a single wash cycle and reconstituted in either H2O or Tris-Gly buffer. The UV-Vis spectrum of each AuNP suspension was nearly identical, with similar z-average diameters and monomodal size distributions (Fig. S4, Table S2). Given the similarities in absorbance and size distribution, the Tris-Gly buffer had little impact on the characteristics of the AuNPs. Consequently, all further characterization data were performed on AuNPs suspended in Tris-Gly buffer, with the “stock” AuNPs reconstituted in Tris-Gly buffer used as control samples.
To test if the initial citrate concentration hindered functionalization of the AuNPs by the thiol ligands, a pre-wash step was performed on the stock AuNP suspension prior to incubation with the HT and HD ligands (as described in the experimental section). From earlier experiments (Fig. S1), it was shown that this wash procedure removes most of the citrate from the solution. Thus, when exposed to the thiol ligands, the AuNPs that have been pre-washed before functionalization contain far less citrate than the stock AuNPs. Both pre-washed and stock AuNPs were exposed to thiol solutions (HT and HD) and incubated for at least 40 h to induce surface functionalization. The investigated physicochemical properties of each of the four thiol-functionalized samples (pre-washed and unwashed HT- and HD-AuNPs) were essentially identical, with the SPR bands of all samples having slight red shifts compared to the stock AuNPs (from 520 nm to 523 nm) and with negligible differences in the hydrodynamic diameter and zeta potential (Fig. S5, Table S3). With no observable differences in the pre-washed and unwashed thiol-functionalized samples, all future experiments were conducted without a pre-wash step in order to minimize sample preparation time.
Prior to protein affinity analysis, the physicochemical properties of the various AuNPs were measured under the optimized preparation conditions, which included two wash cycles and final constitution in Tris-Gly buffer. Stock AuNPs reconstituted in Tris-Gly buffer were used as a control to be compared to citrate, HT, and HD functionalized AuNPs prepared using the optimized protocol. The SPR band of the control and cit-AuNPs had a maximum absorbance at 520 nm, as expected based on the manufacturer specifications (Fig. 1). The SPR band of HT-AuNPs and HD-AuNPs was slightly red shifted to 523 nm, which illustrates their functionalization by the thiol ligands (Fig. 1) [20, 33]. Further, the DLS obtained z-average diameters of the HT-AuNPs and HD-AuNPs were ≈ 3 nm larger than the control and cit-AUNPs and also had smaller PDIs, suggesting the AuNPs were successfully functionalized and that the ligands increased the particle stability (Table 1). The measured ZPs of the AuNPs were ≈ −40 mV (except the HD-AuNPs), which confirmed a negatively charged corona surrounding the NPs, presumably from residual citrate (or other negatively charged ions present in the Tris-Gly buffer; Table 1). Interestingly, the ZP of the HD-AuNPs is −31 mV ± 1 mV, significantly less negative than the other AuNPs, including the HT-AuNPs (−40 mV ± 1 mV; Table 1). Given the similarities of the HT and HD ligands, the underlying cause behind this change likely arises from differences in their chemical structures. While HT is a monothiol, HD is a dithiol, which could result in different packing densities and surface coverage when the ligands form monolayers on the AuNPs, as seen in previous work involving planar Au surfaces [40]. In previous studies, it was shown that HT did not attain the highest possible surface coverage on planar Au substrates, while HD filled almost all the possible Au occupancy sites [40]. A similar phenomenon is likely occurring in the present study with AuNPs; whereby the HT-AuNPs have fewer surface sites occupied by the thiol ligand making it possible for more citrate (or other negatively charged ions in solution) to access the surface and leading to a more negative ZP relative to HD-AuNPs. These measurements suggest that the AuNPs are functionalized by the thiol ligands, which induce differences in their physicochemical properties. Previous work and ZP measurements suggest that the nature of surface modification by the thiol ligands varies due to their chemical structures, which could strongly influence the interactions of the functionalized AuNPs with proteins.

