
Abstract
In this study, four extraction methods of perfluoroalkyl substances (PFASs) in soils and sediments were validated and compared in order to select the one that provides the best recoveries and the highest sensitivity. The determination of PFASs was carried out by liquid chromatography-tandem mass spectrometry. The extraction methods compared were based on (i) an aqueous solution of acetic acid and methanol (recoveries 44–125 %, relative standard deviation (RSD) <25 %), (ii) methanol (34–109 %, <25 %), (iii) sodium hydroxide digestion (24–178 %, <49 %), and (iv) ion pair (35–179 %, <31 %). The best results were obtained with methanol extraction, which recovered a greater number of PFASs and provided values between 45–103 % in sediment and 34–109 % in soil with RSDs <25 % and limits of quantification (LOQs) between 0.02–0.31 and 0.01–6.00 ng g−1, respectively. The selected method was successfully applied to Segura River sediments and soil samples taken near the Turia River. This study demonstrates the presence of PFASs in the studied rivers of the Valencian Community (0.07–14.91 ng g−1 in Segura River sediments; 0.02–64.04 ng g−1 in Turia River soils).

Selected matrices and extraction methods for determination of perfluoroalkyl substances
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Since 1950, perfluoroalkyl substances (PFASs) are used in a number of industrial and commercial applications as surfactants and stain repellents [1]. Examples of products containing PFASs or precursors are antifire foam, alkaline detergents, paints, nonstick cookware, carpets, upholstery, textile fibers, shampoos, floor polish, smoke inhibitors, semiconductors, pesticide formulations, food packaging, tapes, denture cleaners, etc. [2, 3]. PFASs are in the environment due to (i) industrial use and release, (ii) consumer products containing them, and (iii) biotic or abiotic degradation of larger derivatives and polymers containing perfluoroalkylated moieties. The precursors are widely used commercially and reach the environment through the raw materials used in factories or by-products containing them [2]. Another problem with PFASs is that conventional wastewater treatments show a limited efficiency to eliminate them, so they accumulate in sludge or are released into the water via the effluent [4, 5].
During the last decades, numerous studies have detected its presence in food [6], water [7, 8], sediment [9], sewage sludge [5, 10], animals, and humans [11, 12]. Consequently, concern has increased because of the stability, persistence, and bioaccumulative characteristics of these compounds, which can have adverse effects on humans and wildlife [13]. Data from human studies reveal that PFASs are present in breast milk [14], semen [15], umbilical cord [16], urine [17], blood, and serum [18]. Some toxicological studies show that PFASs can cause liver cancer and affect reproduction or weight of newborns [19, 20, 3]. Furthermore, once PFASs enter the human body, they are barely removed [21].
Because of its remarkable ubiquity, perfluorooctanesulfonate (PFOS) was included in 2009, Annex B of the Stockholm Convention on persistent organic pollutants (POPs) to limit their production [22], and in the Annex III of substances subject to review for possible identification as priority substances or priority hazardous substances of Directive 2008/105/EC [23]. Other institutions such as the United States Environmental Protection Agency [24] and Canadian environmental authorities [25] have signed agreements with companies to restrict the use of perfluorooctanoic acid (PFOA). The use of PFASs is only limited in some developed areas like the USA and Europe and its production is moving to other countries such as China, where they had a PFOS production level of 100 t in 2012 [26]. If these regulatory efforts are not coordinated, economic factors may shift the production of these materials to countries that prioritize economic development to environmental concerns [27]. Although lower blood levels of PFASs are being observed in populations of the countries where they have been regulated (e.g., the American Red Cross data indicate a 75 % decline in PFOS concentrations from 2000–2001 to 2010 [28]), these are being replaced by short-chain PFASs that are now found at increasing levels in the environment and humans [29].
At present, there are few well-documented cases about how soil or sediment can play a critical role in the distribution of PFASs in the environment and subsequent human exposure. The main limiting factor to expand the number of studies is the complexity of these matrices, the low PFAS concentrations [30], and the interferences from fluoro-containing materials that compromise quantification. These shortcomings make necessary to develop standardized extraction methods with broad applicability. Promising methods such as microwave-assisted extraction (MAE) and pressurized liquid extraction (PLE) are not always appropriate for PFASs. The vessels where the sample is digested in MAE are mainly made with Teflon (source of PFASs). Moreover, conventional PLE system, even that was proposed several times for the determination of PFASs [31], has also some parts of the instruments made of Teflon, like the rings of the stain steel cells. The replacement of these pieces by homologous made of other nonfluorinated materials is complicated. Common extraction procedures are based on four different methods: (i) acetic acid and methanol extraction [32, 33], (ii) only methanol extraction [34], (iii) sodium hydroxide (NaOH) digestion [35], and (iv) ion pairing with tetrabutylammonium hydrogen sulfate [36]. Methods using acetic acid and/or methanol were developed for application in environmental samples, whereas those applying NaOH digestion and the ion pair were initially designed for biological matrices and later adapted for the environmental ones. The choice of a suitable sample preparation technique is essential for the accurate and reliable characterization of PFASs in trace or ultratrace concentrations. Because of the chemical peculiarities of these compounds, a number of important factors must be considered: (i) background contamination (laboratory materials made of or containing perfluoroethylene or perfluoroalkyl compounds) that is a source of interferences for the analysis of PFASs, (ii) selection of the analyte isolation and preconcentration technique, as well as (iii) careful optimization of the corresponding operational parameters. Other difficulties that complicate the analysis of soil and sediment are the long extraction and purification steps, as well as matrix effects that can make the quantification of some compounds very complicated. Only one report is available comparing ion-pairing and methanol methods [36].
Thus, there is a need to undertake a systematic study to generate data that can be valuable for monitoring the occurrence of PFASs in the environment. The objective of this study was to carry out this systematic evaluation for 20 PFASs including perfluoroalkyl carboxylic acids (PFCAs), perfluoroalkane sulfonic acids (PFSAs), and fluorotelomer unsaturated carboxylic acids (FTUCAs) in two environmental abiotic matrices (soil and sediment). To our knowledge, this is the first time that the performance of the four methods is compared for such a wide range of PFASs. The target compounds have been determined by ultra high-performance liquid chromatography system (UHPLC) coupled to tandem mass spectrometry (MS/MS). Recoveries, precision, sensitivity, and matrix effects of the four extraction methods were assessed. The methods that provide the best results were implemented to carry out a survey on the presence of PFASs in soil and sediment samples.
