
Abstract
We describe a sensitive aptamer-based sandwich assay for protein detection on microplate by using rolling circle amplification (RCA) coupled with thrombin catalysis. This assay takes advantage of RCA generating long DNA oligonucleotides with repeat thrombin-binding aptamer sequence, specific aptamer affinity binding to achieve multiple thrombin labeling, and enzyme activity of thrombin for signal generation. Protein target is specifically captured by antibody-coated microplate. Then, an oligonucleotide containing an aptamer for protein and a primer sequence is added to form a typical sandwich structure. Following a template encoded with complementary sequence of aptamer for thrombin, RCA reaction extends the primer sequence into a long oligonucleotide. Many thrombin molecules bind with the RCA product. Thrombin catalyzes the conversion of its chromogenic or fluorogenic peptide substrates into detectable products for final quantification of protein targets. We applied this strategy to the detection of a model protein target, platelet-derived growth factor-BB (PDGF-BB). Due to double signal amplifications from RCA and thrombin catalysis, this assay enabled the detection of PDGF-BB as low as 3.1 pM when a fluorogenic peptide substrate was used. This assay provides a new way for signal generation in RCA-involved assay through direct thrombin labeling, circumventing time-consuming preparation of enzyme-conjugate and affinity probes. This method has promise for a variety of analytical applications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Aptamers originate from a nucleic acid library through in vitro SELEX process (selective evolution of ligands by exponential enrichment), showing high specificity and affinity to targets [1–3]. As nucleic acid affinity reagents, aptamers possess many advantages in analytical applications and biosensing, such as easy chemical synthesis with low-cost, high-stability, being easy-to-stock, and amplifiable property [4–6]. Numerous assays have been developed for protein detections by using aptamers since the emergence of aptamers [4–9]. The amplifiable property of aptamers enables amplification of nucleic acid sequence to achieve sensitive detection of protein targets in aptamer-based assays by converting protein detection to detection of nucleic acids [4, 10, 11]. Among these assays, polymerase chain reaction (PCR) and rolling circle amplification (RCA) are two popular amplification techniques to enhance the sensitivity of aptamer-based assays [10–12].
RCA is one simple amplification strategy conducted in isothermal condition [10–13]. In a typical RCA reaction, a primer hybridizes with a circle template, which is extended around the template with DNA polymerase. RCA produces a long, single-strand DNA molecule complementary to the circle template. Hence, hundreds to thousands of repeat units are generated from each template. Through a rational design, the obtained long oligonucleotide can combine with versatile reporters (e.g., fluorescence dyes, electrochemical tag, nanoparticles, enzymes) to generate signals, allowing for signal amplification [10–13]. RCA shows advantages over PCR in simplicity and no need to change temperature [10–13]. RCA strategy has been applied in a variety of formats by combining different detection technology, such as colorimetry, fluorescence, electrochemistry, chemiluminescence, diffractometry, etc. [10–12, 14–20]. In the RCA-based aptamer assay, primer sequence can be easily conjugated with aptamer sequence through chemical synthesis [10–17]. By designing the templates, the DNA products of RCA can be readily customized to contain DNA aptamer, DNAzymes, or other specific DNA sequences, making RCA flexible for many applications [12, 14, 16, 21].
Thrombin-binding aptamers are widely used for detection of thrombin, an important enzyme molecule in blood [22–24]. The binding of aptamer to thrombin does not inhibit the activity of thrombin cleaving small peptide substrate because the aptamer binding site is distinct from the active site of thrombin [25, 26]. Taking advantage of aptamer binding and thrombin catalysis, sensitive assays for thrombin have been developed through affinity capture of thrombin by aptamer and the enzyme activity of captured thrombin in cleaving small peptide substrates to generate detectable products [27–30].
