
Abstract
Nanoparticles (NPs) entering water systems are an emerging concern as NPs are more frequently manufactured and used. Single particle inductively coupled plasma-mass spectrometry (SP-ICP-MS) methods were validated to detect Zn- and Ce-containing NPs in surface and drinking water using a short dwell time of 0.1 ms or lower, ensuring precision in single particle detection while eliminating the need for sample preparation. Using this technique, information regarding NP size, size distribution, particle concentration, and dissolved ion concentrations was obtained simultaneously. The fates of Zn- and Ce-NPs, including those found in river water and added engineered NPs, were evaluated by simulating a typical drinking water treatment process. Lime softening, alum coagulation, powdered activated carbon sorption, and disinfection by free chlorine were simulated sequentially using river water. Lime softening removed 38–53 % of Zn-containing and ZnO NPs and >99 % of Ce-containing and CeO2 NPs. Zn-containing and ZnO NP removal increased to 61–74 % and 77–79 % after alum coagulation and disinfection, respectively. Source and drinking water samples were collected from three large drinking water treatment facilities and analyzed for Zn- and Ce-containing NPs. Each facility had these types of NPs present. In all cases, particle concentrations were reduced by a minimum of 60 % and most were reduced by >95 % from source water to finished drinking water. This study concludes that uncoated ZnO and CeO2 NPs may be effectively removed by conventional drinking water treatments including lime softening and alum coagulation.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
As nanoparticles (NPs) are increasingly incorporated into commercial products, the risk of environmental exposure increases [1]. Zinc oxide (ZnO) and cerium dioxide (CeO2) are among the most commonly used NPs with applications in personal care products, paints, and catalysts that will lead to their release via wastewater or runoff into natural water bodies. Studies have suggested that ZnO NPs have a relatively high acute toxicity and result in oxidative stress and oxidative damage [2, 3]. Uptake of CeO2 NPs has been demonstrated by human intestinal epithelial cells [4] by diverse biological systems, and to be toxic to these different systems [5], including human peripheral blood monocytes and human lung cancer cells [4–6]. CeO2 NPs have been shown in an in vivo study to induce oxidative stress in Caenorhabditis elegans at environmentally relevant concentrations [7]. Another study, however, suggested that ZnO induced toxicity in mammalian cells while CeO2 suppressed reactive oxygen species (ROS) production and even protected the cell [8]. Given the uncertainty of human and environmental toxicity of ZnO and CeO2 NPs, it is imperative to analyze their fate and transport through water treatment processes. Work has been done to evaluate the fate of ZnO and CeO2 NPs during wastewater treatment, reporting up to 8 % of Zn in influent wastewater left in the effluent [9, 10] and 6 % of the initial CeO2 NPs left in the effluent [11, 12]. There are many interactions that dictate the behavior of NPs during wastewater treatment, including sorption onto debris and particles, dissolution, settling, and interactions with microorganisms [13]. Among the most important considerations for NP stability in aqueous media are aggregation and dissolution.
Major factors affecting NP aggregation and dissolution in aqueous solution include ionic strength, pH, and organic matter in the system. Low ionic strength and high organic matter content are associated with increased NP stability in water [14, 15]. Natural organic matter (NOM) plays a major role in NP stability. It has been shown that NOM can prevent the dissolution of ZnO NPs in aquatic matrices as well as prevent aggregation [16] and has been demonstrated to keep up to 88 % initially added CeO2 NPs in suspension after 12 d of settling [17]. With typical fresh water NOM concentrations between 0.1 and 20 mg/L, it is likely that NPs released into water systems will not aggregate or dissolve significantly, indicating that humans, mammals, and aquatic life may be exposed to these NPs [18]. Furthermore, if the exposed water system is used as source water for drinking water treatment it will be a direct route for human consumption of NPs if they are not removed during drinking water treatment processes.