Material and methods
Chemicals
PFCAs (PFBA, PFHxA, PFHpA, PFOA, ipPFNA, PFNA, PFDA, PFUnDA, PFDoDA, PFTrDA, PFTeDA, PFHxDA, PFODA), PFSAs (PFBS, PFHxS, PFHpS, PFOS, ipPFNS, PFDS), and FTUCAs (FOUEA), as well as internal standards isotopically labeled (MPFASs) with 13C and 18O (MPFBA, MPFHxA, MPFHxS, MPFOA, MPFOS, MPFNA, MPFDA, MPFUnDA, MPFDoDA), were used. All were from Wellington Laboratories (Guelph, Ontario, Canada) at concentration of 50 μg mL−1 in methanol, with the exception of ipPFNA that was at 45 μg mL−1. The meaning of the acronyms and other useful information are detailed in the Electronic Supplementary Material (ESM) (Tables S1, S2, and S3). Stock standard and solutions were prepared in methanol and stored in polypropylene tubes at 4 °C.
Glacial acetic acid (C2H4O2, 99.9 %) was purchased from Sigma-Aldrich (St. Louis, MO, USA); hydrochloric acid (HCl, 37 %) was from Merck, KGaA (Darmstadt, Germany); formic acid (CH2O2, 94.5 %) was from Amresco (Solon, OH, USA); anhydrous ammonia (NH3, 99.99 %) and tetrabutylammonium hydrogen sulfate (TBAS, 97 %) were from Sigma-Aldrich (St. Louis, MO, USA); ammonium formate (CH5NO2, 97 %) was from Alfa Aesar (Karlsruhe Germany); and sodium hydroxide micropills (NaOH, 98.8 %) were from Poch (Gliwice, Poland). Methyl tert-butyl ether (MTBE, 99 %) and sodium carbonate (Na2CO3, 100.2 %) were obtained from VWR (Radnor, PA, USA). Deionized water was from a Milli-Q SP Reagent Water System (Millipore, Bedford, MA, USA) and LC-MS grade methanol was purchased from Panreac (Darmstadt, Germany).
Sample collection and pretreatment
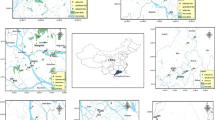
Soil and sediment samples were collected from riverine areas of the Valencian Community (East of Spain). A total of 21 soil samples from the Turia River basin collected in 2012 and 26 in 2013 were analyzed. Soil samples of the upper 20-cm horizon layer were collected. From each sampling point, of 1 m2, two subsamples were taken. Once in the laboratory, soil samples were dried and passed through a 2-mm Ø sieve, and then, the subsamples of each sampling point were homogenized to create a composite one. The composite soil samples were extended in a layer of approximately 1 cm thickness on polypropylene trays and air-dried in darkness at 20 °C to a moisture content of approximately 3 % water. Then, soil samples were stored in a sealed plastic bag at 4 °C. Sediments were taken with a Van Veen Grab sampler from the lower part of the Segura River in December 2013. A total of 12 samples were collected at six sampling points (2 samples per point). Sediment samples were transported in boxes packed with ice and stored at −20 °C in a freezer upon arrival at the laboratory. In the following 48 h after the collection, the sediment samples were freeze-dried (−75 °C, 10 mTorr, 48 h) in a VirTis Sentry 2.0 Freeze Dryer from SP Scientific (Warminster, PA, EEUU), sieved (125 μm), and stored in aluminum containers at −20 °C until analysis. The materials were carefully checked to prevent introduction of contamination. The location of the sampling points can be found in Fig. 1. The coordinates of all sampling points and a brief description of them are given in the ESM (Table S4, Fig. S1a, b).

Location of the sampling points
Extraction methods
Procedural and instrumental blank contamination is a major challenge in most of the laboratories performing trace analysis of PFASs and the possible sources of contamination as well as techniques for reducing the contamination are not well-established yet [37]. In the present study, strict controls were carried out to ensure that the material and reagents are free of PFAS contamination. Fluoropolymer parts of the instrument were exchanged and background signals of the analytes were not observed in solvent blank injections. Procedural blank contamination was reduced by avoiding the use of fluoropolymer materials in the lab during sample preparation and extraction and by rigorously rinsing all equipment with methanol before use. Very low levels of procedural blank contamination were occasionally observed for PFOS. However, the blank contamination was negligible compared to quantified PFOS concentrations in the soil and sediment. MPFASs were added to soil and sediment samples as internal standards to obtain a concentration in the final extract of 25 ng mL−1.
Matrix effects and sensitivity are also important issues associated to the amount of sample processed in each extraction procedure. As the optimization of the different methods was not carried out in the laboratory, the amount of sample used in each method was that reported as optimal by other authors [32–36]. These differences have an effect in sensitivity and matrix effect that is discussed in the “Results and discussion” section.
Acetic acid and methanol extraction
This method is based as previously described by Higgins et al. [32] and has already been detailed elsewhere [33]. Briefly, the homogenized sediment or soil samples (1 g) were transferred to 50 mL polypropylene centrifuge tubes and 10 mL of 1 % acetic acid solution was added. Each tube was vortexed, placed in a 40 °C ultrasonic bath for 15 min, and centrifuged at 956 rcf for 2 min. The supernatant solution was transferred to a second polypropylene tube. The extraction was repeated twice with 2.5 mL methanol (MeOH) and 1 % acetic acid mixture 90:10 (v/v) and with 10 mL of 1 % acetic acid solution. All extracts were combined in the second tube, adjusted to a volume of 250 mL with Milli-Q water, and cleaned up by solid-phase extraction (SPE) (see “Cleanup method”).
Methanol extraction
In this procedure, 5 g of soil or sediment was extracted three times using 10 mL of methanol, vortexed, sonicated for 15 min, and centrifuged at 956 rcf for 15 min. Finally, after reducing the volume to 5 mL under nitrogen purging, 20 μL of formic acid and 100 mL Milli-Q were added and the sample was cleaned up by SPE (see “Cleanup method”).
