
Abstract
N,N-Diallyltryptamine (DALT) and 5-methoxy-DALT (5-MeO-DALT) are synthetic tryptamine derivatives commonly referred to as so-called new psychoactive substances (NPS). They have psychoactive effects that may be similar to those of other tryptamine derivatives. The objectives of this work were to study the metabolic fate and detectability, in urine, of DALT and 5-MeO-DALT. For metabolism studies, rat urine obtained after high-dose administration was prepared by precipitation and analyzed by liquid chromatography–high-resolution mass spectrometry (LC–HR–MS–MS). On the basis of the metabolites identified, several aromatic and aliphatic hydroxylations, N-dealkylation, N-oxidation, and combinations thereof are proposed as the main metabolic pathways for both compounds. O-Demethylation of 5-MeO-DALT was also observed, in addition to extensive glucuronidation or sulfation of both compounds after phase I transformation. The cytochrome P450 (CYP) isoenzymes predominantly involved in DALT metabolism were CYP2C19, CYP2D6, and CYP3A4; those mainly involved in 5-MeO-DALT metabolism were CYP1A2, CYP2C19, CYP2D6, and CYP3A4. For detectability studies, rat urine was screened by GC–MS, LC–MSn, and LC–HR–MS–MS after administration of low doses. LC–MSn and LC–HR–MS–MS were deemed suitable for monitoring consumption of both compounds. The most abundant targets were a ring hydroxy metabolite of DALT, the N,O-bis-dealkyl metabolite of 5-MeO-DALT, and their glucuronides. GC–MS enabled screening of DALT by use of its main metabolites only.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
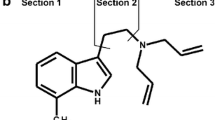
Synthetic tryptamines belong to new psychoactive substances (NPS), a class which also includes synthetic cannabinoids, phenethylamines, synthetic cathinones, and others [1, 2]. They interact with the serotonergic system, resulting in activation of 5-HT1A and 5-HT2A receptor subtypes [3] and increased serotonin release [4–8]. It has been suggested that active oral doses of N,N-diallyltryptamine (DALT) and 5-methoxy-DALT (5-MeO-DALT) range from >40 mg for DALT and from 12–20 mg for 5-MeO-DALT [9–11]. Their chemical structures are depicted in Fig. 1. The control status of both substances differs across the globe. Misuse of 5-MeO-DALT and its association with adverse effects have recently been reported [12, 13]; this serves as a reminder that the ability to perform suitable toxicological analysis is urgently needed. No data are available on misuse of DALT.

Structure of DALT (a) and 5-MeO-DALT (b) with corresponding division into sections to aid identification of the metabolites
Analytical methods used for characterization of N,N-dialkylated tryptamines [14] and naturally-occurring N,N-dimethylated tryptamines found in psychoactive beverages [15] have been reviewed but without reference to determination in biofluids. Information on metabolism in urine is important so that appropriate targets can be identified. Katagi et al. reviewed the general metabolic steps and toxicological analysis of several 5-methoxy-N,N-dialkyltryptamines [16]. Metabolism and detectability of DALT and 5-MeO-DALT in relevant matrices have not yet been reported. Therefore, the purpose of this work was to identify phase I and II metabolites of DALT and 5-MeO-DALT in rat urine and in pooled human liver microsomes (pHLM) by liquid chromatography (LC)–high resolution (HR)–MS–MS and to identify the human cytochrome-P450 (CYP) isoenzymes involved in the main metabolic steps. The detectability of DALT and 5-MeO-DALT in urine by use of the authors’ GC–MS, LC–MSn, and LC–HR–MS–MS standard urine-screening approaches (SUSA) was tested.
Experimental
Chemicals and reagents
DALT, DALT-d 4, and 5-MeO-DALT were synthesized by use of established methods [17]. Isocitrate and isocitrate dehydrogenase were from Sigma (Taufkirchen, Germany), NADP+ from Biomol (Hamburg, Germany), acetonitrile (LC–MS grade), ammonium formate (analytical grade), formic acid (LC–MS grade), methanol (LC–MS grade), and all other chemicals and biochemicals from VWR (Darmstadt, Germany). Baculovirus-infected insect cell microsomes (Supersomes) containing 1 nmol mL−1 human cDNA-expressing CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 (2 nmol mL−1), CYP3A4, and CYP3A5 (2 nmol mL−1), and pooled human liver microsomes (pHLM; 20 mg microsomal protein mL−1, 400 pmol total CYP mg−1 protein) were obtained from BD Biosciences (Heidelberg, Germany). After delivery, the microsomes were thawed at 37 °C, divided into aliquots, snap-frozen in liquid nitrogen, and stored at −80 °C until use.
Urine
Investigations were performed on rat urine from male Wistar rats (Charles River, Sulzfeld, Germany) for toxicological diagnostic reasons. Compounds were administered once in an aqueous suspension by gastric intubation in a single 20 mg kg−1 body mass (BM) dose for identification of the metabolites and once in a single 1 mg kg−1 BM dose for toxicological analysis, in accordance with the German animal-protection act.
The rats were housed in metabolism cages for 24 h, with water ad libitum. Urine was collected separately from feces over a 24-h period. Blank urine was collected before drug administration to confirm the absence of interfering compounds. The samples were directly analyzed and then stored at −20 °C.
Sample preparation for identification of phase I and II metabolites by LC–HR–MS–MS
As described elsewhere [18], 500 μL acetonitrile was added to 100 μL urine. The mixture was shaken on a rotary shaker for 2 min. After centrifugation for 2 min at 10,000×g, 500 μL was transferred to a glass vial and evaporated to dryness under a gentle stream of nitrogen at 70 °C. The residue was dissolved in 50 μL methanol then 10 μL was injected for LC–HR–MS–MS analysis.
Microsomal incubations for pHLM and initial CYP activity screening studies
The conditions used for microsomal incubation of each isomer have been published elsewhere [19]. Briefly, drugs (50 μmol L−1) were incubated with the CYP isoenzymes (50 pmol mL−1) CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4, or CYP3A5, or pHLM (1 mg protein mL−1) for 30 min. Reactions were initiated by addition of the substrate and stopped by addition of 50 μL ice-cold acetonitrile containing DALT-d4 (1 mg L−1) as internal standard. The solution was centrifuged for 2 min at 10,000×g, 50 μL of the supernatant phase was transferred to a glass vial, and 10 μL was injected for LC–HR–MS–MS analysis.