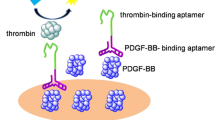
Inspired by the previous work [14, 16, 27–30], here, we demonstrated an aptamer assay for protein detection through RCA coupled with thrombin catalysis by using RCA reaction to produce an oligonucleotide binding with multiple thrombin molecules and using thrombin to generate signals. Figure 1 shows the principle of the strategy for protein detection in a sandwich format on the microplate. The protein target is captured by antibody-coated microplate, and then it is bound with an oligonucleotide containing the aptamer for protein target and a primer sequence for RCA reaction. Complementary sequence of the aptamer for thrombin is encoded in the RCA template. The template hybridizes with primer and is circularized by ligase. Following the circular template, RCA reaction extends the primer into a long oligonucleotide that contains many aptamer sequence units for thrombin. The generated DNA product binds with many thrombin molecules, so multiple thrombin molecules are attached on the sandwich complex. As an enzyme molecule, thrombin catalyzes the cleavage of chromogenic or fluorogenic small peptide substrates into detectable products, to achieve final detection of protein targets by absorbance analysis or fluorescence analysis. This strategy provides a new way for signal generation in RCA-involved assay by using thrombin as multiple enzyme labels through direct thrombin-aptamer binding. It does not need tedious and time-consuming process to prepare enzyme-conjugate and affinity probes for enzyme labeling in RCA. Double signal amplification can be achieved in this assay through RCA and thrombin catalysis.

Schematic of the aptamer assay for protein detection using RCA coupled with thrombin catalysis. Protein target is captured by antibody coated on microplate, and then it is bound with the oligonucleotide (aptamer-primer) containing an aptamer for protein target and a primer sequence to form a sandwich complex. The template encoded with a complementary sequence of the aptamer for thrombin hybridizes with the primer, and it is circularized by ligase. Following the circular template, RCA reaction extends the primer sequence into a long single-stranded DNA sequence for thrombin. The generated long DNA binds with many thrombin molecules, achieving multiple thrombin labeling in sandwich complex. Thrombin catalyzes the cleavage of small peptide substrates into detectable product
To show the proof of principle of the demonstrated strategy, here, we developed an assay for a model protein target, platelet-derived growth factor-BB (PDGF-BB), which is a potential cancer marker and is known to be related to tumor growth, progression, and transformation [31, 32]. Sensitive detection of PDGF-BB with our assay using absorbance analysis or fluorescence analysis was achieved. As low as 3.1 pM PDGF-BB could be detected in our assay using fluorogenic substrate. The demonstrated strategy shows promise in a variety of analytical applications. RCA-combined thrombin labeling can be applied to other versatile RCA assay formats.
Materials and methods
Reagents and apparatus
The DNA-BIND microplates were obtained from the Corning Inc. (NY, USA). Recombinant human PDGF-BB, PDGF-AB, and PDGF-AA were purchased from Invitrogen. Anti-PDGF-BB antibody was obtained from R&D system (Minneapolis, MN, USA). 10× phi 29 DNA polymerase reaction buffer (RCA buffer) was bought from New England Biolabs (Beijing, China). The phi 29 DNA polymerase and dNTPs were provided by the Epicenter (Madison, WI, USA). The Escherichia coli DNA ligase was purchased from Takara Biotechnology Co. Ltd. (Dalian, China). Bovine serum albumin (BSA), hemoglobin (Hb), human immunoglobulin G (IgG), lysozyme (Lys), and human serum sample were obtained from Sigma. Human α-thrombin was ordered from Haematologic Technologies Inc. The chromogenic substrate of thrombin, N-p-tosyl-Gly-Pro-Arg-p-nitroanilide acetate and the fluorogenic substrate of thrombin, N-p-tosyl-Gly-Pro-Arg-7-amido-4-methylcoumarin hydrochloride, were purchased from Sigma. Ultrapure water was obtained through a Purelab Ultra Elga Labwater system for solution preparation. A microplate reader (Varioskan Flash, Thermo Fisher Scientific, Inc.) was used to record the absorbance and fluorescence signals. Oligonucleotides were synthesized and purified by Sangon Biotech Co., Ltd. (Shanghai, China), and sequences were shown in Table 1.