Methanolic NaOH digestion
In the optimized method, 1 g of soil or sediment was mixed with 2 mL of 200 mM NaOH solution in MeOH and ultrasonically extracted for 30 min. Then, 20 mL of MeOH was added to the mixture, shaken for 30 min and added with 0.05 mL of 4 M HCl. The mixture was centrifuged (956 rcf) for 15 min and the supernatant transferred to a second tube of 50 mL polypropylene. The process is repeated again but adding 10 mL methanol instead of 20 mL. The total volume of the final extract was 30 mL of MeOH. For analysis, an aliquot of 10 mL of the final extract was taken and reduced under nitrogen purging to 3 mL and adjusted to a volume of 250 mL with Milli-Q water for SPE cleanup (see “Cleanup method”).
Ion-pair extraction
Briefly, 0.5 mL of 0.5 M TBAS solution and 4 mL of 0.25 M sodium carbonate buffer (pH 10) were added into a 15-mL polypropylene tube containing 0.5 g of dried soil or sediment. After a thorough mixing, 5 mL of MTBE was added to the solution and was vigorously shaken for 20 min and centrifuged at 956 rcf for 8 min. The supernatant was transferred to a second tube. The addition of MTBE was repeated once. Again the supernatant was transferred to the second tube and then evaporated to dryness under a gentle stream of N2 and redissolved with 5 mL of MeOH. Finally, it was adjusted to a volume of 250 mL with Milli-Q water and SPE cleanup was performed (see “Cleanup method”).
Cleanup method
The extracts were cleaned up by SPE to eliminate acids, salts, and other compounds that can potentially cause matrix-induced ion suppression or enhancement during the PFAS analysis. This was performed by passing the samples through a Phenomenex Strata™ C-18 cartridge according to the procedure described by Taniyasu et al. [38]. In the four methods, the same process was performed, and the cartridges were preconditioned with 4 mL of 0.1 % ammonium hydroxide in MeOH (v/v), 4 mL of MeOH, and 4 mL of Milli-Q water. Then, the samples were passed through the cartridges by vacuum and the vacuum was kept during 15 min to dry the cartridge. Finally, PFASs were eluted with 4 mL of 0.1 % ammonium hydroxide in MeOH (v/v) and were recovered in 15 mL polypropylene tubes. Each cartridge was used only once. The tubes were evaporated to dryness under N2 (2–5 h), redissolved in 250 μL of MeOH, sonicated for 2 min, and transferred to a vial to be injected into the liquid chromatography-tandem mass spectrometer (LC/MS-MS).
Liquid chromatography-tandem mass spectrometry
A 1260 Infinity Ultra High-Performance Liquid Chromatograph (UHPLC) combined with a 6410 Triple Quadrupole (QqQ) Mass Spectrometer (MS/MS) of Agilent Technologies (Santa Clara, CA, USA) with electrospray ionization (ESI) was used. Data were processed using MassHunter Workstation Software for qualitative and quantitative analysis (GL Sciences, Tokyo, Japan).
PFASs were separated with a Kinetex C18 (50 × 2.1 mm, 1.7 μm) from Phenomenex (Torrance, CA, EEUU). The mobile phases consisted of (A) water and (B) methanol, both containing 10 mM ammonium formate. The following gradients were applied: 0 min (30 % B), 0.5 min (30 % B), 12 min (95 % B), and 20 min (95 % B) and return to the initial conditions. An equilibration time of 12 min was applied to stabilize the column conditions for a new injection. The flow rate was kept at 0.2 mL min−1 throughout the run, and the sample volume injected was 5 μL. Analysis was performed in negative ion mode. Data acquisition was carried out in selected reaction monitoring (SRM) to identify and quantify using two precursor → product ion transitions (except for PFBA), retention times, and the ratio of intensities between the two product ions. Fragmentor and collision energies were optimized for each compound individually. Information related to instrumental determination and the optimal conditions are reported in ESM (Tables S5 and S6). Separation achieved is shown in Fig. 2.

(a) Standard chromatogram of spiked sediment (25 ng mL−1) with PFASs. (b) Standard chromatogram of spiked sediment (25 ng mL−1) with MPFASs
Method validation
The validation of the instrumental parameters was performed by determining linearity, instrumental limits of detection (LOD) and quantification (LOQ), and intraday (repeatability) and interday (reproducibility) precision. The linearity was evaluated using eight different concentrations (from the LOQ to 75 ng mL−1) of PFAS standard solutions in methanol. The correlation coefficients (R 2) were superior to 0.99 for each PFAS.
Validation experiments were performed by spiking soil and sediment samples with all selected PFASs at different concentrations. Few microliters of a methanolic solution of PFASs at the appropriate concentrations were thoroughly spread onto the sample with a GL syringe. After homogenization, the spiked samples were left to balance for 20 min. Then, the samples were processed as reported in “Extraction methods,” “Cleanup method,” and “Liquid chromatography-tandem mass spectrometry” sections. For the assessment of all mentioned parameters, the analyte response was always related to the internal standard responses to compensate for undesirable matrix effects and losses during the extraction step (except in the evaluation of the matrix effect).
Among the different soil and sediment samples, those that have lower levels of PFASs were chosen. In the case of the soil sample, PFASs were not detected. As in the sediment, the sample used had a very low amount of PFOS, visible at levels lower than LOQ. The selected soil is characterized by pH >7, loamy texture, and low levels of organic matter (≈4 %). The sediment sample was also characterized by pH >7 and a percentage of carbonates >30 %. See ESM for more information about the selected soil and sediment sample characteristics (ESM Table S7). These samples are representatives of those analyzed in this study.
The LOD was calculated as the mass of analyte required to produce a signal-to-noise ratio (S/N) of 3:1, where the noise is calculated as three times the standard deviation of the background signal. The LOQ (i.e., the lowest concentration at which the analyte can be reliably detected meeting some predefined bias and imprecision goals [39]) was also established as that value whose S/N was 10:1. Instrumental LOD values were in the range from 0.11 to 1.11 ng mL−1 and LOQs were between 0.33 and 3.33 ng mL−1 (see ESM Table S8).
The precision was determined by calculating the interday and intraday precision as relative standard deviations (RSD). Repeatability (intraday) was measured as the RSD of the standard concentration of 25 ng mL−1 obtained in five consecutive injections performed on the same day, while the reproducibility (interday) was calculated by measuring the concentration of the standards on five different days. Repeatability provided RSDs lower than 9.6 %. In terms of reproducibility, as expected, the RSD was higher, although it was within acceptable limits (<12 %). All values are specified in ESM (Table S8).
To determine the total effect on the signal (matrix effect × recovery), the peak areas of the internal standards in the matrix (soil or sediment) were compared with those obtained in methanol. Recoveries were calculated by spiking both soil and sediment samples with PFASs. To verify the accuracy of the method, five replicates (n = 5) of each matrix were performed.