LC–HR–MS–MS apparatus for identification of phase I and II metabolites in urine and microsomal incubations
In accordance with published procedures [20], the extracts were analyzed by use of a Accela LC system consisting of an HTC PAL autosampler, a degasser, two 1250 quaternary pumps, an Aria Transcend TLX-I HTLC system, and a valve interface module with built-in switching valves, all controlled by Aria software version 1.6.3, coupled to a Q-Exactive system equipped with a heated electrospray ionization (HESI)-II source and Xcalibur 2.2 SP1.48 software (all from ThermoFisher, Dreieich, Germany). Mass calibration was conducted in accordance with the manufacturer’s recommendations every 72 h, by external mass calibration. In accordance with Helfer et al. (submitted for publication), the LC conditions were: ThermoFisher Accucore phenylhexyl column (100 mm × 2.1 mm I.D., 2.6 μm), guarded by a UHP filter cart (0.5 μm). Chromatography was performed at 35 °C maintained by an analytical column heater (HotDog 5090; Prolab, Reinach, Switzerland). The mobile phases were 2 mmol L−1 aqueous ammonium formate containing 0.1 % formic acid (pH 3, eluent A) and 2 mmol L−1 aqueous ammonium formate containing acetonitrile–methanol (50:50, v/v; 1 % water) plus 0.1 % formic acid (eluent B). The flow rate was 0.5 mL min−1 for 10 min and 0.8 mL min−1 from 10–13.5 min. The gradient program was: 0–1 min 99 % A, 1–10 min to 1 % A, 10–11.5 min hold at 1 % A, 11.5–13.5 min hold at 99 % A. The HESI-II source conditions were: heater temperature, 320 °C; sheath gas, 60 arbitrary units (AU); auxiliary gas, 10 AU; spray voltage, 3.00 kV; capillary temperature, 320 °C; and S-lens RF level, 60.0. Mass spectrometry was performed in positive-ionization full-scan mode and a subsequent data-dependent acquisition (DDA) mode. The settings for full-scan mode were: scan range, m/z 130–1000; resolution, 35,000; microscans, 1; automatic gain control (AGC) target, 1 × 106; and maximum injection time (IT), 120 ms. The settings for DDA mode were: resolution, 17,500; microscans, 1; AGC target, 2 × 105; maximum IT, 250 ms; loop count, 5; isolation window, m/z 1.0; high collision dissociation (HCD) cell stepped normalized collision energy (NCE), 17.5, 35, and 52.5 %; spectrum data type, profile; intensity threshold, 4.0 × 103; dynamic exclusion, 8.0 s.
GC–MS SUSA
In accordance with published procedures [21], urine (5 mL) was divided into two aliquots. One part was submitted to acid hydrolysis followed by extraction of the combined parts with dichloromethane–isopropanol–ethyl acetate. After evaporation, the residue was acetylated with acetic anhydride–pyridine under microwave irradiation, again evaporated, and reconstituted in 100 μL methanol. One microliter was injected for GC–MS analysis. The equipment consisted of a Hewlett–Packard (HP; Agilent, Waldbronn, Germany) 5890 Series II gas chromatograph combined with an HP 5972A MSD mass spectrometer and an HP MS ChemStation (DOS series) with HP G1034C software version C03.00. The GC conditions were: splitless injection mode; column, ThermoFisher TG-1MS capillary (12 m × 0.2 mm I.D.); cross-linked methyl silicone, 330 nm film thickness; injection port temperature, 280 °C; carrier gas, helium; flow rate, 1 mL min−1; column temperature, programmed from 100 to 310 °C at 30° min−1; initial time, 2 min; final time, 5 min. The MS conditions were: full-scan mode, m/z 50–550; electron ionization (EI) mode, ionization energy, 70 eV; ion source temperature, 220 °C; and capillary direct interface, 280 °C [22].
The full-scan data files were evaluated by use of the automated mass spectral deconvolution and identification system (AMDIS) (http://chemdata.nist.gov/mass-spc/amdis/) in simple mode. The target library was a modified version of the Maurer/Pfleger/Weber MPW_2015 library [23]. The deconvolution settings were [24]: width, 32; adjacent peak subtraction, 2; resolution, high; sensitivity, very high; shape requirements, low; minimum match factor, 50.
LC–MSn SUSA
The workup was identical with the procedure described above for identification of phase I and II metabolites. Analysis was performed with a LXQ linear ion trap MS equipped with an HESI II source and coupled to an Accela LC system (all from ThermoFisher). The LC and the MS settings have been described elsewhere [18]. Briefly, DDA was conducted on precursor ions selected from MS1: MS1 was performed in the full-scan mode (m/z 100–800) and MS2 and MS3 were performed in the DDA mode. ThermoFisher Xcalibur 2.2 SP1.48 software was used for data acquisition, NIST MS Search 2.0 (National Institute of Standards and Technology, Gaithersburg, MD, USA) for library generation, and ThermoFisher ToxID 2.1.1 for automatic target screening in the MS2 screening mode. The settings were: retention time (RT) window, 20 min; RT, 0.1 min; signal threshold, 100; search index, 600; and reverse search index, 700. ToxID was run automatically after file acquisition, by using a Xcalibur processing method starting the software tool. The target library was a modified version of the Maurer/Wissenbach/Weber MWW_2014 library [25].
LC–HR–MS–MS SUSA
For identification of phase I and II metabolites the workup and LC–HR–MS–MS method were the same as described above. ThermoFisher TraceFinder Clinical Research 3.2 software was used for data evaluation, as described by Helfer et al. (submitted for publication).
Results and discussion
Identification of phase I and phase II metabolites by LC–HR–MS–MS
The structures of the urinary metabolites were deduced from their MS–MS spectra, by comparison with the spectra of the parent compounds. The fragmentation patterns were postulated on the basis of established rules [26]. The LC–HR–MS–MS spectra of DALT and its phase I metabolites are given in Fig. S1 in the Electronic Supplementary Material (ESM), those of 5-MeO-DALT and its phase I metabolites in ESM Fig. S2. The LC–HR–MS–MS spectra of the phase II metabolites of DALT are given in ESM Fig. S3 and those of 5-MeO-DALT in ESM Fig. S4. In addition, the proposed chemical structures, accurate masses of the ions, calculated elemental formulas, and mass error values in parts per million (ppm) are given in the corresponding spectra.
Proposed fragmentation patterns for identification of the phase I metabolites by LC–HR–MS–MS
For better description of positions, the basic structure was divided into three sections (Fig. 1), the aromatic ring (section 1), the ethyl spacer (section 2), and N,N-diallylamine (section 3). In the following discussion, important fragmentation patterns of the mass spectra of DALT, 5-MeO-DALT, and their phase I metabolites will be discussed in detail. All masses are the calculated exact masses.