The following buffers were used. Coupling buffer contained 50 mM Na2HPO4 (pH 8.5) solution. Blocking buffer contained 50 mM Na2HPO4 and 10 mM Tris–HCl (pH 8.5). PBS buffer (pH 7.5) was composed of 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 2 mM KH2PO4. SSC buffer (pH 7.2) consisted of 0.3 M NaCl and 0.03 M sodium citrate (Na3C6H5O7). Ligation buffer consisted of 30 mM Tris–HCl (pH 7.5), 4 mM MgCl2, 10 mM (NH4)2SO4, 0.1 mM nicotinamide adenine dinucleotide (NAD), 1.2 mM EDTA, and 0.05 mg/mL BSA. Thrombin-binding buffer contained 20 mM Tris–HCl (pH 7.5), 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, and 1 mM CaCl2. Thrombin catalysis buffer contained 50 mM Tris–HCl (pH 8.5) and 1 M NaCl. Five types of buffers were used for washing, including buffer A (10 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.05 % Tween20), buffer B (PBS buffer with 0.1 % Tween 20), buffer C (PBS buffer with 1 mM MgCl2 and 0.1 % Tween 20), buffer D (SSC buffer with 0.05 % Tween 20), and buffer E (thrombin-binding buffer with 0.1 % Tween20).
Assay procedure
Antibody was conjugated on the surface of DNA-BIND microplates by the following procedure. The anti-PDGF-BB antibody was diluted to 2 μg/mL in coupling buffer and added into wells of a clear microplate (for absorbance detection) or a black microplate (for fluorescence detection) (100 μL per well). The microplate was incubated for 1 h to conjugate the antibody on the surface of wells of microplate. The excess of PDGF-BB antibody was removed by decanting the supernatant. The wells were washed three times with buffer A and blocked with blocking buffer containing 10 mg/mL BSA (200 μL per well) for 1 h. After decantation of blocking buffer, the wells were washed once with buffer B.
A series of 100 μL PDGF-BB in PBS buffer containing 1 mM MgCl2 and 4 mg/mL BSA were added into the above-mentioned wells and incubated for 30 min. PDGF-BB was captured on the antibody-coated microplates. After washing with buffer C for three times, 100 μL of 200 nM oligonucleotide probe (named PD44-primer) in PBS containing 1 mM MgCl2 was added to the wells and incubated for 30 min (before addition, the PD44-primer was heated at 85 °C for 3 min and cooled to room temperature). After the wells were rinsed with buffer C for three times, 100 μL of 50 nM template in SSC buffer containing 0.05 % Tween 20 was added into the wells, and incubated for 1 h to hybridize with the PD44-primer. The excess template was removed by washing with buffer D for three times. And then, the template was circularized via ligation by ligase (0.02 U/μL) in ligation buffer (100 μL each well) for 1 h.
RCA reaction was conducted for 1 h in 100 μL of RCA buffer containing 0.4 mM dNTPs, 0.05 U/μL phi29 DNA polymerase, and 0.1 mg/mL BSA. The wells were washed three times with buffer D. One hundred microliters of 20 nM thrombin in thrombin-binding buffer was added and incubated for 30 min. After washing the wells with buffer E for three times, thrombin catalyzed cleavage of the chromogenic substrate (0.375 mM) or fluorogenic substrate (0.059 mM) in 100 μL of thrombin catalysis buffer for 1 h. Finally, the generated product was measured by a plate reader. For assay using chromogenic substrate, the absorbance was measured at the wavelength of 405 nm. For assay using fluorogenic substrate, the fluorescence was measured at 440 nm with the excitation of 370 nm. The incubation temperature was 37 °C in all steps of experiments.
Result and discussion
Design of the RCA-based aptamer assay coupled with thrombin catalysis for PDGF-BB
Figure 1 shows the schematic diagram of the aptamer assay for protein detection using RCA coupled with thrombin catalysis for PDGF-BB detection. Anti-PDGF-BB antibody was coated on the surface of wells of microplate through covalent conjugation. PDGF-BB was captured by the coated antibody on microplate. Next, the oligonucleotide probe (PD44-primer) containing an aptamer sequence (PD44: 5ʹ-TAC TCA GGG CAC TGC AAG CAA TTG TGG TCC CAA TGG GCT GAG TA-3ʹ [33]) for PDGF-BB and a primer sequence for RCA was added and bound with the captured PDGF-BB, forming a sandwich complex. The template encoded with complementary sequence of the aptamer for thrombin was added and hybridized to the primer and this template was circularized by ligase through ligation reaction. Subsequently, in the presence of phi29 DNA polymerase and dNTPs, following the circular template, RCA reaction extended the primer into a long single-stranded DNA containing many copies of aptamer sequences for thrombin. Many thrombin molecules were attached onto the RCA product. Finally, the thrombin catalyzed the cleavage of chromogenic substrate or fluorogenic substrate into detectable products, allowing for the quantification of PDGF-BB.