Recovery was determined by subtracting endogenous PFAS levels from the corresponding spiked samples. The soil and sediment samples were not pre-extracted because the extraction procedures not only extract the analytes but also some matrix components. Then, the effect of the matrix in the analyte extraction can be modified by this previous step. This provides absolute recoveries according to the equation:
The matrix effect in LC-MS analysis was first assessed by Matuszewski et al. [40] who used very simple equations (i), (ii), and (iii) to determine the matrix effect (ME), recovery of extraction (RE), and overall process efficiency (PE) that is the recovery and matrix effect.
In which A is the area of the PFAS(s) recorded for the standard solution, B is the area of the PFAS(s) recorded for the sample spiked with the target compound(s) after extraction, and C is the area of the PFAS(s) recorded for the sample spiked with the target compound(s) before extraction. The use of the recovery and matrix effect has been widely applied in the determination of PFASs [31, 33, 35, 36, 41].
Results and discussion
Extraction by acetic acid and methanol
This method detected all target PFASs in both sediment and soil. As shown in Table 1, the PFCA recoveries were in the range from 44 to 112 % in sediment and 61 to 116 % in soil. Regarding PFSAs, recoveries were from 56 to 125 % in sediment and 63 to 114 % in soil. FTUCAs (FOUEA) showed a recovery of 103 % in sediment and 91 % in soil. As for the accuracy of the method, the RSDs were found between 4 and 21 % in sediment and 5 to 25 % in soil. This method was already used in our laboratory to extract PFASs from sediment and sludge with similar results [4, 32].
Recoveries of this study were compared with those obtained by Higgins et al. [32], who measured PFCAs (PFOA, PFNA, PFDA, PFUnDA, PFDoDA, and PFTeDA) and PFSAs (PFHxS, PFOS, and PFDS) in sediment using a method also based on acidic extraction. Their reported recoveries ranged from 74 to 98 % for PFCAs and from 79 to 85 % for PFSAs. As in our study, recoveries were lower for long-chain compounds. Higgins et al. [32] suggested that it may be due to different reasons such as inefficient removal of environmental solid matrices, insufficient retention and/or elution during SPE as well as the suppression of the signal due to matrix effects during LC-MS/MS analysis (more pronounced in long-chain PFCAs). LOQs were also calculated (Table 1) and they were on the same order of magnitude than those calculated by Higgins et al. [32] for PFCAs (0.46 vs. 0.31 ng g−1) and PFSAs (0.49 vs. 0.19 ng g−1).
An estimate of the total effect on the signal (matrix effect and recovery) was performed comparing the results obtained for different MPFASs in samples and in methanolic standards (Fig. 3). In general, compounds in matrices suffer a negative effect, i.e., present worse recoveries than those in methanol. Suppression ranged from −19 to −79 % for sediment and from −3 to −78 % for soil, with the short- (MPFBA) and long-chain (MPFDoDA) compounds the most affected. Some low intensity signal enhancement was detected in MPFHxA (28 %) and MPFOA (17 %) in soil and MPFHxS (9 %) in sediment. In general, the effects are lower in soil than in sediment.

(a) Total effect on the signal (matrix effect × recovery) in soil. (b) Total effect on the signal (matrix effect × recovery) in sediment
Extraction with methanol
Preliminary experiments were carried out to optimize the method. Several variables such as solvent (acetonitrile and methanol), volume of solvent (2, 5, 10, and 20 mL), and sonication time (5, 15, 25, and 60 min) were optimized. The optimum conditions were those reported in the experimental section. As in the extraction by acetic acid, all target PFASs were recovered in both sediment and soil. Recoveries for PFASs were high, from 69 to 103 % in sediment and from 70 to 109 % in soil, with the exception of PFHxA (59 and 56 % in sediment and soil, respectively) (short-chain PFCA) and PFODA (45 and 34 %) (the longest chain PFCA). Good accuracy in both matrices was obtained: 0.3–21 % RSD in sediment and 1–21 % in soil (Table 2). Methanol was already used as extractant by Beškoski et al. [34] for the analysis of some PFCAs (PFBA, PFHxA, PFHpA, PFOA, PFNA, PFDA, PFUnDA, PFDoDA, PFTrDA, and PFTeDA) and PFSAs (PFBS, PFHxS, PFOS, and PFDS) of sediment samples from a wastewater artificial channel of an industrial complex. Beškoski et al. [34] estimated the LOQ for PFCAs (0.06 ng g−1) and PFSAs (0.12 ng g−1), very close to those obtained in the present study (0.02–0.31 ng g−1). There are no published data about spike tests or recoveries obtained with this method.
The effect on the signal produced in this method is positive in most of the compounds, with values between 23 and 150 % in sediment and from 30 to 188 % in soil. Negative values were recorded for MPFBA (−57 %) in soil and for MPFBA (−60 %), MPFHxA (−9 %), and MPFOS (−11 %) in sediment. The effect in this method is more pronounced due to the quantity of sample used, which is greater than in the rest of methods (5 g) (Fig. 3).
Extraction by methanolic NaOH digestion
NaOH digestion has been widely used in biological matrices because PFASs tend to bind to proteins and NaOH digestion breaks this bond [42–44]. Several variables were optimized such as NaOH concentration (1, 2, and 5 mM), volume (1, 2, and 5 mL), and extraction time (15, 30, and 60 min). The best results were obtained with 2 mL of NaOH 200 mM for 30 min. Of the 20 compounds listed, a total of 18 PFASs in sediment and 13 in soil were detected. Recoveries in sediment were from 24 to 113 % for PFCAs, from 41 to 125 % for PFSAs (except PFBS, with a recovery of 371 %), and 126 % for FOUEA (FTUCAs), whereas in soil, recoveries were from 48 to 109 % for PFCAs, 86 to 134 % for PFSAs, and 178 % for FOUEA (FTUCAs) (Table 3). In soil, short-chain compound of PFSAs (PFBS) and PFCAs (PFBA, PFHxA, and PFHpA) as well as long-chain PFCAs (PFHxDA, PFODA) were not detected. Sediment recoveries were higher (except for PFODA). The recovery of PFBS in sediments was unusually high, which was explained by a possible interference. The precision (RSD) of the method was 2–36 % in sediment and 2–49 % in soil. Yeung et al. [35] measured the concentration of PFCAs (PFBA, PFHxA, PFHpA, PFOA, PFNA, PFDA, PFUnDA, PFDoDA) and PFSAs (PFBS, PFHxS, PFOS, PFDS) obtaining recoveries from 70.5 to 97 % for PFCAs and 73 to 81.5 % for PFSAs. For the compounds mentioned previously, recoveries in this study were from 42 to 109 % for PFCAs and 90 to 125 % for PFSAs (except PFBS). The precision was good, except for some compounds like PFBS in sediments and FOUEA in soils.