DALT
The spectrum of the parent compound is given in ESM Fig. S1, no. 1. In addition to the protonated molecule with m/z 241.1699 (C16H21N2 +), the DALT spectrum showed a characteristic fragment ion with m/z 144.0808 (C10H10N+) representing the ethyl indole residue (sections 1 + 2), a fragment ion with m/z 117.0701 (C9H9 +) representing the indole ring after loss of HCN, and m/z 110.0964 (C7H12N+) representing the N,N-diallylmethanimine residue (section 3) after typical α-cleavage [14].
When section 3 in the metabolites was unchanged, fragment ions could be observed with m/z 110.0964 (nos. 5, 7–13 in ESM Fig. S1); if it was N-deallylated (−40.0313 u), fragment ions could be observed with m/z 70.0651 (2–4 in ESM Fig. S1). When sections 1 and 2 were unmodified, fragment ions with m/z 144.0808 (2, 12 in ESM Fig. S1) appeared, when sections 1 and 2 were monohydroxylated (+15.9949 u) fragment ions with m/z 160.0757 appeared (3–5, 7, 8, 11 in ESM Fig. S1); when the sections were dihydroxylated (+31.9898 u) fragment ions with m/z 176.0706 appeared (9, 10 in ESM Fig. S1), and when the sections were trihydroxylated (+47.9847 u) fragment ions with m/z 192.0655 appeared (13 in ESM Fig. S1). Furthermore, fragment ions with m/z 117.0699 (2–6, 12 in ESM Fig. S1) were observed for N-deallylated metabolites or for those with an unchanged indole ring. After ring monohydroxylation, ions with m/z 115.0542 (C9H7 +) were formed instead (3, 4, 7, 8 in ESM Fig. S1); these represented the hydroxylated indole ring after loss of HCN and further elimination of water. Neither fragment was formed for compounds with multiple hydroxylation sites on the ring system or hydroxylations in section 2. Minor fragment ions with m/z 132.0808 (C9H10N+) representing methyl indole residues were formed by all compounds with either an un-substituted or monohydroxylated indole ring (2–5, 7, 8, 11 in ESM Fig. S1).
The two hydroxy metabolites (7, 8 in ESM Fig. S1) furnished the protonated molecule with m/z 257.1648 (C16H21ON2 +) and identical fragments. Besides the fragment ion with m/z 160.0757 (C10H10ON+), no loss of water (−18.0100 u) could be observed, indicating hydroxylation was in section 1. The spectra of the four dihydroxy metabolites (9–12 in ESM Fig. S1) showed the protonated molecule with m/z 273.1598 (C16H21O2N2 +). Three of the dihydroxy metabolites (9–11 in ESM Fig. S1) showed a fragment ion with m/z 231.1128 (C13H15O2N2 +) resulting from allyl cleavage (see also fragment ion with m/z 261.1234 in spectra 16 and 17 in ESM Fig. S2). Two isomers (9, 10 in ESM Fig. S1) furnished an additional fragment ion with m/z 148.0393 (C8H6O2N+) indicating two hydroxylations had occurred in section 1. The third isomer (11 in ESM Fig. S1) lost two hydrogen atoms (−2.0156 u) to give m/z 174.0550 (C10H8O2N+) instead of a fragment ion with m/z 176.0706 (C10H10O2N+) representing the same structure with a double bond in section 2. A further loss of carbon monoxide (−27.9950 u) leading to m/z 146.0600 (C9H8ON+) was observed. The presence of the fragment ion with m/z 110.0964 (C7H12N+) suggested formation of one arylhydroxy group in section 1 and one alkylhydroxy group in section 2. The fourth isomeric spectrum (12 in ESM Fig. S1) revealed different fragmentation patterns. The main fragment ions with m/z 144.0808 (C10H10N+) and m/z 142.0863 (C7H12O2N+) indicated that both oxygens were introduced at the N,N-diallylic group in section 3 leaving sections 1 and 2 unmodified. Instead of a fragment ion with m/z 231.1128 (9–11 in ESM Fig. S1), a shift of one hydrogen (+1.0078 u) was observed to give a radical cation with m/z 232.1206 (C13H16O2N2 +•). Despite the shift, this fragment ion enabled the positions of the hydroxyl groups to be established. Either both were located on the same allyl group or one hydroxy was located on the nitrogen, forming an N-oxide. Beyond this, the fragment ion with m/z 100.0757 (C5H10ON+) represented a hydroxylated N-allyl-N,N-bis-methylamine with two possible positions. Either oxidation led to an N-oxide or both hydroxy groups were located on different substituents, although this possibility could be excluded by detection of the fragment ion with m/z 232.1206. Thus, the proposed metabolite structure seemed more consistent with an N-oxide. In general, N-oxides elute later than the parent compounds on reversed-phase columns. A corresponding metabolite representing allylic hydroxylation in section 3 was not detected. Of the hydroxy and dihydroxy metabolites detected (7–12 in ESM Fig. S1), the proposed N-oxide had the longest RT (4.5 min); the others were between 3.4 and 4.3 min. This seems to support this suggestion. Among hydroxy and dihydroxy metabolites, the trihydroxy metabolite could also be identified (13 in ESM Fig. S1). The mass spectrum showed a protonated molecule with m/z 289.1547 (C16H21O3N2 +) and a fragment ion with m/z 164.0342 (C8H6O3N+) that corresponded to addition of one oxygen (+15.9949 u) in contrast with the fragment ion with m/z 148.0393 (C8H6O2N+) observed in analysis of aryl dihydroxy metabolites. This represented the threefold arylhydroxylated metabolite.
Protonated molecules with m/z 255.1492 (C16H19ON2 +) revealed two oxo metabolites (5, 6 in ESM Fig. S1). The first isomer spectrum (5 in ESM Fig. S1) showed a fragment ion analogous to the arylhydroxy metabolites (7, 8 in ESM Fig. S1). However, instead of m/z 115.0542 (C9H7 +), a fragment ion with m/z 117.0699 (C9H9 +) was detected; this was indicative of an un-substituted indole ring, as described above. With regard to these fragments, the proposed keto function could only be positioned in section 2. The second isomer spectrum (6 in ESM Fig. S1) had a fragmentation pattern completely different from those of all other spectra. Both main fragment ions with m/z 214.1101 (C13H14ON2 +•) and m/z 173.0709 (C10H9ON2 +) contained the unchanged amine group and calculated chemical formulas could only be explained by ring closure with a suggested structure given in spectrum 6 in ESM Fig. S1. The initial step may reflect hydroxylation in the amine α-position representing a rather instable hemiaminal. This chemical structure could either degrade, be oxidized to an amide, or could engage in nucleophilic attack on the indole 2-position. The product after ring closure could explain both the main fragment ion after loss of the allyl groups (−41.0391 u) and the m/z 146.0600 ion (C9H8ON+) after further loss of hydrocyanic acid (−27.0109 u).