While several studies have been conducted evaluating the fate of NPs during drinking water treatment, there exists a significant research gap regarding specific processes and NP types. CeO2 and ZnO NP removal by filtration processes alone (sand filtration, microfiltration, and/or ultrafiltration) have been studied by several research groups [19–22]. The results of these studies indicated that typical filtration processes efficiently remove uncoated (non-surface functionalized) NPs. Alum coagulation has been shown to result in between 40 and 99 % removal of ZnO under typical conditions, with the highest removal observed using sweep floc dosages in buffered nanopure water [22, 23]. No published papers have been found on CeO2 NPs removal during coagulative drinking water treatments, whereas ZnO NP removal has been evaluated in two studies. In both of these studies, the NP detection methods have only been indirect. Specifically, NP content in effluent water was detected by acid digestion followed by inductively coupled plasma-mass spectrometry (ICP-MS) or graphite furnace atomic absorption (GFAA) resulting in loss of information regarding NP size and aggregation state after treatment. On the other hand, single particle ICP-MS (SP-ICP-MS) has more recently proven to be an emerging and reliable technique for monitoring NPs in aquatic matrices [24–26], including applications to monitor removal of Ag, Au, and TiO2 NPs during typical drinking water treatments [27]. The key advantages of SP-ICP-MS include high sensitivity for environmentally relevant concentrations and simultaneous data acquisition regarding NP size, size distribution, and dissolved element concentration. SP-ICP-MS coupled with an ion exchange resin (IEC) has been used to monitor ZnO NPs in water using a dwell time of 0.5 ms [28]. Using the IEC improved NP signals by removing high background levels of Zn, which was demonstrated in previous work [29]. However, reducing the analysis dwell time has also been shown to improve the resolution between NP signals and dissolved background [30].
In this study, the fate of ZnO and CeO2 NPs during conventional drinking water treatments were evaluated. SP-ICP-MS methods were developed using short dwell times of 0.1 ms to monitor the NPs throughout drinking water treatment process to provide increased resolution between the dissolved background and NP signals without the use of ion exchange resins or columns for potentially high dissolved background element. NP removal was evaluated after lime softening, alum coagulation with simultaneous powdered activated carbon (PAC) sorption, and disinfection by free chlorine. Filtration was not simulated because it has been studied in detail and published for the selected NPs [19, 22]. Water samples were collected from three drinking water treatment facilities (DWTFs) and monitored for Zn- and Ce-containing NPs using the developed SP-ICP-MS method. The removal efficiencies were compared with the simulation results to evaluate the efficacy of the treatments on bench and full scales.
Materials and methods
Materials and instrumentation
CeO2 NPs (30–50 nm diameter, 40 wt% CeO2 dispersed in water, stock no. US7120) and ZnO NPs (80–200 nm diameter, nanopowder, stock no. US3555) were purchased from US Research Nanomaterials, Inc. (Houston, TX, USA). These nanoparticles were selected for this study because the size distributions were all or partially in the nanoscale range (≤100 nm) and they were larger than the particle size detection limits of the SP-ICP-MS. A dilute CeO2 solution was prepared by diluting the stock solution 1000 times into ultra-pure water. A dilute stock solution of ZnO NPs was prepared by dispersing a known amount of nanopowder in ultra-pure water. Particle concentrations were calculated using the density, mass, and particle size range of NPs suspended and the volume of ultra-pure water used. The working particle concentrations were determined by averaging calculated particle concentrations for several sizes over the manufacturer size range. Size calibration standard Au NPs (citrate-capped, 50, 80, and 100 nm diameter) suspended in 2 mM sodium citrate were obtained from nanoComposix, Inc. (San Diego, CA, USA). All purchased NPs were characterized by both scanning electron microscopy (SEM) and SP-ICP-MS. An S-4700 model field emission scanning electron microscope (FESEM) with energy-dispersive X-ray spectroscopy (EDS) capability (Hitachi, Tokyo, Japan) was used to image and confirm elemental composition of NPs. The NPs purchased were original materials without surface modification.
Dissolved Ce and Zn standards (PerkinElmer, Waltham, MA, USA) were used for dissolved element calibrations. Sodium hydroxide (caustic), aluminum sulfate (Al2(SO4)3·14.3H2O), and trace metal grade nitric acid, were purchased from Thermo Fisher Scientific Inc. (Pittsburgh, PA, USA). Hydrodarco B (HDB) powdered activated carbon (PAC) was product of Cabot (Marshall, TX, USA). Sodium hypochlorite solutions and trace metal grade sulfuric acid were purchased from Sigma-Aldrich (St. Louis, MO, USA). A Simplicity185 water purification system from Millipore was used to generate ultra-pure water. Dissolved organic carbon (DOC) was monitored with a TOC-L analyzer with ASI-L liquid autosampler from Shimadzu Scientific Instruments (Columbia, MD, USA) and turbidity was tested using a TB200 Portable Turbidimeter (Orbeco-Hellige, Sarasota, FL, USA). The pH of the water samples was monitored at each step of the simulation treatment.