The LOQs were calculated for soils, being in the range from 0.07 to 1.66 ng g−1 for PFCAs, 0.07 to 0.52 ng g−1 for PFSAs, and 0.77 ng g−1 for FTUCAs (FOUEA). The LOQs for sediment were in the range from 0.08 to 1.72 ng g−1 for PFCAs, 0.07 to 0.33 ng g−1 for PFSAs, and 0.55 ng g−1 for FTUCAs (FOUEA) (Table 3). The effect on the signal of the two shortest chain PFCAs (MPFBA and MPFHxA) could not be calculated. For the rest of the compounds, there were losses between −51 and −89 % in sediment and from −51 to −82 % in soil (Fig. 3). Other authors have used this method successfully for sediment (e.g., Yang et al. [45], who obtained recoveries from 31 to 121 % in sediment for PFCAs (PFHpA, PFOA, PFNA, PFDA, PFUnDA, PFDoDA) and from 50 to 93 % for PFSAs (PFHxS, PFOS, and PFDS), as well as LOQs in the range from 0.02 to 0.05 ng g−1 for both PFCAs and PFSAs).
Extraction using ion pair
The ion-pair method was first developed by Hansen et al. [46] for biological matrices. This extraction procedure is suitable for the analysis of relatively homogenous matrices and is often used when the patterns can be added to similar samples to those under study (e.g., the use of animal sera to quantify PFASs in human sera) (e.g., Llorca et al. [43]). However, when this method is applied to heterogeneous matrices, such as soils and sediments used in this study, it is limited by the complexity of the matrix, since the compounds present in it can lead to deletion or enhancement of the ion signal between samples. Despite the problems, this method has been used in complex matrices, as in the study by Zhang et al. [36], where sewage sludge was analyzed achieving good results through the implementation of previous SPE processes (not used in biological matrices) and the use of isotopically labeled internal standards. Zhang et al. [36] applied the method for PFCAs (PFHxA, PFHpA, PFOA, PFNA, PFDoDA, PFTeDA), PFSAs (PFBS, PFHxS, PFOS), FTUCAs (FOUEA), and others not analyzed in this paper.
Although an intensive optimization of all method variables was carried out including the amount and concentration of the ion-pairing agent, as well as the volume of MTBE and number of extractions, of the 20 possible PFASs, only 11 were recovered in sediment and 12 in soil samples. As shown in Table 4, the recoveries of the compounds detected were between 69 and 178 % in sediment and from 35 to 131 % in soil. van Leeuwen and de Boer [30] pointed out that the variability in the recoveries of this method is one of its main disadvantages, and it may be due to the complexity of the matrix used. The method precision for PFASs detected was between 5 and 31 % for both soil and sediment. Zhang et al. [36] obtained recoveries from 85 to 153 % for PFASs analyzed (except for PFBS, which value was 52 %), similar to our results.
The total effect on the signal, as in the acetic acid and NaOH methods, was mainly negative (−16 to −74 % in sediment and −39 to −81 % in soil). In the work of Zhang et al. [36], the effects on the signal obtained were calculated for MPFBA (−18 %), MPFHxA (15 %), MPFOA (24 %), and MPFOS (−13 %). LOQ reported in Table 4 ranged from 0.21 to 0.95 in sediment and 0.15 to 3.74 ng g−1 in soil. Zhang et al. [36] calculated LOQs for PFCAs (1.4 ng g−1), PFSAs (6 ng g−1), and FTUCAs (1 ng g−1) in sewage sludge, which were clearly higher.
Comparison of methods
The total effect on the compound signal (matrix effect and recovery) was mostly characterized by response suppression (Fig. 3). Suppression effect due to coelution of matrix components has been widely described [47]. Furthermore, recoveries are commonly <100 % that also justifies the lower signals of the internal standards (ISs) in samples than in the methanolic standard. However, the extraction with methanol provides signal enhancement for most of the PFASs in both soil and sediment. The addition of the ISs to the samples corrects for these effects and achieves a proper quantification without the need to use matrix-matched standards.
Just the methods based on methanolic extraction, acidified or not, were able to extract all the PFASs collected. The only methanol extraction was the most sensitive, providing the lowest LOQ for all compounds (except PFHxA in soil), while the ion pairing was the least sensitive. The acetic acid extraction was the second most sensitive method for PFCAs and the NaOH digestion for PFSAs. The smaller number of compounds detected using the NaOH digestion can be explained because the basic pH can promote the binding between PFSAs and soil cations preventing their extraction. Finally, the ion-pair method, despite being widely used, provided the worst results. This method was initially developed for biological matrices and problems with accuracy and variability have been frequently noted [30].
Recoveries obtained using methanol extraction with or without acetic acid are also the best ones. The only methanol method was applied to soil and sediment because it was more sensitive. However, both have certain limitations, such as being labor-intensive in the case of methanol and acetic acid (it takes 6 h to prepare a sample) or the long evaporation steps in the case of methanol that can affect volatile PFASs. Figure 2a, b shows a chromatogram of spiked (25 ng mL−1) sediment with PFASs and MPFASs used, respectively.
Application
Based on the results, methanol extraction method was selected for its application in Segura River sediments and Turia River soils, in order to detect PFAS presence. Each sample was analyzed in triplicate. For the correct determination of PFASs in these matrices, before and after each batch of 25–30 samples, calibration lines were constructed. Furthermore, for each of the 15 samples, a quality control was performed by injecting an experimental blank, a procedural blank, and a positive control.
Segura River sediments
Figure 4 presents the PFAS concentrations detected. PFCAs were found in 100 % of the samples at a concentration range of 0.07 to 14.91 ng g−1, being the highest concentration for the shortest chain PFCA (PFBA). Two long-chain PFCAs (PFTrDA and PFTeDA) were also found in one of the sampling points. For PFSAs, PFOS levels up to 2.29 ng g−1 were found. Note the presence of FOUEA in one sampling point with a concentration of 2.56 ng g−1.