A metabolic N-dealkylation step led to a spectrum (2 in ESM Fig. S1) indicative of a protonated molecule with m/z 201.1386 (C13H17N2 +) and a typical fragment ion with m/z 70.0651 (C4H8N+) similar that described above. Combinations with monohydroxylation led to two isomers (3, 4 in ESM Fig. S1) furnishing protonated molecules with m/z 217.1335 (C13H17ON2 +). Both isomers had similar fragmentation patterns as arylhydroxy metabolites (7, 8 in ESM Fig. S1), except for the allyl shift and an additional fragment ion with m/z 148.0757 (C9H10ON+), indicative of arylhydroxylation of a methylindole species. It should be mentioned that this fragment ion differed from the fragment ion with m/z 148.0393 (C8H6O2N+) detected for aryl dihydroxy metabolites (9, 10 in ESM Fig. S1).
5-MeO-DALT
The 5-MeO-DALT spectrum is given in ESM Fig. S2, no. 1. Basic fragmentation patterns were analogous to those of DALT. Thus, in addition to the protonated molecule with m/z 271.1805 (C17H23ON2 +), the ethylindole residue (sections 1 + 2) was represented by a fragment ion with m/z 174.0913 (C11H12ON+), the indole ring after HCN loss by a fragment ion with m/z 117.0699 (C9H9 +), and the iminium ion (section 3) by a fragment ion with m/z 110.0964 (C7H12N+). Moreover, an additional fragment ion was observed with m/z 159.0679 (C10H9ON+) representing the hydroxy ethylindole part after radical loss of a methyl group (see also spectra 3, 18, 21 in ESM Fig. S2).
When section 3 in the metabolites was unmodified, fragment ions could be observed with m/z 110.0964 (7–11, 13–17, 19, 20 in ESM Fig. S2) and when N-deallylated (−40.0313 u), ions with m/z 70.0651 (2–6 in ESM Fig. S2) were detected. When there was no metabolic change in sections 1 and 2, a fragment ion with m/z 174.0913 ( 3, 18, 21 in ESM Fig. S2) was observed; when monohydroxylated the ion with m/z 190.0863 (5, 6, 10, 11, 13, 14 in ESM Fig. S2) was observed; when dihydroxylated the ion with m/z 206.0812 (16, 17 in ESM Fig. S2) was observed; when trihydroxylated the ion with m/z 222.0761 (20 in ESM Fig. S2) was observed, and when O-demethylated the ion with m/z 160.0757 (2 in ESM Fig. S2) was observed. O-Demethylation in combination with monohydroxylation led to a fragment ion with m/z 176.0706 (4, 8 in ESM Fig. S2) or, when dihydroxylation was involved, to a fragment ion with m/z 192.0655 (15 in ESM Fig. S2). Again, the fragmented indole ring was represented by a fragment ion with m/z 117.0699 (2, 3, 10–12 in ESM Fig. S2) or 115.0542 (2, 7 in ESM Fig. S2). Neither fragment was formed by compounds with multiple hydroxylation of the ring system, with hydroxylations in section 2 (4, 5, 8, 9, 13, 15–21 in ESM Fig. S2), or with arylhydroxymethoxy substituents (6, 14 in ESM Fig. S2). Finally, the methyl indole residues were represented by a fragment ion with m/z 132.0808 (C9H10N+) for all compounds with either an un-substituted or monohydroxylated indole ring (2, 7 in ESM Fig. S2), except for those hydroxylated in section 2 (9 in ESM Fig. S2).
The two hydroxy metabolites (13, 14 in ESM Fig. S2) furnished a protonated molecule with m/z 287.1754 (C17H23O2N2 +). For the first isomer (13 in ESM Fig. S2) metabolic hydroxylation in section 2 occurred, identified by a fragment ion with m/z 158.0600 (C10H8ON+) representing the hydroxy ethylene indole part after subsequent loss of the O-methyl group and water. For the second isomer (14 in ESM Fig. S2), a fragment ion with m/z 162.0550 (C9H8O2N+) represented the dihydroxy methyl indole part after loss of the O-methyl group, which specified the position of hydroxylation in section 1. Three dihydroxy metabolites were detected (16–18 in ESM Fig. S2) with protonated molecules with m/z 303.1703 (C17H23O3N2 +). Two of these (16, 17 in ESM Fig. S2) were consistent with aryl dihydroxy metabolites with fragment ions with m/z 178.0499 (C9H8O3N+: 162.0550 + 15.9949 u) as described above. In contrast, the third isomer (18 in ESM Fig. S2) furnished a fragment ion with m/z 174.0913 (C11H12ON+), indicating loss of both hydroxy groups within section 3. This was confirmed by detection of an ion with m/z 142.0863 (C7H12O2N+: 110.0964 + 31.9899 u). Furthermore, the fragment ion with m/z 100.0757 (C5H10ON+) represented hydroxylated N-allyl-N,N-bis-methylamine, as already described for DALT, and again two options could be considered. Either the two hydroxy groups were located at different N-allyl positions or one was an N-oxide. Considering N-oxidation, a fragmentation pattern similar to that for the proposed DALT N-oxide and a comparable shift in RT (4.4 min) compared with other metabolites would be observed, again suggesting the presence of the N-oxide. Three trihydroxy metabolites (19–21 in ESM Fig. S2) were found with protonated molecules with m/z 319.1652 (C17H23O4N2 +). Isomer one (19 in ESM Fig. S2) furnished only two fragment ions with m/z 110.0964 (C7H12N+) and 178.0499 (C9H8O3N+). The first fragment ion represented a non-hydroxylated section 3, the second fragment ion a trihydroxy methylindole after loss of the O-methyl group, as already described above. Both fragment ions indicated the positions of two arylhydroxy groups present in section 1 and one in section 2. In isomer two (20 in ESM Fig. S2), all the hydroxy groups were positioned in section 1, confirmed by detection of a fragment ion with m/z 194.0448 (C9H8O4N+) representing threefold hydroxylated methyl indole. Isomer three (21 in ESM Fig. S2) had all the hydroxy groups on the alkyl side chain in section 3. Confirmation of this assignment came from a fragment ion with m/z 174.0913 (C11H12ON+) as described for the spectrum of the parent compound and a fragment ion with m/z 158.0812 (C7H12O3N+: 110.0964 + 47.9848 u). The fragmentation patterns and RT (4.4 min) were indicative of the presence of an N-oxide.