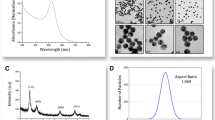
UV-Vis and DLS plots comparing the different preparations of AuNP suspensions, as labeled in each DLS plot and with colors corresponding to the UV-Vis spectra. All suspensions are constituted in Tris-Gly buffer and were prepared as detailed in the Experimental Section. The vertical dotted line on the UV-Vis plot highlights the λ max (520 nm) of the stock AuNPs’ SPR band
TEM images of the AuNPs were also obtained to provide additional information on the various AuNPs (Fig. S6). Size analysis of AuNPs (n = 210) resulted in respective average particle diameters of 29.2 nm ± 2.5 nm (cit-AuNPs), 28.1 nm ± 2.5 nm (HT-AuNPs), and 30.3 nm ± 2.9 nm (HD-AuNPs). These results are in strong agreement with each other and the vendor-reported average particle diameter of 30.3 nm ± 2.7 nm. This suggests that the functionalization of the AuNPs with HT or HD does not alter their size or lead to particle aggregation. The images of the HT- and HD-AuNPs show that the functionalized particles dried on the TEM grids in a less dispersed manner compared to their cit-AuNPs counterparts, suggesting a modified particle surface. Higher magnification (Fig. S7) reveals a thin (≈ 1.5 nm thick) carbonaceous layer on the surfaces of the HT- and HD-AuNPs. It is well-known that electron beam irradiation can induce the graphitization of carbon-based materials in the TEM [66], which would arise from irradiation of the thiol ligands in this particular case. These observations, in addition to the physicochemical characterizations described earlier (and the CE results to follow), support our assertion of the successful functionalization of the AuNPs by HT or HD.
Improved robustness of thiol-functionalized gold nanoparticles
During optimization of the sample preparation protocol, it was observed that HT- and HD-AuNPs could be subjected to successive wash cycles without significant aggregation (Fig. S3, Table 1). To further probe the apparent robustness of the functionalized AuNPs, HT- and HD-AuNPs were subjected to 9 successive wash cycles. After 9 cycles, neither of the functionalized AuNPs exhibited significant change in z-average diameter and the PDIs were below ≈ 0.150 with a monomodal size distribution (Fig. 2, Table S4). In comparison, the cit-AuNPs subjected to 3 wash cycles exhibited a multimodal size distribution and the PDI increased from 0.10 ± 0.02 to 0.46 ± 0.01. It was expected that removal of citrate would induce significant aggregation since citrate is added to AuNP suspensions as a stabilizing agent. Most notable was the tolerance of the thiol-functionalized AuNPs to successive washing, with minimal sample aggregation. This apparent stability of EO-functionalized AuNPs is consistent with other studies conducted with analogous ligands [20, 34].

DLS size distributions of (a) cit-AuNPs subjected to 3 centrifugation cycles, or (b) HT-AuNPs, and (c) HD-AuNPs subjected to 9 centrifugation cycles
Determination of equilibrium and rate constants for cit-AuNPs using NECEEM
Before beginning affinity studies, CE conditions were optimized for distinguishing cit-, HT-, and HD-AuNPs. With a straightforward modification of previously reported buffer conditions for particle separations in CE [67], the optimal buffer for separation of the different AuNPs was a Tris-Gly buffer. Using this buffer, cit- and HT-AuNPs migrated at ≈ 5.1 min while HD-AuNPs migrated at ≈ 4.2 min (Fig. S8). Since the basis for separation by CE is size-to-charge ratio, the migration times are consistent with the observed zeta potentials. Given the similar size of all the AuNPs, the more positive HD-AuNPs (ζ ≈ −31 mV) would migrate earlier than the more negative cit- and HT-AuNPs (ζ ≈ −39 mV) based on the migration principles of normal polarity mode in CE. Some peak broadening is observed for each AuNP under the optimized buffer conditions. Other reports in the literature have utilized surfactants, like sodium dodecyl sulfate (SDS), to improve peak efficiencies of AuNPs in CE [68, 69]. However, to avoid the influence of added surfactant on nanoparticle-protein binding, additives were not used to achieve more narrow peaks.
In order to carry out NECEEM studies, protein titrations were performed to identify suitable concentration ranges of lysozyme and fibrinogen. For example, a single peak was observed for cit-AuNPs alone at ≈ 5.1 min, but with the addition of increasing amounts of lysozyme up to 25 nM a second peak at earlier migration time (≈ 4.1 min) was observed (Fig. 3). The second peak is attributed to protein-bound AuNPs, which are expected to migrate differently than the AuNPs alone due to the difference in size-to-charge ratio caused by the adsorbed protein. At lysozyme concentrations greater than 25 nM, only a single peak at ≈ 4.1 min was observed, suggesting that most of the AuNPs were bound (Fig. 3). The addition of fibrinogen to cit-AuNPs resulted in similar changes although at lower fibrinogen concentrations (beginning at ≈ 1 nM; Fig. S9), presumably due to its significantly larger size (≈ 340 kDa compared to ≈ 14 kDa for lysozyme).