Two thrombin-binding DNA aptamers have been widely used. One has 15 bases with a lower binding affinity (denoted as Apt15, K d ∼100 nM), and the other is a 29-mer aptamer with high binding affinity (denoted as Apt29, K d ∼0.5 nM) (sequence information shown in Table 1) [22–24]. We tried two kinds of templates to generate the 15-mer thrombin-binding aptamer sequence and the 29-mer DNA aptamer, respectively. The template encoded the complementary sequence of Apt15 could be used to generate repeat sequences of Apt15 in RCA product, and it was denoted as Template-Apt15. The other template, called Template-Apt29, could be used to produce tandem Apt29 sequences in the RCA product, as the template contained the complementary sequence of Apt29. We first tested the feasibility of the assay using chromogenic substrate, N-p-tosyl-Gly-Pro-Arg-p-nitroanilide. The labeled thrombin could cleave the chromogenic substrate into p-nitroaniline, and the generated p-nitroaniline was measured at 405 nm [30]. As Fig. 2 shows, we observed great signal increase in the presence of PDGF-BB over the signal from blank sample when either of the templates was used in the assay, showing our strategy is feasible for the detection of PDGF-BB. When Template-Apt29 was used, the obtained signal increase over the blank was much higher than that from the assay using Template-Apt15. The enhanced signal can be attributed to the higher binding affinity of Apt29 than Apt15. Thus, Template-Apt29 was used in our assay.

The absorbance signal obtained in the assay using different templates (Template-Apt15 and Template-Apt29). Experimental conditions: 200 nM PD44-primer, 50 nM template, 0.02 U/μL ligase, 0.1 U/μL polymerase, 0.2 mM dNTPs, 10 nM thrombin
Optimization of experimental conditions
We further investigated experimental conditions of assays for the detection of PDGF-BB, including the amount of primer, template, ligase, polymerase, dNTPs, and thrombin. We measured the absorbance signal from the blank sample and the absorbance signal from PDGF-BB (0.25 or 0.5 nM). We subtracted the absorbance signal of the blank from the absorbance signal of the PDGF-BB sample to get the net increase of absorbance signal (ΔAbs). The effects of experimental conditions were evaluated by measuring the change of ΔAbs.
The obtained ΔAbs increased with increasing concentration of PD44-primer (see Electronic Supplementary Material (ESM) Fig. S1) A high ΔAbs value was obtained when the concentration was higher than 100 nM. Though 400 nM PD44-primer could further increase the signal, 200 nM PD44-primer was selected for subsequent experiments because a low blank signal was observed at this concentration. Furthermore, the obtained ΔAbs increased as the concentration of ligase increased from 0.005 to 0.02 U/μL, and then ΔAbs slightly decreased when the concentration of ligase further increased (see ESM Fig. S2). The results show the use of ligase at the tested concentration range can allow effective ligation of the template to form a circular template. Thus, 0.02 U/μL of ligase was selected due to the maximum ΔAbs was observed at this concentration.
We also tested the influence of template concentration on the obtained ΔAbs. The maximum ΔAbs was obtained with template at 50 nM, so 50 nM template was preferred (see ESM Fig. S3). The amount of dNTPs had a large effect on the obtained ΔAbs. A high ΔAbs was obtained when the concentration of dNTPs ranged from 0.4 to 1.6 mM (Fig. 3). The further increase of dNTPs caused decrease of ΔAbs, and this phenomenon is consistent with the previous report [20] The high concentrations of dNTPs may cause the generated products wrapped with other, which affects the formation of right conformation of aptamer of thrombin and the affinity binding of thrombin. However, the exact reason for the signal decrease caused by higher concentrations of dNTPs is not known yet. dNTPs at 0.4 mM was applied in the assay due to a high signal was obtained at this concentration.