Three oxo metabolites formed protonated molecules with m/z 285.1598 (C17H21O2N2 +) (10–12 in ESM Fig. S2). According to their common fragmentation patterns, the oxygen positions were consistent with section 2 for two of the isomers (10, 11 in ESM Fig. S2) as proved by the presence of a fragment ion with m/z 110.0964 (C7H12N+) and loss of carbon monoxide (−27.9950 u) from fragment ions with m/z 190.0863 (C11H12O2N+) resulting in formation of m/z 162.0913 (C10H12ON+). It should be mentioned that this fragment ion differed from the fragment ion with m/z 162.0550 (C9H8O2N+) representing dihydroxy methylindole with regard to loss of the O-methyl group, as described above (14, 16, 17 in ESM Fig. S2). The third isomer (12 in ESM Fig. S2) had a fragmentation patterns similar to those described for DALT in spectrum no. 6 in ESM Fig. S1. Fragment ions with m/z 244.1206 (C14H16O2N2 +•), 203.0815 (C11H11O2N2 +), and 176.0706 (C10H10O2N+) were regarded as analogous with fragment ions with m/z 214.1101 (C13H14ON2 +•), 173.0709 (C10H9ON2 +), and 146.0600 (C9H8ON+), respectively.
N-Dealkylation led to a spectrum (3 in ESM Fig. S2) with a protonated molecule with m/z 231.1492 (C14H19ON2 +) and a typical fragment ion with m/z 70.0651 (C4H8N+), as already described above. Combinations of N-dealkylation and hydroxylation led to two metabolites with protonated molecules with m/z 247.1441 (C14H19O2N2 +) (5, 6 in ESM Fig. S2) and a fragment ion with m/z 178.0863 (C10H12O2N+). This was degraded either to give a fragment ion with m/z 158.0600 (C10H8ON+), indicating hydroxylation in section 2, or a fragment ion with m/z 162.0550 (C9H8O2N+), indicating arylhydroxylation in section 1.
O-Demethylation (7 in ESM Fig. S2) led to a protonated molecule with m/z 257.1648 (C16H21ON2 +) with fragment ions similar to those described for arylhydroxy DALT (7, 8 in ESM Fig. S1). Two O-demethyl hydroxy metabolites could be postulated (8, 9 in ESM Fig. S2) with protonated molecules with m/z 273.1598 (C16H21O2N2 +). Fragmentation of the first isomer (8 in ESM Fig. S2) did not differ from that of aryl dihydroxy DALT (9, 10 in ESM Fig. S1) with fragment ions with m/z 162.0550 (C9H8O2N+) and hydroxylation in section 1. For the second isomer (9 in ESM Fig. S2), loss of water (−18.0100 u) from the fragment ion with m/z 176.0706 (C10H10O2N+) to give m/z 158.0606 (C10H8ON+) indicated hydroxylation in section 2. An O-demethyl dihydroxy metabolite (15 in ESM Fig. S2) was detected with a protonated molecule with m/z 289.1547 (C16H21O3N2 +) and fragment ions similar to those identified for aryl trihydroxy DALT (13 in ESM Fig. S1). Thus, the two metabolically formed hydroxy groups were located in section 1. The spectrum of the O-demethyl N-dealkyl metabolite (2 in ESM Fig. S2) showed a protonated molecule with m/z 217.1335 (C13H17ON2 +) and fragment ions that did not differ from those of N-dealkyl arylhydroxy DALT (3, 4 in ESM Fig. S1). Further hydroxylation of this metabolite (4 in ESM Fig. S2) led to a protonated molecule with m/z 233.1285 (C13H17O2N2 +). The position of the hydroxy group was believed to be located in section 2, because of the observed fragment ion with m/z 148.0757 (C9H10ON+) that excluded the position of the second hydroxy group on the methylindole ring.
Proposed fragmentation patterns for identification of the phase II metabolites by LC–HR–MS–MS
In the following discussion, important fragmentation patterns of the mass spectra of DALT and 5-MeO-DALT phase II metabolites will be discussed. All masses are the calculated exact masses.
DALT
In general, the conjugates of the aryl monohydroxy metabolites furnished the same fragment ion as the underlying phase I metabolites, following conjugate losses of 79.9568 u for sulfates or 176.0321 u for glucuronides, with additional fragment ions with m/z 240.0325 (3S, 4S, 7S, 8S in ESM Fig. S3) for sulfates or at 336.1078 (3G, 4G, 7G, 8G in ESM Fig. S3) for glucuronides. These fragment ions represented the ethyl indole part (sections 1 + 2) with + 79.9568 u for sulfates or + 176.0321 u for glucuronides. In total, sulfates and glucuronides could be detected for two arylhydroxy isomers (7S, 8S, 7G, 8G in ESM Fig. S3) and two N-dealkyl arylhydroxy isomers (3S, 4S, 3G, 4G in ESM Fig. S3).
5-MeO-DALT
Sulfates and glucuronides were observed for alkylhydroxy (13S, 13G in ESM Fig. S4), arylhydroxy (14S, 14G in ESM Fig. S4), N-dealkyl alkylhydroxy (5S, 5G in ESM Fig. S4), N-dealkyl arylhydroxy (6S, 6G in ESM Fig. S4), O-demethyl (7S, 7G in ESM Fig. S4), O-demethyl N-dealkyl (2S, 2G in ESM Fig. S4), and O-demethyl N-dealkyl alkylhydroxy (4S, 4G in ESM Fig. S4) isomers. For O-demethyl alkylhydroxy (9G in ESM Fig. S4) and O-demethyl arylhydroxy (8G in ESM Fig. S4) metabolites, only glucuronides could be observed. Again, in addition to the fragment ions described for phase I metabolites, fragment ions with m/z 240.0325 (2S, 7S in ESM Fig. S4), 256.0274 (4S in ESM Fig. S4), 270.0431 (5S, 6S, 13S, 14S in ESM Fig. S4), 336.1078 (2G, 7G in ESM Fig. S4), 352.1027 (4G, 8G, 9G in ESM Fig. S4), or 366.1183 (5G, 6G, 13G, 14G in ESM Fig. S4) were detected for sulfates or glucuronides of metabolites with hydroxy groups on the ethylindole moiety (sections 1 + 2).