Vertically offset electropherograms of 1 nM cit-AuNPs titrated with lysozyme (concentrations as indicated above each electropherogram)
Ultimately, affinity calculations were performed for AuNPs (≈ 0.3 nM) combined with 10 nM, 15 nM, or 20 nM lysozyme and 1 nM, 2 nM, or 3 nM fibrinogen. Three replicate CE measurements were recorded at each concentration point and the entire experiment was performed in triplicate (with fresh sample preparation for each replicate). The dissociation constant, K d, and the unimolecular rate constant for dissociation, k off, were determined for each replicate and the bimolecular rate constant for association, k on, was determined using the K d and k off (see ESM). All values are reported as the average (n = 27) and standard deviation. Currently developed NECEEM theory assumes a 1:1 binding stoichiometry. It is known that nanoparticle-protein binding does not follow straightforward 1:1 stoichiometry, so the accuracy of equilibrium and kinetic values reported herein is limited by this theoretical assumption.
Results show that fibrinogen has slightly higher affinity than lysozyme for cit-AuNPs (Table 2). Further, the kinetics for protein-AuNP association (k on) are about 5-fold faster for fibrinogen relative to lysozyme (Table 2). The higher affinity and faster association kinetics for fibrinogen relative to lysozyme are consistent with it being a larger, “sticky” plasma protein. The dissociation constant and kinetic constants for fibrinogen differ significantly from another report in the literature conducted with 17 nm AuNPs (K d ≈ 0.5 μM, k off ≈ 0.002 s−1, k on ≈ 4000 M−1 s−1) [43]. However, these variations could be attributed to the smaller particle size and/or the assay used, where the previous study involved SPR analysis of AuNPs immobilized on a chip. SPR analysis has limitations related to the complexity of the sample matrix and it has further been shown that immobilization of one component in a binding assay (heterogeneous binding) can reduce the binding affinity [67]. In addition, the advantage of using a separation technology, like CE and NECEEM analysis, is that small changes in the AuNP sample due to protein adsorption can be detected based on observable changes in migration time. This allows for the analysis to be conducted at lower concentrations of protein (nanomolar range) compared to previous work (micromolar range).
Improved protein resistance of functionalized gold nanoparticles
Finally, protein titrations were performed with HT- and HD-AuNPs and compared to cit-AuNPs. For both HT- and HD-AuNPs, a second peak was not observed in the electropherogram when increasing amounts of protein were added to the sample, so NECEEM calculations were unable to be performed. However, a qualitative analysis of the electropherograms still provided significant insight as to how the HT and HD ligands modified the protein-AuNP interaction. For HT-AuNPs, slight changes in migration time were observed and for HD-AuNPs a declination of the AuNP peak was observed with no corresponding increase in a second peak.
For cit-AuNPs incubated with ≈ 1 nM fibrinogen, a protein-AuNP peak was observed at ≈ 4 min in addition to the AuNP peak at ≈ 5 min. At a fibrinogen concentration of ≈ 50 nM, only the peak at ≈ 4 min was observed, suggesting all of the AuNPs (initial concentration ≈ 0.3 nM) were bound (Fig. 4). In contrast, the electropherograms of ≈ 25 nM fibrinogen with HT- and HD-AuNPs had only a slight decrease in signal of the AuNP peak and not until a concentration of ≈ 50 nM fibrinogen was a marked decrease in the HT- or HD-AuNP peak observed (Fig. 4). Similarly, ≈ 10 nM lysozyme produced a second peak in the electropherogram for cit-AuNPs, but lysozyme concentrations up to ≈ 25 nM for HT-AuNPs and ≈ 100 nM for HD-AuNPs were required to observe any decrease in signal or change in migration time in the electropherogram (Fig. 5). In fact, HT-AuNPs did not undergo a significant decline in signal until the lysozyme concentration exceeded 50 nM and HD-AuNPs appeared relatively resistant to lysozyme adsorption up to 300 nM (Fig. 5). Overall, based on the concentration at which a decline in signal or change in migration time was first observed, a 2.5-fold and 10-fold improvement in lysozyme binding resistance was observed for HT- and HD-AuNPs, respectively and a 15-fold improvement in fibrinogen binding resistance was observed for both HT- and HD-AuNPs when compared to cit-AuNPs. These improvements in protein binding resistance of EO-functionalized AuNPs are consistent with binding studies conducted with other variations of EO ligands [19, 32, 34, 42]. Importantly, the observation that HD demonstrated greater protein-binding resistance compared to HT and related structural compounds is consistent with previous work conducted on planar gold surfaces [40].