Single particle ICP-MS methods
A PerkinElmer (Shelton, CT, USA) NexION 300D/350 ICP-MS equipped with Syngistix Nano Application module operating in single particle mode was used. The instrument parameters were optimized for detection of ZnO and CeO2 NPs as well as their corresponding ions. The system was equipped with a concentric nebulizer, cyclonic spray chamber, and platinum sampler and skimmer cones. The rf power was set to 1600 W to ensure complete atomization of NPs. Parameters optimized daily included sample flow rate (between 0.26 and 0.29 mL/min) and the transport efficiency (between 7.5 and 8.5 %). Masses of 140Ce and 67Zn were monitored with a dwell time of 100 μs over a 100 s sampling time. 67Zn was monitored to avoid 50Ti16O interferences that occur with the higher abundance 66Zn isotope. Water collected from the Missouri River used in this and our previous study have had high Ti concentrations, rendering the isotope selection necessary [27]. The instrument was calibrated with a filtered matrix blank and three dissolved Ce and Zn matrix spikes (0–20 μg/L spikes). In most cases Ce and Zn were present in the matrix; therefore, a blank subtraction was used. This calibration was used for both dissolved ions and NP sizing.
Water sample collection and water quality parameter measurement
Three DWTFs (referred to as Facilities 1, 2, and 3) were selected for this study. Facility 1 uses a blend of ground water and Missouri River water as source water and the other two DWTFs use Missouri River water as source water. The water samples were collected in duplicate from each facility. Each DWTF employed lime softening, ferric coagulation, PAC sorption, and chlorine/chloramine disinfection treatments. Detailed treatment information can be found in the Supplementary Material of our recent publication [27]. Samples were collected in pre-cleaned 125 mL polypropylene bottles without additives or filtration, and immediately brought back to our laboratory for SP-ICP-MS analysis within 8 h to minimize NP transformations after collection. The other key water quality parameters were also measured for the water samples by using the same methods as those in our recent publication [27]. Missouri River water was collected during the cold season for simulated treatments. The collected water was allowed to settle for at least 24 h before use to remove any noncolloidal materials.
Drinking water treatment simulations
A six-gang stirrer system (Phipps and Bird, Richmond, VA, USA) was used to simulate several major drinking water treatment steps in sequence. Lime softening, alum coagulation with PAC sorption, and disinfection were simulated with and without added CeO2 and ZnO in Missouri River water using six-gang stirrers. The river water was analyzed for Ce- and Zn-containing NPs, dissolved ions, DOC, turbidity, and pH before use in jar tests and after each treatment step during the simulation. NPs (30–50 nm CeO2 and 80–200 nm ZnO) were added to 2-L of Missouri River water at 1 × 106 particles/mL (mass concentrations of ~7 μg/L ZnO and ~4 μg/L CeO2) and dispersed by stirring at 100 rpm for 1 min. Lime was dosed with 260 mg/L as Ca(OH)2 to reach pH 11 before rapid mixing (300 rpm for 30 s), flocculation (10 min each at 58, 42, and 28 rpm), and sedimentation (0 rpm for 180 min). The clear supernatant was decanted into a clean 2-L square beaker, leaving only solids behind. Alum coagulation (60 mg/L as Al2(SO4)3·14.3H2O, Zone 4 coagulation at pH 7–8) with simultaneous PAC sorption by HDB (20 mg/L) was simulated on the softened water using the same stirring parameters as softening. After alum addition, the pH was quickly adjusted to pH 8 using trace metal grade sulfuric acid during flocculation. After settling, 1-L of the clear supernatant was transferred to a clean 2-L square beaker for disinfection. Sodium hypochlorite solution was added to obtain a residual concentration of 2 to 4 mg/L as Cl2 (stirred at 28 rpm for 60 min). The water samples were taken before and after each treatment step, and were analyzed by SP-ICP-MS immediately after the simulation treatments were completed.
Results and discussion
Nanoparticle characterization
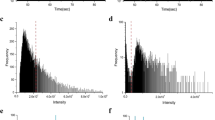
ZnO and CeO2 NPs used for the drinking water treatment simulations were characterized by SP-ICP-MS and SEM imaging before use. Representative SP-ICP-MS size distribution histograms and SEM images can be seen in Fig. 1. In each case, the NPs were suspended in ultra-pure water and sonicated to reduce aggregation before analysis for SP-ICP-MS characterization. ZnO NPs were diluted to 70 μg/L total mass concentration and the CeO2 NPs were diluted to 40 μg/L total mass concentration. The ZnO NPs used (80–200 nm diameter specification by the manufacturer) had irregular shapes and sizes (Fig. 1b), contributing to the wide size distribution histogram obtained by SP-ICP-MS (Fig. 1a). The detected particle concentration for the ZnO NPs was 2.16 × 104 particles/mL and a detected dissolved concentration of 16.4 μg/L. The CeO2 NPs used (30–50 nm diameter specification by the manufacturer) are more regularly shaped and relatively spherical (Fig. 1d), which is also reflected in the SP-ICP-MS size distribution histogram (Fig. 1c), though more large sizes were observed due to aggregation. The detected particle concentration for the CeO2 NPs was 4.83 × 104 particles/mL and the detected dissolved concentration was below the method detection limit.