Proposed metabolic pathways
According to the identified metabolites, metabolic pathways could be proposed as depicted in Fig. 2 for DALT and in Fig. 3 for 5-MeO-DALT.

Proposed metabolic pathways for DALT (numbering in accordance with ESM Fig. S1)

Proposed metabolic pathways for 5-MeO-DALT (numbering in accordance with ESM Fig. S2)
DALT
Three hydroxylations were expected on the indole ring although only two could be detected (7, 8 in Fig. 2), most probably because of insufficient separation or low abundance of the third isomer. These monohydroxy metabolites were further hydroxylated to two isomeric dihydroxy metabolites (9, 10) and these finally to a trihydroxy metabolite (13). Aliphatic hydroxylation in section 2 formed a precursor for the corresponding oxo metabolite (5) and the ring-rearranged metabolite (6). N-dealkylation (2) and N-oxidation were also observed. The following combinations could be proposed from these observations: aromatic and aliphatic hydroxylation in section 2 (11), N-oxidation with aliphatic hydroxylation in section 3 (12), and N-dealkylation with aromatic hydroxylation (3, 4). Glucuronidation and sulfation could be observed for both isomers of the arylhydroxylated metabolites with (3, 4) and without (7, 8) N-dealkylation.
5-MeO-DALT
The metabolic pathways were similar to those observed for DALT, including aromatic and aliphatic hydroxylation at different sites. Again, the indole ring was expected to bear three different hydroxylation sites although only one could be detected (14 in Fig. 3), presumably for reasons similar to those mentioned above. These monohydroxy metabolites were further hydroxylated to two isomeric dihydroxy metabolites (16, 17) and finally to a trihydroxy metabolite (20). Aliphatic hydroxylation in section 2 (13) formed a precursor for the corresponding two oxo metabolites (10, 11) and the ring-rearranged metabolite (12). N-Dealkylation (3), N-oxidation, or O-demethylation (7) were also observed. The following combinations could be proposed: aromatic and aliphatic hydroxylation in section 2 (19), N-oxidation with aliphatic mono or dihydroxylation in section 3 (18, 21), N-dealkylation with aliphatic (5) or aromatic (6) hydroxylation, O-demethylation with aliphatic (9) or aromatic mono (8) or dihydroxylation (15), with N-dealkylation (2), or N-dealkylation with aliphatic hydroxylation (4). Glucuronidation and sulfation was observed for alkyl (13) or arylhydroxy (14) metabolites, for O-demethyl (7) or N-dealkyl (3) metabolites, and for O-demethyl hydroxy metabolites (2, 4–6). For O-demethyl alkyl (9) or arylhydroxy metabolites (8), only glucuronides were detected.
If the pathways for both drugs are compared, it is obvious that one of the dihydroxy metabolites of DALT (9 in Fig. 2) is identical to the O-demethyl hydroxy metabolite of 5-MeO-DALT (8 in Fig. 3). Similarly, the trihydroxy metabolite of DALT (13 in Fig. 2) is identical with the O-demethyl dihydroxy metabolite of 5-MeO-DALT (15 in Fig. 3), thus explaining identical spectra and retention times. The O-demethyl metabolite of 5-MeO-DALT (7 in Fig. 3) could be identical either with hydroxy metabolite 7 (Fig. 2) of DALT, with slightly different spectra and retention times, or with the undetected hydroxy metabolite.
Microsomal incubations of DALT and 5-MeO-DALT
Unfortunately, no human urine samples were available to enable confirmation that the metabolites in rat urine were also present in human urine. According to many former studies [16, 20, 27–34], combination of rat urine studies with incubation of new drugs with pHLM is a technique enabling good prediction of phase I metabolites for detection in human urine. Katagi et al. discussed transferability for other N,N-di-alkyl tryptamines [16]. After pHLM incubation, all phase I metabolites were detected for DALT, and metabolites 3, 5–7, 10–14, 16–18, and 21 (Fig. 3) for 5-MeO-DALT. Therefore, it seems reasonable to assume that at least some of these metabolites might also be found in human urine.
For initial CYP activity screening, the metabolites were detected after incubation with individual CYPs. The relative involvement of the single CYPs in the metabolic pathways of DALT are listed in Table 1; those for 5-MeO-DALT are listed in Table 2. The relative involvement of individual CYPs was defined in relation to the highest peak abundances during precursor ion monitoring of the metabolites formed. A CYP was defined as mainly involved if the corresponding relative peak abundance values were >50 %. According to these findings, the isoenzymes mainly involved in given metabolic steps were: for DALT, CYP3A4 in all types of hydroxylation, CYP2D6 in oxidation to the oxo metabolite, and CYP2C19 and CYP3A4 in N-dealkylations; for 5-MeO-DALT, CYP3A4 in all types of hydroxylation, CYP1A2 and CYP2D6 in oxidation to the oxo metabolites, CYP2C19 and CYP3A4 in N-dealkylations, and CYP2D6 in O-demethylations. Given that several CYP enzymes were involved in the metabolism of both drugs, one might expect CYP enzyme polymorphisms might be of minor toxicological relevance.
Toxicological detection of DALT and 5-MeO-DALT by GC–MS, LC–MSn, and LC–HR–MS–MS
The doses of 1 mg kg−1 BM administered to rats corresponded to low human single doses of approximately 10 mg scaled by the dose-by-factor approach of Sharma et al. [35]. As described above, these doses corresponded to the suggested dose for 5-MeO-DALT [9–11]. The corresponding urine samples were used for detectability studies. As already discussed, no human urine samples were available and thus, the main target for human urine analysis could not be elucidated. However, the SUSA cover all metabolites detected in rat urine including those identified in pHLM with their conjugates. From the experience gained in recent years, the possibility that consumption of these drugs and identification of their metabolites could be overlooked seems rather low.
By use of GC–MS, only DALT could be screened for via the metabolites given in Table 3 with the corresponding fragment ions, their relative abundances, and their retention indices. 5-MeO-DALT and its metabolites could be monitored in rat urine only after high dose administration; it might, therefore, be detectable in overdose cases only. The corresponding GC–MS data have been published elsewhere [23].