Electropherograms comparing the effect of AuNP surface functionality on fibrinogen adsorption. AuNPs are as indicated in the upper right corner of the electropherograms and fibrinogen concentrations are as indicated in the figure legend

Electropherograms comparing the effect of AuNP surface functionality on lysozyme adsorption. AuNPs are as indicated in the upper right corner of the electropherograms and lysozyme concentrations are as indicated in the figure legend
Conclusions
We have demonstrated the functionalization of 30 nm citrate-stabilized AuNPs with short-chain EO oligomers, as confirmed by UV-Vis, DLS, and zeta potential measurements. We have further demonstrated the successful application of NECEEM for determining protein-AuNP equilibrium and rate constants for citrate-stabilized AuNPs. Semi-quantitative analysis of protein binding to HT- and HD-functionalized AuNPs reveals improvements in protein binding resistance (decreases in protein affinity) ranging from 2.5-fold to 15-fold. In summary, this work indicates that short-chain EO oligomers, which have previously shown improved protein-binding resistance on planar Au surfaces, have significant potential toward improving biocompatibility of spherical AuNPs. Future work that quantifies EO ligand surface coverage and explores the effect of particle size (and thereby curvature) on coverage is in progress. Further, improvements in CE separation efficiency are needed to determine protein binding affinities and kinetics for HT- and HD-AuNPs by NECEEM.
Notes
Certain commercial equipment, instruments, and materials are identified in this paper to specify an experimental procedure as completely as possible. In no case does the identification of particular equipment or materials imply a recommendation or endorsement by the National Institute of Standards and Technology nor does it imply that the materials, instruments, or equipment are necessarily the best available for the purpose.
References
Daniel MC, Astruc D. Gold nanoparticles: assembly, supramolecular chemistry, quantum-size-related properties, and applications toward biology, catalysis, and nanotechnology. Chem Rev. 2004;104(1):293–346. https://doi.org/10.1021/cr030698+.
Eustis S, el-Sayed MA. Why gold nanoparticles are more precious than pretty gold: noble metal surface plasmon resonance and its enhancement of the radiative and nonradiative properties of nanocrystals of different shapes. Chem Soc Rev. 2006;35(3):209–17. https://doi.org/10.1039/b514191e.
Han G, Ghosh P, Rotello VM. Functionalized gold nanoparticles for drug delivery. Nanomedicine (Lond). 2007;2(1):113–23. https://doi.org/10.2217/17435889.2.1.113.
Giljohann DA, Seferos DS, Daniel WL, Massich MD, Patel PC, Mirkin CA. Gold nanoparticles for biology and medicine. Angew Chem Int Ed Engl. 2010;49(19):3280–94. https://doi.org/10.1002/anie.200904359.
Murphy CJ, Gole AM, Stone JW, Sisco PN, Alkilany AM, Goldsmith EC, et al. Gold nanoparticles in biology: beyond toxicity to cellular imaging. Acc Chem Res. 2008;41(12):1721–30. https://doi.org/10.1021/ar800035u.
Jain PK, Huang X, El-Sayed IH, El-Sayed MA. Noble metals on the nanoscale: optical and photothermal properties and some applications in imaging, sensing, biology, and medicine. Acc Chem Res. 2008;41(12):1578–86. https://doi.org/10.1021/ar7002804.
Dreaden EC, Alkilany AM, Huang X, Murphy CJ, El-Sayed MA. The golden age: gold nanoparticles for biomedicine. Chem Soc Rev. 2012;41(7):2740–79. https://doi.org/10.1039/C1CS15237H.
Klein J. Probing the interactions of proteins and nanoparticles. Proc Natl Acad Sci U S A. 2007;104(7):2029–30. https://doi.org/10.1073/pnas.0611610104.