SP-ICP-MS and SEM characterizations of NPs. (a) SP-ICP-MS size distribution histogram of ZnO with 70 μg/L total mass concentration (56 μg/L Zn), (b) SEM image of ZnO NP, (c) SP-ICP-MS size distribution histogram of CeO2 with 40 μg/L total mass concentration (33 μg/L Ce), (d) SEM image of CeO2 NP
SP-ICP-MS method performance
Methods for ZnO and CeO2 were developed and validated for simultaneous analysis of dissolved ion and NPs by SP-ICP-MS. The instrument was calibrated using 0.45 μm nylon-membrane-filtered matrix water over 0 to 20 μg/L dissolved Zn and Ce using a blank subtraction when the ions were present in the matrix water. Transport efficiency, density, mass fraction, ionization efficiency, and other parameters can be seen in Table 1. The dissolved calibration curve was used to convert NP signals to diameter based on the mass fraction and density of the material using calculations described elsewhere [24, 31]. The NP detection limits (DLs) were determined to be five times the standard deviation above the background intensity in ultrapure water when analyzed against the dissolved calibration curve in the matrix [32]. This was calculated to be 35 to 40 nm for ZnO and 18 to 20 nm for CeO2, depending on daily instrument optimization. The ZnO DL is similar to previously reported values, but the CeO2 DL is ~10 nm higher than previously reported values, most likely due to differences in instrument optimization and matrix effects when calibrating in the sample matrix [32]. DLs of dissolved ion were determined to be 0.20 and 0.10 μg/L for Zn and Ce, respectively. These detection limits were determined in ultra-pure water and validated in calibrations in the sample matrix. Dissolved DLs were higher than traditional ICP-MS because of the short dwell times used.
Fate of ZnO and CeO2 NPs during simulated drinking water treatments
Three typical drinking water treatment steps (lime softening, alum coagulation with PAC sorption, and disinfection by free chlorine) were simulated sequentially to determine the fate of ZnO and CeO2 NPs. DOC, pH, turbidity, NP concentration, and size distribution were characterized at each treatment step to ensure the treatments were effective and to monitor NP status. The changes in pH, DOC, and turbidity after each treatment step are summarized in Table 2. The river water used had initial DOC, pH, and turbidity of 2.38 mg/L ± 0.25, 8.31 ± 0.02, and 4.04 NTU ± 0.29, respectively, after aliquoted into six samples at room temperature [n = 6, relative standard deviation (RSD)]. DOC and turbidity were on the low range for river water because they were collected during the cold season when turbidity and organic content are low. After lime softening, the pH increased to 11.35 ± 0.04 (RSD) and DOC and turbidity decreased to 16 % and 61 % of the original value, respectively. After alum and PAC treatment, the pH decreased to 8.23 ± 0.08 (RSD), and DOC and turbidity decreased to 51 % and 93 % of the original value, respectively. The pH was 8.21 ± 0.09 (RSD) after sodium hypochlorite disinfection, with no significant change from the pH before disinfection [8.23 ± 0.08 (RSD)]. DOC and turbidity were unchanged after disinfection. At the pH of each step (pH between 8 and 12), it has been reported that dissolved Zn2+ and Ce3+ exist as ionic oxides [33], indicating that dissolved constituents would not result in NP signals during analysis as the pH increased. If, however, these dissolved constituents would form colloidal hydroxides at high pH, the resulting colloidal materials would be formed during drinking water treatment that would be effected by the treatment process.