The effect of dNTPs concentration on net increase of absorbance signal (ΔAbs) obtained from PDGF-BB sample. Experimental conditions: 0.25 nM PDGF-BB, 200 nM PD44-primer, 25 nM template, 0.02 U/μL ligase, 0.1 U/μL polymerase, 10 nM thrombin
The amount of polymerase also had significant influence on the obtained ΔAbs. (Fig. S4 in ESM) When the concentration of polymerase was lower than 0.05 U/μL, the obtained ΔAbs was low. When the concentration of polymerase increased from 0.05 to 0.4 U/μL, the obtained ΔAbs decreased. A similar phenomenon was also observed in a previous study [20]. The real reason for the decrease of signals caused by high concentration of polymerase is still not known; 0.05 U/μL of polymerase was applied in the assay. The amount of thrombin was also optimized (Fig. 4). The use of thrombin at 5 nM gave low ΔAbs, while the ΔAbs sharply increased when thrombin at 10 nM was used; 20 nM of thrombin enabled the assay to give the maximum ΔAbs. Further increase of thrombin cause ΔAbs decreased. We do not know what caused the signal decreasing when thrombin at concentrations higher than 20 nM was applied; 20 nM thrombin was finally used in the assay.

The effect of thrombin concentration on net increase of absorbance signal (ΔAbs) obtained from PDGF-BB sample. Experimental conditions: 0.25 nM PDGF-BB, 200 nM PD44-primer, 50 nM template, 0.02 U/μL ligase, 0.4 mM dNTPs, and 0.05 U/μL polymerase
In addition, we also did a control experiment without using RCA. In the control experiment, after sandwiching the PDGF-BB with the capture antibody coated on microplate and PD44-primer, we added thrombin into the wells. After incubation and washing, the chromogenic substrate of thrombin was added and was incubated. In this control experiment, the obtained absorbance signal was close to that from the blank sample. The result shows that the thrombin could not bind with oligonucleotide probe PD44-primer in the sandwich complex because PD44-primer did not contain the thrombin-binding aptamer sequence.
Detection of PDGF-BB
Under the optimized conditions, our assay allowed successful detection of varying concentrations of PDGF-BB. Figure 5a shows that ΔAbs increased with the increasing PDGF-BB concentration. A good linear relationship between the ΔAbs and PDGF-BB concentration was obtained in the range from 0.031 to 0.5 nM (y = 1.31x, R 2 = 0.998). The detection limit was 0.031 nM, according to signal increase higher than three times of the standard deviation of the blank. When PDGF-BB concentration is higher than 0.5 nM, the saturated signals were observed, which can be attributed to that the chromogenic substrate was totally converted into product by thrombin. The limited antibody on the surface of microplate can also cause the saturated signals.

a Detection of PDGF-BB by the assay using chromogenic substrate. b Detection of PDGF-BB by the assay using fluorogenic substrate
To evaluate the efficiency of RCA reaction, we also did a control experiment, in which only one thrombin molecule was labeled on the sandwich complex by using a DNA probe (5ʹ-TAC TCA GGG CAC TGC AAG CAA TTG TGG TCC CAA TGG GCT GAG TA TTTTTT AGT CCG TGG TAG GGC AGG TTG GGG TGA CT-3ʹ) containing the aptamer for PDGF-BB (PD44) and the aptamer for thrombin (Apt29) to replace PD44-primer. When PDGF-BB was captured by antibody on the microplate, then the DNA probe was added to form a sandwich complex. After that, single thrombin bound with the probe through binding between thrombin and Apt29. Thrombin then catalyzed the hydrolysis of its chromogenic substrate for signal generation. In this control experiment, except without using RCA reaction steps, other experimental conditions were the same as that used in the RCA assays. In this control experiment, PDGF-BB ranging from 0.5 to 32 nM was detected with a linear relationship between the ΔAbs and PDGF-BB concentration (y = 0.021x, R 2 = 0.997) in the assay using single thrombin labeling (Fig. S5 in ESM). The slope of the linear fitting equations corresponding to multiple thrombin labeling obtained by RCA is 62-fold of the slope of the linear fitting equation corresponding to single thrombin labeling. The result shows RCA caused 62-fold signal amplification by multiple thrombin labeling. More than 62 repeat aptamer sequences for thrombin binding may be generated through RCA. The RCA amplification efficiency is close to the reported value at similar experimental conditions [14].