Lundqvist M, Stigler J, Elia G, Lynch I, Cedervall T, Dawson KA. Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts. Proc Natl Acad Sci U S A. 2008;105(38):14265–70. https://doi.org/10.1073/pnas.0805135105.
Lundqvist M, Stigler J, Cedervall T, Berggard T, Flanagan MB, Lynch I, et al. The evolution of the protein corona around nanoparticles: a test study. ACS Nano. 2011;5(9):7503–9. https://doi.org/10.1021/nn202458g.
Lynch I, Cedervall T, Lundqvist M, Cabaleiro-Lago C, Linse S, Dawson KA. The nanoparticle-protein complex as a biological entity; a complex fluids and surface science challenge for the 21st century. Adv Colloid Interf Sci. 2007;134-135:167–74. https://doi.org/10.1016/j.cis.2007.04.021.
Treuel L, Nienhaus GU. Toward a molecular understanding of nanoparticle–protein interactions. Biophys Rev. 2012;4(2):137–47. https://doi.org/10.1007/s12551-012-0072-0.
Monopoli MP, Walczyk D, Campbell A, Elia G, Lynch I, Bombelli FB, et al. Physical-chemical aspects of protein corona: relevance to in vitro and in vivo biological impacts of nanoparticles. J Am Chem Soc. 2011;133(8):2525–34. https://doi.org/10.1021/ja107583h.
Monopoli MP, Aberg C, Salvati A, Dawson KA. Biomolecular coronas provide the biological identity of nanosized materials. Nat Nanotechnol. 2012;7(12):779–86. https://doi.org/10.1038/nnano.2012.207.
Fleischer CC, Payne CK. Nanoparticle-cell interactions: molecular structure of the protein corona and cellular outcomes. Acc Chem Res. 2014;47(8):2651–9. https://doi.org/10.1021/ar500190q.
Walkey CD, Olsen JB, Song F, Liu R, Guo H, Olsen DW, et al. Protein corona fingerprinting predicts the cellular interaction of gold and silver nanoparticles. ACS Nano. 2014;8(3):2439–55. https://doi.org/10.1021/nn406018q.
Treuel L, Brandholt S, Maffre P, Wiegele S, Shang L, Nienhaus GU. Impact of protein modification on the protein corona on nanoparticles and nanoparticle-cell interactions. ACS Nano. 2014;8(1):503–13. https://doi.org/10.1021/nn405019v.
Arvizo RR, Giri K, Moyano D, Miranda OR, Madden B, McCormick DJ, et al. Identifying new therapeutic targets via modulation of protein corona formation by engineered nanoparticles. PLoS One. 2012;7(3):e33650. https://doi.org/10.1371/journal.pone.0033650.
Zheng M, Davidson F, Huang X. Ethylene glycol monolayer protected nanoparticles for eliminating nonspecific binding with biological molecules. J Am Chem Soc. 2003;125(26):7790–1. https://doi.org/10.1021/ja0350278.
Zhang F, Skoda MW, Jacobs RM, Zorn S, Martin RA, Martin CM, et al. Gold nanoparticles decorated with oligo(ethylene glycol) thiols: protein resistance and colloidal stability. J Phys Chem A. 2007;111(49):12229–37. https://doi.org/10.1021/jp074293v.
Deng ZJ, Liang M, Toth I, Monteiro MJ, Minchin RF. Molecular interaction of poly(acrylic acid) gold nanoparticles with human fibrinogen. ACS Nano. 2012;6(10):8962–9. https://doi.org/10.1021/nn3029953.
Larson TA, Joshi PP, Sokolov K. Preventing protein adsorption and macrophage uptake of gold nanoparticles via a hydrophobic shield. ACS Nano. 2012;6(10):9182–90. https://doi.org/10.1021/nn3035155.
Boulos SP, Davis TA, Yang JA, Lohse SE, Alkilany AM, Holland LA, et al. Nanoparticle-protein interactions: a thermodynamic and kinetic study of the adsorption of bovine serum albumin to gold nanoparticle surfaces. Langmuir. 2013;29(48):14984–96. https://doi.org/10.1021/la402920f.
Murthy AK, Stover RJ, Hardin WG, Schramm R, Nie GD, Gourisankar S, et al. Charged gold nanoparticles with essentially zero serum protein adsorption in undiluted fetal bovine serum. J Am Chem Soc. 2013;135(21):7799–802. https://doi.org/10.1021/ja400701c.