Fate of Zn-containing NP without ZnO NP addition
Zn-containing NPs were present in the river water initially. The real form of the Zn-containing NPs could not be determined by SP-ICP-MS. It was assumed that the NPs were present as ZnO NPs, and measured size was calculated by ZnO NPs. The most frequent ZnO NP sizes were centered on 75 nm with a wide particle size distribution arranged from DL to several hundred nanometers, as shown in Fig. 2a. Dissolved Zn (or ZnO NP at size smaller than the particle sized detection limit) was also found in the river water at a concentration of 1.11 μg/L. It has been demonstrated that ZnO NPs are toxic [2, 3]. If the real forms of the Zn-containing NPs are present as ZnO NPs, the health risk deserved to be evaluated if it not removed during water treatment process Therefore, their fate and removal were evaluated by simulated drinking water treatment first without the addition of the ZnO NPs. Control tests were performed in parallel with the treatment tests by adding NPs but not adding treatment materials. The size distribution and concentration of the spiked ZnO NPs in the control samples did not change in the time period of the simulated treatment and until analysis. This indicated that the ZnO NPs in the selected surface water were stable during 12 h for experiment, presumably stabilized by the water matrix. When the river water was treated by excess lime softening to pH 11, the Zn-containing NPs exhibited a shift in size distribution to the most frequent size of 35 nm as ZnO and the particle concentration (particles/mL) was reduced by 38 % (Fig. 2a, “After lime softening”). This indicated that the ZnO NPs may have been partially dissolved during the treatment, that the larger NPs were removed but smaller ZnO NPs were remained in suspension after softening, or that the formerly soluble zinc in solution transformed into insoluble colloids as the pH increased. After lime softening, the dissolved Zn concentration was reduced from 1.11 μg/L ± 0.08 (n = 6, RSD) to 0.43 μg/L ± 0.08 (n = 3, RSD), so dissolution or aggregation could not be confirmed by dissolved concentration and NP results. Alum coagulation with PAC sorption was performed on the softened water and resulted in a particle concentration decrease of 74 % relative to the original concentration while maintaining a similar size distribution, indicating that the NPs were not physically changed during this step of treatment but were removed during this step of treatment. The dissolved Zn content was also not significantly changed during this treatment step (0.48 μg/L ± 0.02, n = 3, RSD). Minimal dissolution of ZnO was observed after disinfection by free chlorine as evidenced by further reduction in frequency in the size distribution histogram and particle concentration reduction to 77 % of the original concentration in the water (Fig. 2a “After disinfection”) and the dissolved Zn concentration (0.52 μg/L ± 0.08, n = 3, RSD). Overall, disinfection had little effect on the Zn-containing NP distribution and dissolved Zn content.

Change in size distribution histograms after sequential simulated drinking water treatments in Missouri River water for (a) ZnO without NP addition (n = 2), (b) ZnO with 80–200 nm ZnO addition (7 μg/L mass concentration) (n = 2), (c) CeO2 without NP addition (n = 2), and (d) CeO2 with 30–50 nm CeO2 addition (4 μg/L mass concentration) (n = 2)
Fate of ZnO NPs with ZnO NP addition
To evaluate the removal of engineered ZnO NPs during drinking water treatments, 80 to 200 nm diameter ZnO NPs were added to the river water and subjected to the same treatments previously mentioned. The change in size distribution histograms for the ZnO NPs can be seen in Fig. 2b. After the NPs were added, the size distribution shifted to the most frequent size of 120 nm and the dissolved Zn concentration increased from 1.11 μg/L ± 0.08 (n = 6, RSD) to 16.45 μg/L ± 0.83 [n = 2, relative percent difference (RPD)], indicating either the smaller (smaller than the size detection limit) ZnO NPs were detected as “dissolved” Zn, the NPs partially dissolved after addition to the water, or insoluble colloids formed from the dissolved zinc ions in solution. This result is similar to dissolved concentration when added to ultra-pure water (16.38 μg/L). The water was then treated by lime softening to pH 11, which resulted in a reduction in size distribution to around 50 nm, total particle concentration reduction by 53 % and dissolved Zn to 0.36 μg/L ± 0.95 (n = 2, RPD). Again, the NPs may have dissolved during lime softening, but was not confirmed due to the reduction in dissolved Zn. The softened water was then treated with alum and PAC, and resulted in a further reduction in NP concentration by 79 % of the original value (Fig. 2b “After alum+PAC”) and in dissolved Zn concentration of 0.46 μg/L ± 6.38 (n = 2, RPD). No further changes were observed after disinfection. Overall, the behavior of the added engineered ZnO NPs was similar to the Zn-containing NPs in the river water.
Fate of Ce-containing NP without NP addition
Ce-containing NPs were also present in the river water collected for the removal experiments and were studied without the addition of engineered CeO2 NPs. The actual form of the CeO2-containing NPs could also not be determined by SP-ICP-MS. It was assumed that the NPs were present as CeO2 NPs, and measured size was calculated as CeO2 NPs. The water contained a size distribution histogram with a maximum at 24 nm assumed as CeO2 with a particle size distribution arranged from DL to ~60 nm, as shown in Fig. 2c. The dissolved concentration of Ce was close or bellow detection limit (0.10 μg/L). The fate and removal of CeO2 NPs during the water treatment are also important because of the toxicity of this type of NPs. For the duration of the simulation treatment experiments, the dissolved Ce levels were below the DL. After lime softening to pH 11, over 99 % of the particles were removed from the water. Because the Ce-containing particle content was below the DL after lime softening, no changes were observed after alum coagulation and PAC sorption.