To further improve the sensitivity of the assay, we used fluorogenic substrate instead of chromogenic substrate in the assay. In this case, the labeled thrombin catalyzed the cleavage of the fluorogenic substrate, N-p-tosyl-Gly-Pro-Arg-7-amido-4-methylcoumarin, into a fluorescent product. The product was measured at emission wavelength of 440 nm with an excitation at 370 nm. Figure 5b shows the detection of PDGF-BB with the assay using fluorogenic substrate. Fluorescence intensity was shown with relative fluorescence unit (RFU). Net fluorescence intensity increase (ΔRFU) was obtained by subtracting the obtained fluorescence intensity of blank sample from the obtained fluorescence signal of PDGF-BB sample. Due to the sensitive fluorescence measurement, the detection limit of the assay was further put down to 3.1 pM, a good linear relationship between ΔRFU and concentration of PDGF-BB in the range from 3.1 to 150 pM was obtained (y = 10.2x, R 2 = 0.998). When PDGF-BB with concentrations higher than 150 pM, the obtained signals were not further significantly increased because the used fluorogenic substrate was all converted into the fluorescent product at this condition. The increase of fluorogenic substrate may extend the upper detection range. The obtained sensitivity of our assay is higher or comparable to some previously reported aptamer-based assays for PDGF-BB [17, 18, 34–39]. Although the sensitivity of our assay was lower than that obtained from assays using multiple signal amplification or highly sensitive analysis approach [14, 16, 20], our assay provides a simple way to generate enhanced signals from RCA by introducing many thrombin molecules as enzyme labels. Through direct binding with enzyme molecule of thrombin, our assay greatly reduces the tedious and time-consuming steps to prepare enzyme-conjugates and affinity probes for enzyme labeling in RCA.
Specificity test
We tested a few non-target proteins in place of PDGF-BB with the same experimental procedures to assess the specificity of our assay using fluorogenic substrate for PDGF-BB detection. As shown in Fig. 6, the addition of the high concentrations of the non-target proteins (Hb, IgG, Lys at 100 nM and thrombin at 20 nM) did not cause interference with detection of PDGF-BB. In addition, we also tested two common PDGF variants (PDGF-AA and PDGF-AB). Compared with the blank signal, PDGF-AB (50 pM) could cause a slight signal increase, but much lower than that from PDGF-BB (50 pM). The signal from PDGF-AA (50 pM) was also close to the blank signal. The results could be explained by that the aptamer was originally selected to bind to the B chain. PDGF-AB consists of both A and B chains meaning the PDGF-AB can also bind to the aptamer with lower affinity, while PDGF-AA does not bind with aptamer [38, 39]. The results show that our assay has a good selectivity for discrimination of PDGF-BB from other proteins. The result for specificity test of the assay using chromogenic substrate was shown in Fig. S5 in ESM, and PDGF-BB could also be selectively tested.

Specificity test for detection of PDGF-BB by the assay using fluorogenic substrate; 50 pM PDGF-BB, 50 pM PDGF-AB, 50 pM PDGF-AA, 100 nM Hb, 100 nM IgG, 100 nM Lys, and 20 nM thrombin were tested
We further assessed the performance of the assay for the detection of PDGF-BB in a complex sample matrix. PDGF-BB spiked in the 100-fold diluted human serum was analyzed. As shown in Fig. S6A in ESM, PDGF-BB in the range of 0.031–0.5 nM in the 100-fold diluted serum sample could still be detected (y = 1.35x, R 2 = 0.993) when the assay using chromogenic substrate was applied. When the assay using fluorogenic substrate was applied, PDGF-BB ranging from 3.1 to 150 pM could be detected in the 100-fold diluted human serum sample (y = 9.75x, R 2 = 0.995) (Fig. S6B in ESM). The result indicates that our assay can be used for detection of PDGF-BB in complex sample matrix.