Khan S, Gupta A, Verma NC, Nandi CK. Kinetics of protein adsorption on gold nanoparticle with variable protein structure and nanoparticle size. J Chem Phys. 2015;143(16):164709. https://doi.org/10.1063/1.4934605.
Pensa E, Cortes E, Corthey G, Carro P, Vericat C, Fonticelli MH, et al. The chemistry of the sulfur-gold interface: in search of a unified model. Acc Chem Res. 2012;45(8):1183–92. https://doi.org/10.1021/ar200260p.
Niidome T, Yamagata M, Okamoto Y, Akiyama Y, Takahashi H, Kawano T, et al. PEG-modified gold nanorods with a stealth character for in vivo applications. J Control Release. 2006;114(3):343–7. https://doi.org/10.1016/j.jconrel.2006.06.017.
Shenoy D, Fu W, Li J, Crasto C, Jones G, DiMarzio C, et al. Surface functionalization of gold nanoparticles using hetero-bifunctional poly(ethylene glycol) spacer for intracellular tracking and delivery. Int J Nanomedicine. 2006;1(1):51–7. https://doi.org/10.2147/nano.2006.1.1.51.
Pelaz B, del Pino P, Maffre P, Hartmann R, Gallego M, Rivera-Fernandez S, et al. Surface functionalization of nanoparticles with polyethylene glycol: effects on protein adsorption and cellular uptake. ACS Nano. 2015;9(7):6996–7008. https://doi.org/10.1021/acsnano.5b01326.
Jokerst JV, Lobovkina T, Zare RN, Gambhir SS. Nanoparticle PEGylation for imaging and therapy. Nanomedicine (Lond). 2011;6(4):715–28. https://doi.org/10.2217/nnm.11.19.
Kenausis GL, Voros J, Elbert DL, Huang NP, Hofer R, Ruiz-Taylor L, et al. Poly(L-lysine)-g-poly(ethylene glycol) layers on metal oxide surfaces: attachment mechanism and effects of polymer architecture on resistance to protein adsorption. J Phys Chem B. 2000;104(14):3298–309. https://doi.org/10.1021/jp993359m.
Zheng M, Li Z, Huang X. Ethylene glycol monolayer protected nanoparticles: synthesis, characterization, and interactions with biological molecules. Langmuir. 2004;20(10):4226–35. https://doi.org/10.1021/la035981i.
Zhang F, Dressen DG, Skoda MW, Jacobs RM, Zorn S, Martin RA, et al. Gold nanoparticles decorated with oligo(ethylene glycol) thiols: kinetics of colloid aggregation driven by depletion forces. Eur Biophys J. 2008;37(5):551–61. https://doi.org/10.1007/s00249-007-0255-y.
Schollbach M, Zhang F, Roosen-Runge F, Skoda MW, Jacobs RM, Schreiber F. Gold nanoparticles decorated with oligo(ethylene glycol) thiols: surface charges and interactions with proteins in solution. J Colloid Interface Sci. 2014;426:31–8. https://doi.org/10.1016/j.jcis.2014.03.052.
Vanderah DJ, Valincius G, Meuse CW. Self-assembled monolayers of methyl 1-thiahexa(ethylene oxide) for the inhibition of protein adsorption. Langmuir. 2002;18(12):4674–80. https://doi.org/10.1021/la025720t.
Vanderah DJ, La H, Naff J, Silin V, Rubinson KA. Control of protein adsorption: molecular level structural and spatial variables. J Am Chem Soc. 2004;126(42):13639–41. https://doi.org/10.1021/ja047744n.
Jeon SI, Andrade JD. Protein—surface interactions in the presence of polyethylene oxide. J Colloid Interface Sci. 1991;142(1):159–66. https://doi.org/10.1016/0021-9797(91)90044-9.
Love JC, Estroff LA, Kriebel JK, Nuzzo RG, Whitesides GM. Self-assembled monolayers of thiolates on metals as a form of nanotechnology. Chem Rev. 2005;105(4):1103–69. https://doi.org/10.1021/cr0300789.
Vanderah DJ, Vierling RJ, Walker ML. Oligo(ethylene oxide) self-assembled monolayers with self-limiting packing densities for the inhibition of nonspecific protein adsorption. Langmuir. 2009;25(9):5026–30. https://doi.org/10.1021/la803896a.