Fate of CeO2 NP with CeO2 NP addition
To determine the behavior of engineered CeO2 NPs under the same treatment conditions, 30 to 50-nm diameter CeO2 NPs were added to the river water and subjected to the same treatments. After NP addition, the size distribution histogram showed most frequent size at 38 nm and size distribution from detection limit to about 60 nm (Fig. 2d). The dissolved Ce content was still below the DL after the NPs were added, which indicated that the NPs were not dissolving. Like the Ce-containing NPs, the CeO2 NPs were completely (>99 %) removed after lime softening. No changes were observed in subsequent treatments. The CeO2 results were similar to those from our previous study of TiO2, Ag, and Au NPs removal during the simulated drinking water treatment process [27]. CeO2, Ag, Au, and TiO2 NPs had removal after lime softening higher than 90 % without showing significant dissolution. ZnO NPs were significantly reduced after lime softening treatment (38–53 %) but not completely removed. After alum coagulation and PAC sorption, ZnO removal increased to 61–79 %. Zeta potentials for similar uncoated NPs were negative in previous reports [34, 35], indicating stabilization due to a negative surface charge at high pH for both NPs. Under these conditions, the cations added during lime softening may neutralize the surface charge on the NPs, causing them to settle during sedimentation. These two studies suggest that lime softening is effective in removing the selected NPs from surface water. Other studies have shown that alum coagulation can remove up to 99 % of ZnO NPs [22] and up to 70 % of TiO2 and/or Ag NPs depending on treatment conditions without preceding lime softening treatment [22, 23, 36]. Thus, with a combination of lime softening and coagulation, it is likely that most uncoated CeO2 NPs at low concentrations in source water of drinking water would be removed based on the results of this work.
ZnO and CeO2 NP occurrence in water from three drinking water treatment facilities
Samples were taken from three large DWTFs from source water and drinking water to evaluate the occurrence of Zn- and Ce-containing NPs and their fate after full-scale drinking water treatments. DOC, pH, and turbidity measured for each water sample are presented in Table 3. The NP and dissolved ion detection results are presented in Table 4. All field blanks were below the DLs for each analyte and all dissolved element spike recoveries were between 76 and 123 % and the reproducibility of duplicated samples was good, indicating good method performance in the collected samples. Zn-containing NPs and dissolved Zn ions were present in the source water from each facility with concentrations at around 1.5 × 105 particles/mL for NPs and 5.49, 5.36, and 32.2 μg/L Zn ions at Facilities 1, 2, and 3, respectively. Ce-containing NPs were present in source water from each facility at around 5.5 × 105 particles/mL and Ce ions were present between 0.16 and 0.75 μg/L at the three facilities. Facility 1 source water had 87 nm Zn-containing NPs (as ZnO) at ~105 particles/mL. After treatment, the size increased to 114 nm as ZnO, but the particle concentration decreased to ~104 particles/mL. It is likely that some of the particles were removed and that the remaining particles aggregated resulting in the size increase. Ce-containing NPs were in Facility 1 source water at ~5 × 105 particles/mL and 30 nm as CeO2. After treatment, these particles were removed to below the DL. Facility 2 had Zn- and Ce-containing NPs in source water. Zn-containing NPs were reduced from 81 nm at ~105 particles/mL to 60 nm at ~5 × 105 particles/mL during treatment. Ce-containing NPs were reduced from 44 nm at ~5 × 105 particles/mL to 17 nm at ~5 × 104 particles/mL. For this facility, it is likely that the NPs were partially dissolved during treatment. Facility 3 had Zn-containing NPs at 114 nm and ~105 particles/mL as ZnO and Ce-containing NPs at 41 nm and ~5 × 105 particles/mL as CeO2. After treatment, Zn-containing NPs were reduced to 54 nm and ~104 particles/mL and Ce-containing NPs were reduced to below the DL. The results showed that the treatment combination in each facility was effective in removing Zn- and Ce-containing NPs from surface water using a combination of softening and ferric coagulation. Although further study is needed under other conditions, these results suggest the protective nature of combined softening and ferric coagulation for general control of uncoated CeO2 and ZnO NPs in drinking water treatment systems.