By use of LC–MSn, both drugs could be screened for via the metabolites given in Table 4, and using the protonated precursor ions, characteristic MS2 and MS3 fragment ions, and retention times. The LC–HR–MS–MS SUSA enabled monitoring of intake of both drugs by detecting the following metabolites by use of the given HR-MS–MS spectra: for DALT, spectra nos 1, 4, 7, 10, and 13 in ESM Fig. S1 and 3S, 4S, 8S, 3G, 4G, 7G, and 8G in ESM Fig. S3; for 5-MeO-DALT, spectra nos 2 in ESM Fig. S2 and 7S, 13S, 5G, 6G, 7G, 9G, 13G, and 14G in ESM Fig. S4. Reconstructed LC–HR–MS–MS ion chromatograms with the given m/z for the most abundant metabolites after work-up of a rat urine sample (low dose) are depicted in Fig. 4. The metabolites of DALT (part A) with the most abundant peak areas were arylhydroxy glucuronide isomer 1 (7G in ESM Fig. S3), its isomer 2 (8G in ESM Fig. S3), arylhydroxy isomer 1 (7 in ESM Fig. S1), and aryl trihydroxy metabolite (13 in ESM Fig. S1). The metabolites of 5-MeO-DALT (part B) with the most abundant peak areas were O-demethylglucuronide (7G in ESM Fig. S4), alkylhydroxyglucuronide (13G in ESM Fig. S4), N-dealkyl-arylhydroxyglucuronide (6G in ESM Fig. S4), arylhydroxyglucuronide (14G in ESM Fig. S4), and N-dealkyl alkyl hydroxyglucuronide metabolite (5G in ESM Fig. S4). Fortunately, because different targets could be detected, the risk of false negative results caused by ion suppression of a particular target was limited.

Reconstructed LC–HR–MS ion chromatograms with the corresponding protonated molecular exact masses (±5 ppm) indicating the given DALT metabolites (a) and 5-MeO-DALT metabolites (b) in rat urine samples after low-dose administration
To assess the general performance of the SUSA, the limits of detection (LOD) for the parent compounds were determined by use of the particular approaches, although they were not the main targets chosen for urine analysis. Urine samples spiked with the corresponding drugs in decreasing concentrations were analyzed with all SUSA and a signal-to-noise ratio of 3 was defined as the LOD. For GC–MS, LC–MSn, and LC–HR–MS–MS SUSA, the LOD were, respectively, 10, 10, and 1 ng mL−1 for DALT in urine and 50, 10, and 1 ng mL−1 for 5-MeO-DALT. Considering that for GC–MS 5 mL urine was processed for work-up and for both LC–MS approaches only 0.1 mL urine was processed, the LODs for DALT were comparable for GC–MS and LC–MSn but, as expected, much lower for LC–HR–MS–MS. The rather high LOD for 5-MeO-DALT in GC–MS might be caused by reduced ionization, because the peak area for the reference standards also differed in the same range.
Conclusions
The two new psychoactive substances DALT and 5-MeO-DALT were extensively metabolized in rats. It can be assumed the same metabolites would also be found in human urine considering the results obtained from pHLM incubations. CYP2C19, CYP2D6, and CYP3A4 were mainly involved in DALT and 5-MeO-DALT metabolism. The LC–MSn and LC–HR–MS–MS SUSA should be suitable for reliable monitoring of the intake of both drugs. The GC–MS SUSA should be able to monitor DALT intake, but 5-MeO-DALT only in overdose cases.
References
United Nations Office on Drugs and Crime (UNODC) (2014) World Drug Report 2014. http://www.unodc.org/documents/data-and-analysis/WDR2014/World_Drug_Report_2014_web.pdf
Brandt SD, King LA, Evans-Brown M (2014) The new drug phenomenon. Drug Test Anal 6:587–597
Meyer MR, Caspar A, Brandt SD, Maurer HH (2014) A qualitative/quantitative approach for the detection of 37 tryptamine-derived designer drugs, 5 beta-carbolines, ibogaine, and yohimbine in human urine and plasma using standard urine screening and multi-analyte approaches. Anal Bioanal Chem 406:225–237
Blough BE, Landavazo A, Decker AM, Partilla JS, Baumann MH, Rothman RB (2014) Interaction of psychoactive tryptamines with biogenic amine transporters and serotonin receptor subtypes. Psychopharmacology (Berl) 231:4135–4144
Cozzi NV, Gopalakrishnan A, Anderson LL, Feih JT, Shulgin AT, Daley PF, Ruoho AE (2009) Dimethyltryptamine and other hallucinogenic tryptamines exhibit substrate behavior at the serotonin uptake transporter and the vesicle monoamine transporter. J Neural Transm 116:1591–1599
Halberstadt AL, Geyer MA (2011) Multiple receptors contribute to the behavioral effects of indoleamine hallucinogens. Neuropharmacology 61:364–381
Halberstadt AL, Koedood L, Powell SB, Geyer MA (2011) Differential contributions of serotonin receptors to the behavioral effects of indoleamine hallucinogens in mice. J Psychopharmacol 25:1548–1561
Nichols DE (2012) Structure-activity relationships of serotonin 5-HT2A agonists. WIREs Membr Transport Signaling 1:559–579
Shulgin AT (2003) Basic pharmacology and effects. In: Laing R R, Halluinogens A Forensic drug handbook. Elsevier Science, London, 67–137
Shulgin AT, Shulgin A (2004) 5-MeO-DALT. http://isomerdesign.com/PiHKAL/read.php?domain=tk&id=56
Shulgin AT, Shulgin A (2004) DALT. http://isomerdesign.com/PiHKAL/read.php?domain=tk&id=57
Corkery JM, Durkin E, Elliott S, Schifano F, Ghodse AH (2012) The recreational tryptamine 5-MeO-DALT (N, N-diallyl-5-methoxytryptamine): a brief review. Prog Neuropsychopharmacol Biol Psychiatry 39:259–262
Jovel A, Felthous A, Bhattacharyya A (2014) Delirium due to intoxication from the novel synthetic tryptamine 5-MeO-DALT. J Forensic Sci 59:844–846
Brandt SD, Martins CPB (2010) Analytical methods for psychoactive N, N-dialkylated tryptamines. Trends Anal Chem 29:858–869
Gaujac A, Navickiene S, Collins MI, Brandt SD, de Andrade JB (2012) Analytical techniques for the determination of tryptamines and beta-carbolines in plant matrices and in psychoactive beverages consumed during religious ceremonies and neo-shamanic urban practices. Drug Test Anal 4:636–648