Vaish A, Vanderah DJ, Vierling R, Crawshaw F, Gallagher DT, Walker ML. Membrane protein resistance of oligo(ethylene oxide) self-assembled monolayers. Colloids Surf B Biointerfaces. 2014;122(0):552–8. https://doi.org/10.1016/j.colsurfb.2014.07.031.
Ostuni E, Grzybowski BA, Mrksich M, Roberts CS, Whitesides GM. Adsorption of proteins to hydrophobic sites on mixed self-assembled monolayers. Langmuir. 2003;19(5):1861–72. https://doi.org/10.1021/la020649c.
Zheng M, Huang X. Nanoparticles comprising a mixed monolayer for specific bindings with biomolecules. J Am Chem Soc. 2004;126(38):12047–54. https://doi.org/10.1021/ja047029d.
Patra A, Ding T, Engudar G, Wang Y, Dykas MM, Liedberg B, et al. Component-specific analysis of plasma protein corona formation on gold nanoparticles using multiplexed surface Plasmon resonance. Small. 2016;12(9):1174–82. https://doi.org/10.1002/smll.201501603.
Cedervall T, Lynch I, Lindman S, Berggard T, Thulin E, Nilsson H, et al. Understanding the nanoparticle-protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proc Natl Acad Sci U S A. 2007;104(7):2050–5. https://doi.org/10.1073/pnas.0608582104.
Sasidharan A, Riviere JE, Monteiro-Riviere NA. Gold and silver nanoparticle interactions with human proteins: impact and implications in biocorona formation. J Mater Chem B. 2015;3(10):2075–82. https://doi.org/10.1039/c4tb01926a.
Treuel L, Malissek M, Grass S, Diendorf J, Mahl D, Meyer-Zaika W, et al. Quantifying the influence of polymer coatings on the serum albumin corona formation around silver and gold nanoparticles. J Nanopart Res. 2012;14(9):1102. https://doi.org/10.1007/s11051-012-1102-3.
Lacerda SH, Park JJ, Meuse C, Pristinski D, Becker ML, Karim A, et al. Interaction of gold nanoparticles with common human blood proteins. ACS Nano. 2010;4(1):365–79. https://doi.org/10.1021/nn9011187.
Bekdemir A, Stellacci F. A centrifugation-based physicochemical characterization method for the interaction between proteins and nanoparticles. Nat Commun. 2016;7:13121. https://doi.org/10.1038/ncomms13121.
Galievsky VA, Stasheuski AS, Krylov SN. Capillary electrophoresis for quantitative studies of biomolecular interactions. Anal Chem. 2015;87(1):157–71. https://doi.org/10.1021/ac504219r.
Ban E, Song EJ. Recent developments and applications of capillary electrophoresis with laser-induced fluorescence detection in biological samples. J Chromatogr B Analyt Technol Biomed Life Sci. 2013;929:180–6. https://doi.org/10.1016/j.jchromb.2013.04.028.
Righetti PG, Candiano G. Recent advances in electrophoretic techniques for the characterization of protein biomolecules: a poker of aces. J Chromatogr A. 2011;1218(49):8727–37. https://doi.org/10.1016/j.chroma.2011.04.011.
Guihen E. Nanoparticles in modern separation science. Trends Anal Chem. 2013;46:1–14. https://doi.org/10.1016/j.trac.2013.01.011.
Pyell U. Characterization of nanoparticles by capillary electromigration separation techniques. Electrophoresis. 2010;31(5):814–31. https://doi.org/10.1002/elps.200900555.
Surugau N, Urban PL. Electrophoretic methods for separation of nanoparticles. J Sep Sci. 2009;32(11):1889–906. https://doi.org/10.1002/jssc.200900071.
Aleksenko SS, Shmykov AY, Oszwaldowski S, Timerbaev AR. Interactions of tumour-targeting nanoparticles with proteins: potential of using capillary electrophoresis as a direct probe. Metallomics. 2012;4(11):1141–8. https://doi.org/10.1039/c2mt20141k.
Li N, Zeng S, He L, Zhong W. Probing nanoparticle--protein interaction by capillary electrophoresis. Anal Chem. 2010;82(17):7460–6. https://doi.org/10.1021/ac101627p.