Conclusion
We reported an aptamer assay for protein detection by combining rolling circle amplification (RCA) and thrombin catalysis, using the detection of PDGF-BB as an example. In this strategy, we achieved multiple thrombin molecules attached on the target protein by using RCA to generate oligonucleotide containing aptamer sequences for thrombin. The final detection of target was obtained by cleaving small peptide substrates into detectable products with thrombin. The double amplification approaches from RCA and thrombin catalysis improved the sensitivity. With this strategy, we achieved detection of PDGF-BB in a sandwich format on PDGF-BB antibody-coated microplate by using an oligonucleotide containing aptamer for PDGF-BB and a primer of RCA. This method provides a simple way for signal generation in RCA assay with multiple thrombin labels through thrombin-aptamer binding. This strategy is not limited in sandwich assays using aptamer as affinity ligands, other affinity ligands (e.g., antibody) can be used when RCA primer can be labeled on the affinity ligands to generate aptamer sequences for thrombin. It shows promise in a variety of analytical applications.
References
Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346(6287):818–22.
Tuerk C, Gold L. Systemic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249(4968):505–10.
Hermann T, Patel DJ. Adaptive recognition by nucleic acid aptamers. Science. 2000;287(5454):820–5.
Li F, Zhang H, Wang Z, Newbigging AM, Reid MS, Li XF, et al. Aptamers facilitating amplified detection of biomolecules. Anal Chem. 2015;87(1):274–92.
Liu J, Cao Z, Lu Y. Functional nucleic acids sensors. Chem Rev. 2009;109(5):1948–98.
Juskowiak B. Nucleic acid-based fluorescent probes and their analytical potential. Anal Bioanal Chem. 2011;399(9):3157–76.
Cho EJ, Lee JW, Ellington AD. Applications of aptamers as sensors. Annu Rev Anal Chem. 2009;2:241–64.
Zhou J, Battig MR, Wang Y. Aptamer-based molecular recognition for biosensor development. Anal Bioanal Chem. 2010;398(6):2471–80.
Deng N, Liang Z, Liang Y, Sui Z, Zhang L, Wu Q, et al. Aptamer modified organic–inorganic hybrid silica monolithic capillary columns for highly selective recognition of thrombin. Anal Chem. 2012;84(23):10186–90.
Zhang H, Li F, Dever B, Li XF, Le XC. DNA-mediated homogeneous binding assays for nucleic acids and proteins. Chem Rev. 2013;113(4):2812–41.
Zhao Y, Chen F, Li Q, Wang L, Fan C. Isothermal amplification of nucleic acids. Chem Rev. 2015;115(22):12491–545.
Ali MM, Li F, Zhang ZQ, Zhang KX, Kang DK, Ankrum JA, et al. Rolling circle amplification: a versatile tool for chemical biology, materials science and medicine. Chem Soc Rev. 2014;43(10):3324–41.
Schweitzer B, Wiltshire S, Lambert J, O’Malley S, Kukanskis K, Zhu Z, et al. Immunoassays with rolling circle DNA amplification: a versatile platform for ultrasensitive antigen detection. Proc Natl Acad Sci U S A. 2000;97(18):10113–9.
Zhou L, Ou LJ, Chu X, Shen GL, Yu RQ. Aptamer-based rolling circle amplification: a platform for electrochemical detection of protein. Anal Chem. 2007;79(19):7942–500.
Bi S, Li L, Zhang SS. Triggered polycatenated DNA scaffolds for DNA sensors and aptasensors by a combination of rolling circle amplification and DNAzyme amplification. Anal Chem. 2010;82(22):9447–54.
Tang LH, Liu Y, Ali MM, Kang D, Zhao WA, Li JH. Colorimetric and ultrasensitive bioassay based on a dual-amplification system using aptamer and DNAzyme. Anal Chem. 2012;84(11):4711–7.
Lee J, Icoz K, Roberts A, Ellington AD, Savran CA. Diffractometric detection of proteins using microbead-based rolling circle amplification. Anal Chem. 2010;82(1):197–202.
Yang LT, Fung CW, Cho EJ, Ellington AD. Real-time rolling circle amplification for protein detection. Anal Chem. 2007;79(9):3320–9.
Wang QP, Zheng HY, Gao XY, Lin ZY, Chen GN. A label-free ultrasensitive electrochemical aptameric recognition system for protein assay based on hyperbranched rolling circle amplification. Chem Commun. 2013;49(97):11418–20.
Cao ZJ, Peng QW, Qiu X, Liu CY, Lu JZ. Highly sensitive chemiluminescence technology for protein detection using aptamer-based rolling circle amplification platform. J Pharm Anal. 2011;1(3):159–65.