Katagi M, Kamata T, Zaitsu K, Shima N, Kamata H, Nakanishi K, Nishioka H, Miki A, Tsuchihashi H (2010) Metabolism and toxicologic analysis of tryptamine-derived drugs of abuse. Ther Drug Monit 32:328–331
Brandt SD, Tirunarayanapuram SS, Freeman S, Dempster N, Barker SA, Daley PF, Cozzi NV, Martins CPB (2008) Microwave-accelerated synthesis of psychoactive deuterated N, N-dialkylated-[alpha, alpha, beta, beta-d(4)]-tryptamines. J Label Compd Radiopharm 51:423–429
Wissenbach DK, Meyer MR, Remane D, Weber AA, Maurer HH (2011) Development of the first metabolite-based LC–MSn urine drug screening procedure - exemplified for antidepressants. Anal Bioanal Chem 400:79–88
Meyer MR, Vollmar C, Schwaninger AE, Maurer HH (2012) New cathinone-derived designer drugs 3-bromomethcathinone and 3-fluoromethcathinone: studies on their metabolism in rat urine and human liver microsomes using GC–MS and LC-high-resolution MS and their detectability in urine. J Mass Spectrom 47:253–262
Helfer AG, Turcant A, Boels D, Ferec S, Lelievre B, Welter J, Meyer MR, Maurer HH (2015) Elucidation of the metabolites of the novel psychoactive substance 4-methyl-N-ethyl-cathinone (4-MEC) in human urine and pooled liver microsomes by GC–MS and LC–HR–MS–MS techniques and of its detectability by GC–MS or LC–MSn standard screening approaches. Drug Test Anal 7:368–375
Maurer HH, Pfleger K, Weber AA (2011) Mass spectral and GC data of drugs, poisons, pesticides, pollutants and their metabolites. Wiley-VCH, Weinheim (Germany)
Welter J, Kavanagh P, Maurer HH (2014) GC–MS and LC-(high-resolution)-MSn studies on the metabolic fate and detectability of camfetamine in rat urine. Anal Bioanal Chem 406:3815–3829
Maurer HH, Pfleger K, Weber AA (2015) Mass spectral library of drugs, poisons, pesticides, pollutants and their metabolites. Wiley-VCH, Weinheim
Meyer MR, Peters FT, Maurer HH (2010) Automated mass spectral deconvolution and identification system for GC–MS screening for drugs, poisons, and metabolites in urine. Clin Chem 56:575–584
Maurer HH, Wissenbach DK, Weber AA (2014) Maurer/Wissenbach/Weber MWW LC–MSn Library of drugs, poisons, and their metabolites. Wiley-VCH, Weinheim
Niessen WMA (2011) Fragmentation of toxicologically relevant drugs in positive-ion liquid chromatography-tandem mass spectrometry. Mass Spectrom Rev 30:626–663
Caspar AT, Helfer AG, Michely JA, Auwaerter V, Brandt SD, Meyer MR, Maurer HH (2015) Studies on the metabolism and toxicological detection of the new psychoactive designer drug 2-(4-iodo-2,5-dimethoxyphenyl)-N-[(2-methoxyphenyl)methyl]ethanamine (25I-NBOMe) in human and rat urine using GC–MS, LC–MSn, and LC–HR–MS–MS. Anal Bioanal Chem. doi:10.1007/s00216-015-8828-6
Welter J, Kavanagh P, Meyer MR, Maurer HH (2015) Benzofuran analogues of amphetamine and methamphetamine: studies on the metabolism and toxicological analysis of 5-APB and 5-MAPB in urine and plasma using GC–MS and LC-(HR)-MSn techniques. Anal Bioanal Chem 407:1371–1388
Wink CSD, Meyer MR, Braun T, Turcant A, Maurer HH (2015) Biotransformation and detectability of the designer drug 2,5-dimethoxy-4-propylphenethylamine (2C-P) studied in urine by GC–MS, LC–MSn and LC-high resolution-MSn. Anal Bioanal Chem 407:831–843
Meyer MR, Mauer S, Meyer GMJ, Dinger J, Klein B, Westphal F, Maurer HH (2014) The in vivo and in vitro metabolism and the detectability in urine of 3',4'-methylenedioxy-alpha-pyrrolidinobutyrophenone (MDPBP), a new pyrrolidinophenone-type designer drug, studied by GC–MS and LC–MSn. Drug Test Anal 6:746–756
Welter J, Meyer MR, Wolf E, Weinmann W, Kavanagh P, Maurer HH (2013) 2-Methiopropamine, a thiophene analogue of methamphetamine: studies on its metabolism and detectability in the rat and human using GC–MS and LC-(HR)-MS techniques. Anal Bioanal Chem 405:3125–3135
Meyer MR, Wilhelm J, Peters FT, Maurer HH (2010) Beta-keto amphetamines: studies on the metabolism of the designer drug mephedrone and toxicological detection of mephedrone, butylone, and methylone in urine using gas chromatography-mass spectrometry. Anal Bioanal Chem 397:1225–1233
Philipp AA, Wissenbach DK, Weber AA, Zapp J, Zoerntlein SW, Klein ON, Kanogsunthornrat J, Maurer HH (2010) Use of liquid chromatography coupled to low- and high-resolution linear ion trap mass spectrometry for studying the metabolism of paynantheine, an alkaloid of the herbal drug Kratom in rat and human urine. Anal Bioanal Chem 396:2379–2391
Philipp AA, Wissenbach DK, Weber AA, Zapp J, Maurer HH (2010) Phase I and II metabolites of speciogynine, a diastereomer of the main Kratom alkaloid mitragynine, identified in rat and human urine by liquid chromatography coupled to low and high resolution linear ion trap mass spectrometry. J Mass Spectrom 45:1344–1357
Sharma V, McNeill JH (2009) To scale or not to scale: the principles of dose extrapolation. Br J Pharmacol 157:907–921
Kovats E (1958) Gaschromatographische Charakterisierung organischer Verbindungen. Teil 1. Retentionsindices aliphatischer Halogenide, Alkohole, Aldehyde und Ketone. Helv Chim Acta 41:1915–1932
Acknowledgments
The authors thank Achim T. Caspar, Julia Dinger, Golo M. J. Meyer, Jessica Welter, Carina S. D. Wink, Carsten Schröder, Gabriele Ulrich, and Armin A. Weber for their support and/or helpful discussion.
Conflict of interest
The authors declare there are no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 4945 kb)
Rights and permissions
About this article

Cite this article
Michely, J.A., Helfer, A.G., Brandt, S.D. et al. Metabolism of the new psychoactive substances N,N-diallyltryptamine (DALT) and 5-methoxy-DALT and their detectability in urine by GC–MS, LC–MSn, and LC–HR–MS–MS. Anal Bioanal Chem 407, 7831–7842 (2015). https://doi.org/10.1007/s00216-015-8955-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-015-8955-0