Berezovski M, Krylov SN. Nonequilibrium capillary electrophoresis of equilibrium mixtures--a single experiment reveals equilibrium and kinetic parameters of protein-DNA interactions. J Am Chem Soc. 2002;124(46):13674–5. https://doi.org/10.1021/ja028212e.
Berezovski M, Nutiu R, Li Y, Krylov SN. Affinity analysis of a protein−aptamer complex using nonequilibrium capillary electrophoresis of equilibrium mixtures. Anal Chem. 2003;75(6):1382–6. https://doi.org/10.1021/ac026214b.
Krylov SN, Berezovski M. Non-equilibrium capillary electrophoresis of equilibrium mixtures - appreciation of kinetics in capillary electrophoresis. Analyst. 2003;128(6):571–5. https://doi.org/10.1039/b212913b.
Okhonin V, Krylova SM, Krylov SN. Nonequilibrium capillary electrophoresis of equilibrium mixtures, mathematical model. Anal Chem. 2004;76(5):1507–12. https://doi.org/10.1021/ac035259p.
Berezovski M, Drabovich A, Krylova SM, Musheev M, Okhonin V, Petrov A, et al. Nonequilibrium capillary electrophoresis of equilibrium mixtures: a universal tool for development of aptamers. J Am Chem Soc. 2005;127(9):3165–71. https://doi.org/10.1021/ja042394q.
Berezovski MV, Musheev MU, Drabovich AP, Jitkova JV, Krylov SN. Non-SELEX: selection of aptamers without intermediate amplification of candidate oligonucleotides. Nat Protoc. 2006;1(3):1359–69. https://doi.org/10.1038/nprot.2006.200.
Hackley VA, Clogston JD (2010) NIST - NCL Joint Assay Protocol, PCC-1 Version 1.1 Measuring the Size of Nanoparticles in Aqueous Media Using BatchMode Dynamic Light Scattering. https://ncl.cancer.gov/sites/default/files/protocols/NCL_Method_PCC-1.pdf.
Light TS, Kingman B, Bevilacqua AC (1995) The Conductivity of Low Concentrations of CO2 Dissolved in Ultrapure Water from 0–100°C. Paper presented at the 209th American Chemical Society National Meeting, Anaheim.
Haiss W, Thanh NT, Aveyard J, Fernig DG. Determination of size and concentration of gold nanoparticles from UV-vis spectra. Anal Chem. 2007;79(11):4215–21. https://doi.org/10.1021/ac0702084.
Egerton RF, Li P, Malac M. Radiation damage in the TEM and SEM. Micron. 2004;35(6):399–409. https://doi.org/10.1016/j.micron.2004.02.003.
Riley KR, Liu S, Yu G, Libby K, Cubicciotti R, Colyer CL. Using capillary electrophoresis to characterize polymeric particles. J Chromatogr A. 2016;1463:169–75. https://doi.org/10.1016/j.chroma.2016.08.017.
Qu H, Mudalige TK, Linder SW. Capillary electrophoresis/inductively-coupled plasma-mass spectrometry: development and optimization of a high resolution analytical tool for the size-based characterization of nanomaterials in dietary supplements. Anal Chem. 2014;86(23):11620–7. https://doi.org/10.1021/ac5025655.
Liu F-K, Wei G-T. Adding sodium dodecylsulfate to the running electrolyte enhances the separation of gold nanoparticles by capillary electrophoresis. Anal Chim Acta. 2004;510(1):77–83. https://doi.org/10.1016/j.aca.2003.12.064.
Acknowledgements
KRR and CMS acknowledge funding and support from the National Academy of Sciences - National Research Council Postdoctoral Research Associateship Program. ITW recognizes the support of the NIST Summer Undergraduate Research Fellowship (SURF) Program.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
M. L. Walker and D. J. Vanderah conceived the initial project; I. T. Wood and M. L. Walker performed initial studies; K.R. Riley and C.M. Sims refined the project and contributed equally to essential experimentation and composition of the completed work.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any research with human or animal subjects.
Electronic supplementary material
ESM 1
(PDF 9.52 mb)
Rights and permissions
About this article

Cite this article
Riley, K.R., Sims, C.M., Wood, I.T. et al. Short-chained oligo(ethylene oxide)-functionalized gold nanoparticles: realization of significant protein resistance. Anal Bioanal Chem 410, 145–154 (2018). https://doi.org/10.1007/s00216-017-0704-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-017-0704-0