References
Boxall A, Chaudhry Q, Sinclair C, Jones A, Aitken R, Jefferson B, et al. Current and future predicted environmental exposure to ENPs. Central Science Laboratory, Department of the Environement and Rural Affairs, London, UK 89; 2007.
Lin W, Xu Y, Huang C-C, Ma Y, Shannon KB, Chen D-R, et al. Toxicity of nano- and micro-sized ZnO particles in human lung epithelial cells. J Nanoparticle Res. 2008;11(1):25–39.
Ma H, Williams PL, Diamond SA. Ecotoxicity of manufactured ZnO nanoparticles—a review. Environ Pollut. 2013;172:76–85.
Hussain S, Al-Nsour F, Rice AB, Marshburn J, Yingling B, Ji Z, et al. Cerium dioxide nanoparticles induce apoptosis and autophagy in human peripheral blood monocytes. ACS Nano. 2012;6(7):5820–9.
Gaiser BK, Fernandes TF, Jepson MA, Lead JR, Tyler CR, Baalousha M, et al. Interspecies comparisons on the uptake and toxicity of silver and cerium dioxide nanoparticles. Environ Toxicol Chem. 2012;31(1):144–54.
Lin W, Huang YW, Zhou XD, Ma Y. Toxicity of cerium oxide nanoparticles in human lung cancer cells. Int J Toxicol. 2006;25(6):451–7.
Zhang H, He X, Zhang Z, Zhang P, Li Y, Ma Y, et al. Nano-CeO2 exhibits adverse effects at environmental relevant concentrations. Environ Sci Technol. 2011;45(8):3725–30.
Xia T, Kovochich M, Long M, Mädler L, Gilbert B, Shi H, et al. Comparison of the mechanism of toxicity of zinc oxide and cerium oxide nanoparticles based on dissolution and oxidative stress properties. ACS Nano. 2008;2(10):2121–34.
Lombi E, Donner E, Tavakkoli E, Turney TW, Naidu R, Miller BW, et al. Fate of zinc oxide nanoparticles during anaerobic digestion of wastewater and post-treatment processing of sewage sludge. Environ Sci Technol. 2012;46(16):9089–96.
Ma R, Levard C, Judy JD, Unrine JM, Durenkamp M, Martin B, et al. Fate of zinc oxide and silver nanoparticles in a pilot wastewater treatment plant and in processed biosolids. Environ Sci Technol. 2014;48(1):104–12.
Gomez-Rivera F, Field JA, Brown D, Sierra-Alvarez R. Fate of cerium dioxide (CeO2) nanoparticles in municipal wastewater during activated sludge treatment. Bioresource Technol. 2012;108:300–4.
Limbach LK, Bereiter R, Müller E, Krebs R, Gälli R, Stark WJ. Removal of oxide nanoparticles in a model wastewater treatment plant—influence of agglomeration and surfactants on clearing efficiency. Environ Sci Technol. 2008;42(15):5828–33.
Brar SK, Verma M, Tyagi RD, Surampalli RY. Engineered nanoparticles in wastewater and wastewater sludge—evidence and impacts. Waste Manag. 2010;30(3):504–20.
Keller AA, Wang H, Zhou D, Lenihan HS, Cherr G, Cardinale BJ, et al. Stability and aggregation of metal oxide nanoparticles in natural aqueous matrices. Environ Sci Technol. 2010;44(6):1962–7.
Van Hoecke K, De Schamphelaere KA, Van der Meeren P, Smagghe G, Janssen CR. Aggregation and ecotoxicity of CeO2 nanoparticles in synthetic and natural waters with variable pH, organic matter concentration and ionic strength. Environ Pollut. 2011;159(4):970–6.
Li M, Lin D, Zhu L. Effects of water chemistry on the dissolution of ZnO nanoparticles and their toxicity to Escherichia coli. Environ Pollut. 2013;173:97–102.
Quik JT, Lynch I, Van Hoecke K, Miermans CJ, De Schamphelaere KA, Janssen CR, et al. Effect of natural organic matter on cerium dioxide nanoparticles settling in model fresh water. Chemosphere. 2010;81(6):711–5.
Volk C, Wood L, Johnson B, Robinson J, Zhu HW, Kaplan L. Monitoring dissolved organic carbon in surface and drinking waters. J Environ Monit. 2002;4(1):43–7.
Hassan AA, Li Z, Sahle-Demessie E, Sorial GA. Computational fluid dynamics simulation of transport and retention of nanoparticle in saturated sand filters. J Hazardous Mater. 2013;244(245):251–8.