Wang L, Tram K, Ali MM, Salena BJ, Li J, Li Y. Arrest of rolling circle amplification by protein-binding DNA aptamers. Chem Eur J. 2014;20(9):2420–4.
Bock LC, Griffin LC, Latham JA, Vermaas EH, Toole JJ. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature. 1992;355(6360):564–6.
Tasset DM, Kubik MF, Steiner W. Oligonucleotide inhibitors of human thrombin that bind distinct epitopes. J Mol Biol. 1997;272(5):688–98.
Deng B, Lin Y, Wang C, Li F, Wang Z, Zhang H, et al. Aptamer binding assays for proteins: the thrombin example—a review. Anal Chim Acta. 2014;837:1–15.
Wu Q, Tsiang M, Sadler JE. Localization of the single-stranded DNA binding site in the thrombin anion-binding exosite. J Bio Chem. 1992;267(34):24408–12.
Zhou G, Huang X, Qu Y. The binding effect of aptamers on thrombin. Biochem Eng J. 2010;52(2–3):117–22.
Mir M, Vreeke M, Katakis I. Different strategies to develop an electrochemical thrombin aptasensor. Electrochem Commun. 2006;8(3):505–11.
Muller J, Becher T, Braunstein J, Berdel P, Gravius S, Rohrbach F, et al. Profiling of active thrombin in human blood by supramolecular complexes. Angew Chem Int Ed. 2011;50(27):6075–8.
Zhao Q, Li XF, Le XC. Aptamer capturing of enzymes on magnetic beads to enhance assay specificity and sensitivity. Anal Chem. 2011;83(24):9234–6.
Zhao Q, Wang XF. An aptamer-capture based chromogenic assay for thrombin. Biosens Bioelectron. 2012;34(1):232–7.
Westermark B, Heldin CH. Platelet-derived growth factor. Structure, function and implications in normal and malignant cell growth. Acta Oncol. 1993;32(2):101–5.
Heldin CH. Structural and functional studies on platelet-derived growth factor. The EMBO J. 1992;11(12):4251–9.
Green LS, Jellinek D, Jenison R, Ostman A, Heldin CH, Janjic N. Inhibitory DNA ligands to platelet-derived growth factor B-chain. Biochemistry. 1996;35(45):14413–24.
Jin X, Zhao JJ, Zhang LL, Huang Y, Zhao SL. An enhanced fluorescence polarization strategy based on multiple protein–DNA–protein structures for sensitive detection of PDGF-BB. RSC Adv. 2014;4(13):6850–3.
Li H, Zhu Y, Dong SY, Qiang WB, Sun L, Xu DK. Fast functionalization of silver decahedral nanoparticles with aptamers for colorimetric detection of human platelet-derived growth factor-BB. Anal Chim Acta. 2014;829:48–53.
Wang P, Song YH, Zhao YJ, Fan AP. Hydroxylamine amplified gold nanoparticle-based aptameric system for highly selective and sensitive detection of platelet-derived growth factor. Talanta. 2013;103:392–7.
Chang CC, Wei SC, Wu TH, Lee CH, Lin CW. Apatamer-based colorimetric detection of platelet-derived growth factor using unmodified gold nanoparticles. Biosens Bioelectron. 2013;42:119–23.
Fang XH, Li JJ, Tan WH. Using molecular beacons to probe molecular interactions between lactate dehydrogenase and single-stranded DNA. Anal Chem. 2000;72(14):3280–5.
Zhang H, Li XF, Le XC. Differentiation and detection of PDGF isomers and their receptors by tunable aptamer capillary electrophoresis. Anal Chem. 2009;81(18):7795–800.
Acknowledgments
We thank the support from National Natural Science Foundation of China (Grant No. 21222503) and Outstanding Youth Talents Program of Shanxi Province.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 205 kb)
Rights and permissions
About this article

Cite this article
Guo, L., Hao, L. & Zhao, Q. An aptamer assay using rolling circle amplification coupled with thrombin catalysis for protein detection. Anal Bioanal Chem 408, 4715–4722 (2016). https://doi.org/10.1007/s00216-016-9558-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-016-9558-0