Accumulated concentration (ng g−1) of PFASs in sediment at different sampling points of Segura River
As shown in Fig. 4, in Segura River sediments, PFBA and PFOS were the most frequently detected (100 % of the samples). PFOA was found in 50 % of the samples and the other compounds were detected only in one sampling point. As pointed out in some recent studies [3, 48], PFBA is the dominant perfluoroalkyl substance replacing PFOA because of production restrictions. The presence of these compounds can be explained by their release in WWTP effluents during the last decades, industrial waste (chemical and electrical industries located at the upper part of Segura River basin) and even agricultural (as they may be aids of some pesticides formulations). See ESM (Table S9) for more information about PFAS concentrations in Segura River.
Comparing the concentrations in sediments of the Segura River with those reported in the literature, comparable values were found. A similar study by Campo et al. [49] in sediments from the Llobregat River (Catalonia, Spain) presented similar mean concentrations, although slightly higher for PFOA and PFOS (0.74 and 2.76 ng g−1, respectively). Like in Segura River, PFTrDA (0.19 ng g−1) was found in one sampling point. In both studies, PFBA is the compound found at a higher concentration, although in the Llobregat River (3.67 ng g−1), the average is 3 times lower than that detected in the Segura River (10.47 ng g−1). Other studies in sediments measured concentrations of some PFASs such in L’Albufera of Valencia, with values ranging from 0.03 to 10.9 ng g−1 for PFOA and 0.10 to 4.80 ng g−1 for PFOS [33]. PFOA values were 10 times higher in L’Albufera than in the Segura River, while PFOS levels were similar in both places. However, other authors have found lower concentrations, such as Yang et al. [45] who measured concentrations of PFOA and PFOS in sediments of Liao River (China) 4 and 8 times lower, respectively, than those measured in the Segura River. Thompson et al. [50] also found concentrations of PFOA 10 times lower and PFOS values slightly higher.
Turia River soils (2012 and 2013)
In 2012, PFCAs values from 0.02 to 19.97 ng g−1 were obtained and from 0.82 to 2.74 ng g−1 for PFSAs, and FTUCA was not detected. Based on occurrence in sediments, PFBA is the compound at higher concentrations (maximum of 17.96 ng g−1), followed by PFOA (3.08 ng g−1) and PFOS (2.74 ng g−1). PFHxA was also detected at concentrations below the LOQ of the method (Fig. 5a). PFBA was found in a larger number of sampling points (77 %), followed by PFOA (59 %) and PFOS (14 %). In 2013, slightly higher values than those of the previous year were obtained for PFCAs (from 0.06 to 64.04 ng g−1) and PFSAs (0.69 to 4.15 ng g−1). Again PFBA was the compound at the highest concentration (64.04 ng g−1), followed by PFOA (6.96 ng g−1) and PFOS (4.15 ng g−1) (Fig. 5b). PFBA was also the prevalent (42 %), followed by PFOA (31 %) and PFOS (15 %). However, the occurrence frequency was lower than that obtained in 2012.

(a) Accumulated concentration (ng g−1) of PFASs in soil at different sampling points of Turia River in 2012. (b) Accumulated concentration (ng g−1) of PFASs in soil at different sampling points of Turia River in 2013
There are few articles about PFAS concentrations in soil. The published data only focused on the concentrations of PFOA and PFOS. Li et al. [41], in agricultural soils of Shanghai, found PFOA in the range from 3.3 to 44 ng g−1 and 9.2 to 10.4 ng g−1 for PFOS. Strynar et al. [51] conducted a pilot study to analyze soil PFASs in six countries and they obtained variable ranges from 0.95 to 12.4 ng g−1 for PFHxA, 0.76 to 31.7 ng g−1 for PFOA, and 0.58 to 10.1 ng g−1 for PFOS. However, no studies have been found in river basins that can be compared with this study. See SI for more information about PFAS concentrations in 2012 (ESM Table S10) and 2013 (ESM Table S11).
Conclusions
Of the four extraction methods tested, the extraction with methanol (recoveries between 34 and 109 %, RSD <25 %, and LOQ 0.01–6.00 ng g−1) and with acetic acid (44–125 %, <25 %, 0.06–8.00 ng g−1) gave appropriate results. Digestion with sodium hydroxide (NaOH) and the ion-pair extraction showed worse recoveries (some PFASs were not extracted) and less sensitivity. The total effect on the signal (matrix effect × recovery) showed that the matrix effect produced in the ionization source is still a challenge in the analysis of PFASs in solid matrices, despite being corrected by the use of internal standards (MPFASs). The quantification obtained by both methods was appropriate and the scope of their application can be widened in the future to other similar matrices like sludge samples.
In the monitoring conducted in sediment samples from the Segura River, PFOA, PFBA, PFOS, PFTeDA, PFTrDA, and FOUEA were detected. PFBA, PFOA, and PFOS were detected in soil samples taken at the Turia River basin in 2012 and 2013. The highest concentrations were in both rivers for PFBA, confirming the growing presence of short-chain compounds that replace traditional PFASs. This is the first study about PFAS concentrations in the Segura and Turia Rivers and the first time FOUEA has been detected in sediments.