Li Z, Aly Hassan A, Sahle-Demessie E, Sorial GA. Transport of nanoparticles with dispersant through biofilm coated drinking water sand filters. Water Res. 2013;47(17):6457–66.
Li Z, Sahle-Demessie E, Hassan AA, Sorial GA. Transport and deposition of CeO2 nanoparticles in water-saturated porous media. Water Res. 2011;45(15):4409–18.
Chalew TEA, Ajmani GS, Huang H, Schwab KJ. Evaluating nanoparticle breakthrough during drinking water treatment. Environ Health Perspect. 2013;121(10):1161–6.
Zhang Y, Chen Y, Westerhoff P, Hristovski K, Crittenden JC. Stability of commercial metal oxide nanoparticles in water. Water Res. 2008;42(8):2204–12.
Dan Y, Shi H, Stephan C, Liang X. Rapid analysis of titanium dioxide nanoparticles in sunscreens using single particle inductively coupled plasma-mass spectrometry. Microchem J. 2015;122:119–26.
Dan Y, Zhang W, Xue R, Ma X, Stephan C, Shi H. Characterization of gold nanoparticles uptake by tomato plants using enzymatic extraction followed by single particle inductively coupled plasma-mass spectrometry. Environ Sci Technol. 2015. doi:10.1021/es506179e.
Mitrano DM, Ranville JF, Bednar A, Kazor K, Hering AS, Higgins CP. Tracking dissolution of silver nanoparticles at environmentally relevant concentrations in laboratory, natural, and processed waters using single particle ICP-MS (spICP-MS). Environ Sci Nano. 2014;1(3):248–59.
Donovan AR, Adams CD, Ma Y, Stephan C, Eichholz T, Shi H. Single particle ICP-MS characterization of titanium dioxide, silver, and gold nanoparticles during drinking water treatment. Chemosphere. 2015;144:148–53.
Hadioui M, Merdzan V, Wilkinson KJ. Detection and characterization of ZnO nanoparticles in surface and waste waters using single particle ICPMS. Environ Sci Technol. 2015;49(10):6141–8.
Hadioui M, Peyrot C, Wilkinson KJ. Improvements to single particle ICPMS by the online coupling of ion exchange resins. Anal Chem. 2014;86(10):4668–74.
Montaño MD, Badiei HR, Bazargan S, Ranville JF. Improvements in the detection and characterization of engineered nanoparticles using spICP-MS with microsecond dwell times. Environ Sci Nano. 2014;1(4):338.
Degueldre C, Favarger PY. Colloid analysis by single particle inductively coupled plasma-mass spectroscopy: a feasibility study. Colloid Surf A Physicochem Eng Aspect. 2003;217(1/3):137–42.
Lee S, Bi X, Reed RB, Ranville JF, Herckes P, Westerhoff P. Nanoparticle size detection limits by single particle ICP-MS for 40 elements. Environ Sci Technol. 2014;48(17):10291–300.
Takeno N. Atlas of Eh-pH diagrams. Geological survey of Japan open file report. 2005;419:102.
Degen A, Kosec M. Effect of pH and impurities on the surface charge of zinc oxide in aqueous solution. J Eur Ceramic Soc. 2000;20:667–73.
Berg JM, Romoser A, Banerjee N, Zebda R, Sayes CM. The relationship between pH and zeta potential of ~30 nm metal oxide nanoparticle suspensions relevant to in vitro toxicological evaluations. Nanotoxicology. 2009;3(4):276–83.
Wang H, Qi J, Keller AA, Zhu M, Li F. Effects of pH, ionic strength, and humic acid on the removal of TiO2 nanoparticles from aqueous phase by coagulation. Colloid Surf A Physicochem Eng Aspect. 2014;450:161–5.
Acknowledgments
The authors acknowledge funding received from the Missouri Department of Natural Resources for this study. The NexION 300D/350D ICP-MS was provided by PerkinElmer, Inc. The authors appreciate the support from the Center for Single Nanoparticle, Single Cell, and Single Molecule Monitoring (CS3M) at Missouri University of Science and Technology. They also thank Qingbo Yang for assisting with SEM imaging.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Published in the topical collection Single-particle-ICP-MS Advances with guest editors Antonio R. Montoro Bustos and Michael R. Winchester.
Rights and permissions
About this article

Cite this article
Donovan, A.R., Adams, C.D., Ma, Y. et al. Detection of zinc oxide and cerium dioxide nanoparticles during drinking water treatment by rapid single particle ICP-MS methods. Anal Bioanal Chem 408, 5137–5145 (2016). https://doi.org/10.1007/s00216-016-9432-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-016-9432-0