References
Onghena M, Moliner-Martinez Y, Picó Y, Campíns-Falcó P, Barceló D (2012) Analysis of 18 perfluorinated compounds in river waters: comparison of high performance liquid chromatography–tandem mass spectrometry, ultra-high-performance liquid chromatography–tandem mass spectrometry and capillary liquid chromatography–mass spectrometry. Journal of Chromatography A 1244:88–97. doi:10.1016/j.chroma.2012.04.056
Buck RC, Franklin J, Berger U, Conder JM, Cousins IT, de Voogt P, Astrup Jensen A, Kannan K, Mabury SA, van Leeuwen SPJ (2011) Perfluoroalkyl and polyfluoroalkyl substances in the environment: terminology, classification, and origins. Integrated Environmental Assessment and Management 7(4):513–541. doi:10.1002/ieam.258
Rahman MF, Peldszus S, Anderson WB (2014) Behaviour and fate of perfluoroalkyl and polyfluoroalkyl substances (PFASs) in drinking water treatment: a review. Water Research 50:318–340. doi:10.1016/j.watres.2013.10.045
Campo J, Masiá A, Picó Y, Farré M, Barceló D (2014) Distribution and fate of perfluoroalkyl substances in Mediterranean Spanish sewage treatment plants. Science of The Total Environment 472:912–922. doi:10.1016/j.scitotenv.2013.11.056
Gómez-Canela C, Barth JC, Lacorte S (2012) Occurrence and fate of perfluorinated compounds in sewage sludge from Spain and Germany. Environmental Science and Pollution Research 19(9):4109–4119. doi:10.1007/s11356-012-1078-7
Vestergren R, Berger U, Glynn A, Cousins IT (2012) Dietary exposure to perfluoroalkyl acids for the Swedish population in 1999, 2005 and 2010. Environment International 49:120–127. doi:10.1016/j.envint.2012.08.016
Appleman TD, Higgins CP, Quiñones O, Vanderford BJ, Kolstad C, Zeigler-Holady JC, Dickenson ERV (2014) Treatment of poly- and perfluoroalkyl substances in U.S. full-scale water treatment systems. Water Research 51:246–255. doi:10.1016/j.watres.2013.10.067
Flores C, Ventura F, Martin-Alonso J, Caixach J (2013) Occurrence of perfluorooctane sulfonate (PFOS) and perfluorooctanoate (PFOA) in N.E. Spanish surface waters and their removal in a drinking water treatment plant that combines conventional and advanced treatments in parallel lines. Science of The Total Environment 461–462:618–626. doi:10.1016/j.scitotenv.2013.05.026
Zhu Z, Wang T, Wang P, Lu Y, Giesy JP (2014) Perfluoroalkyl and polyfluoroalkyl substances in sediments from South Bohai coastal watersheds, China. Marine Pollution Bulletin 85(2):619–627. doi:10.1016/j.marpolbul.2013.12.042
Arvaniti OS, Ventouri EI, Stasinakis AS, Thomaidis NS (2012) Occurrence of different classes of perfluorinated compounds in Greek wastewater treatment plants and determination of their solid–water distribution coefficients. Occurrence and fate of emerging contaminants in municipal wastewater treatment systems J Mater Sci 239–240:24–31. doi:10.1016/j.jhazmat.2012.02.015
Naile JE, Khim JS, Wang T, Chen C, Luo W, Kwon B-O, Park J, Koh C-H, Jones PD, Lu Y, Giesy JP (2010) Perfluorinated compounds in water, sediment, soil and biota from estuarine and coastal areas of Korea. Environmental Pollution 158(5):1237–1244. doi:10.1016/j.envpol.2010.01.023
Sundström M, Ehresman DJ, Bignert A, Butenhoff JL, Olsen GW, Chang S-C, Bergman Å (2011) A temporal trend study (1972–2008) of perfluorooctanesulfonate, perfluorohexanesulfonate, and perfluorooctanoate in pooled human milk samples from Stockholm, Sweden. Environment International 37(1):178–183. doi:10.1016/j.envint.2010.08.014
Llorca M, Farré M, Picó Y, Müller J, Knepper TP, Barceló D (2012) Analysis of perfluoroalkyl substances in waters from Germany and Spain. Science of The Total Environment 431:139–150. doi:10.1016/j.scitotenv.2012.05.011
Lankova D, Lacina O, Pulkrabova J, Hajslova J (2013) The determination of perfluoroalkyl substances, brominated flame retardants and their metabolites in human breast milk and infant formula. Talanta 117:318–325. doi:10.1016/j.talanta.2013.08.040
Raymer JH, Michael LC, Studabaker WB, Olsen GW, Sloan CS, Wilcosky T, Walmer DK (2012) Concentrations of perfluorooctane sulfonate (PFOS) and perfluorooctanoate (PFOA) and their associations with human semen quality measurements. Reproductive Toxicology 33(4):419–427. doi:10.1016/j.reprotox.2011.05.024
Hanssen L, Dudarev AA, Huber S, Odland J, Nieboer E, Sandanger TM (2013) Partition of perfluoroalkyl substances (PFASs) in whole blood and plasma, assessed in maternal and umbilical cord samples from inhabitants of arctic Russia and Uzbekistan. Science of The Total Environment 447:430–437. doi:10.1016/j.scitotenv.2013.01.029
Jurado-Sánchez B, Ballesteros E, Gallego M (2014) Analytical method for biomonitoring of perfluoroalkyl acids in human urine. Talanta 128:141–146. doi:10.1016/j.talanta.2014.04.071
Calafat AM, Wong L-Y, Kuklenyik Z, Reidy JA, Needham LL (2007) Polyfluoroalkyl chemicals in the U.S. population: data from the National Health and Nutrition Examination Survey (NHANES) 2003–2004 and comparisons with NHANES 1999–2000. Environmental Health Perspectives 115:1596–1602. doi:10.1289/ehp.10598
Bull S, Burnett K, Vassaux K, Ashdown L, Brown T, Rushton L (2014) Extensive literature search and provision of summaries of studies related to the oral toxicity of perfluoroalkylated substances (PFASs), their precursors and potential replacements in experimental animals and humans. Area 1: data on toxicokinetics (absorption, distribution, metabolism, excretion) in in vitro studies, experimental animals and humans. Area 2: data on toxicity in experimental animals. Area 3: data on observations in humans. EFSA supporting publication, EN-572, 345 pp
Joensen UN, Bossi R, Leffers H, Jensen AA, Skakkebæk NE, Jørgensen N (2009) Do perfluoroalkyl compounds impair human semen quality? Environmental Health Perspectives 117(6):923–927. doi:10.1289/ehp.0800517
Seals R, Bartell SM, Steenland K (2010) Accumulation and clearance of perfluorooctanoic acid (PFOA) in current and former residents of an exposed community. Environmental Health Perspectives 119:119–124. doi:10.1289/ehp.1002346
UNEP (2010) New POPs SC-4/17: listing of perfluorooctane sulfonic acid, its salts and perfluorooctane sulfonyl fluoride. United Nations Environment Programme: Stockholm Convention on Persistent Organic Pollutants (POPs), Génova, Suiza
European Parlament D (2008) Directive 2008/105/EC of the European Parliament and of the Council of 16 December 2008 on environmental quality standards in the field of water policy, amending and subsequently repealing Council Directives 82/176/EEC, 83/513/EEC, 84/156/EEC, 84/491/EEC, 86/280/EEC and amending Directive 2000/60/EC of the European Parliament and of the Council. Official Journal of the European Union 348/84
USEPA (2006) US Environmental Protection Agency. 2010/2015 PFOA Stewardship Program
Environment Canada (2010) Environmental performance agreement respecting perfluorinated carboxylic acids (PFCAs) and their precursors in perfluorochemical products sold in Canada (http://ec.gc.ca/epe-epa/default.asp?lang=En&n=0D8C879E-1#X-2013092511492112). Accessed 24 Apr 2015
Zhang L, Liu J, Hu J, Liu C, Guo W, Wang Q, Wang H (2012) The inventory of sources, environmental releases and risk assessment for perfluorooctane sulfonate in China. Chemicals Management and Environmental Assessment of Chemicals in China 165:193–198. doi:10.1016/j.envpol.2011.09.001
Lindstrom AB, Strynar MJ, Libelo EL (2011) Polyfluorinated compounds: past, present, and future. Environmental Science & Technology 45(19):7954–7961. doi:10.1021/es2011622
Olsen GW, Lange CC, Ellefson ME, Mair DC, Church TR, Goldberg CL, Herron RM, Medhdizadehkashi Z, Nobiletti JB, Rios JA, Reagen WK, Zobel LR (2012) Temporal trends of perfluoroalkyl concentrations in American Red Cross adult blood donors, 2000–2010. Environmental Science & Technology 46(11):6330–6338. doi:10.1021/es300604p
Glynn A, Berger U, Bignert A, Ullah S, Aune M, Lignell S, Darnerud PO (2012) Perfluorinated alkyl acids in blood serum from primiparous women in Sweden: serial sampling during pregnancy and nursing, and temporal trends 1996–2010. Environmental Science & Technology 46(16):9071–9079. doi:10.1021/es301168c
van Leeuwen SPJ, de Boer J (2007) Extraction and clean-up strategies for the analysis of poly- and perfluoroalkyl substances in environmental and human matrices. Journal of Chromatography A 1153(1–2):172–185. doi:10.1016/j.chroma.2007.02.069
Llorca M, Farré M, Picó Y, Barceló D (2011) Analysis of perfluorinated compounds in sewage sludge by pressurized solvent extraction followed by liquid chromatography–mass spectrometry. Journal of Chromatography A 1218(30):4840–4846. doi:10.1016/j.chroma.2011.01.085
Higgins CP, Field JA, Criddle CS, Luthy RG (2005) Quantitative determination of perfluorochemicals in sediments and domestic sludge. Environmental Science & Technology 39(11):3946–3956. doi:10.1021/es048245p
Picó Y, Blasco C, Farré M, Barceló D (2012) Occurrence of perfluorinated compounds in water and sediment of L’Albufera Natural Park (València, Spain). Environmental Science and Pollution Research 19(4):946–957. doi:10.1007/s11356-011-0560-y
Beškoski VP, Takemine S, Nakano T, Slavković Beškoski L, Gojgić-Cvijović G, Ilić M, Miletić S, Vrvić MM (2013) Perfluorinated compounds in sediment samples from the wastewater canal of Pančevo (Serbia) industrial area. Chemosphere 91(10):1408–1415. doi:10.1016/j.chemosphere.2012.12.079
Yeung LWY, De Silva AO, Loi EIH, Marvin CH, Taniyasu S, Yamashita N, Mabury SA, Muir DCG, Lam PKS (2013) Perfluoroalkyl substances and extractable organic fluorine in surface sediments and cores from Lake Ontario. Environment International 59:389–397. doi:10.1016/j.envint.2013.06.026
Zhang T, Sun H, Gerecke AC, Kannan K, Müller CE, Alder AC (2010) Comparison of two extraction methods for the analysis of per- and polyfluorinated chemicals in digested sewage sludge. Journal of Chromatography A 1217(31):5026–5034. doi:10.1016/j.chroma.2010.05.061
Yamashita N, Kannan K, Taniyasu S, Horii Y, Okazawa T, Petrick G, Gamo T (2004) Analysis of perfluorinated acids at parts-per-quadrillion levels in seawater using liquid chromatography-tandem mass spectrometry. Environ Sci Technol 38(21):5522–5528. doi:10.1021/es0492541
Taniyasu S, Kannan K, Yeung LWY, Kwok KY, Lam PKS, Yamashita N (2008) Analysis of trifluoroacetic acid and other short-chain perfluorinated acids (C2–C4) in precipitation by liquid chromatography–tandem mass spectrometry: comparison to patterns of long-chain perfluorinated acids (C5–C18). Analytica Chimica Acta 619(2):221–230. doi:10.1016/j.aca.2008.04.064
Armbruster DA, Pry T (2008) Limit of blank, limit of detection and limit of quantitation. Clin Biochem Rev 29(Suppl 1):S49–S52
Matuszewski BK, Constanzer ML, Chavez-Eng CM (1998) Matrix effect in quantitative LC/MS/MS analyses of biological fluids: a method for determination of finasteride in human plasma at picogram per milliliter concentrations. Anal Chem 70(5):882–889
Li F, Zhang C, Qu Y, Chen J, Chen L, Liu Y, Zhou Q (2010) Quantitative characterization of short- and long-chain perfluorinated acids in solid matrices in Shanghai, China. Science of The Total Environment 408(3):617–623. doi:10.1016/j.scitotenv.2009.10.032
Delinsky AD, Strynar MJ, Nakayama SF, Varns JL, Ye X, McCann PJ, Lindstrom AB (2009) Determination of ten perfluorinated compounds in bluegill sunfish (Lepomis macrochirus) fillets. Environmental Research 109:975–984. doi:10.1016/j.envres.2009.08.013
Llorca M, Farré M, Picó Y, Barceló D (2009) Development and validation of a pressurized liquid extraction liquid chromatography–tandem mass spectrometry method for perfluorinated compounds determination in fish. Journal of Chromatography A 1216(43):7195–7204. doi:10.1016/j.chroma.2009.06.062
Ye X, Schoenfuss HL, Jahns ND, Delinsky AD, Strynar MJ, Varns J, Nakayama SF, Helfant L, Lindstrom AB (2008) Perfluorinated compounds in common carp (Cyprinus carpio) fillets from the Upper Mississippi River. Environment International 34:932–938. doi:10.1016/j.envint.2008.02.003
Yang L, Zhu L, Liu Z (2011) Occurrence and partition of perfluorinated compounds in water and sediment from Liao River and Taihu Lake, China. Chemosphere 83(6):806–814. doi:10.1016/j.chemosphere.2011.02.075
Hansen KJ, Clemen LA, Ellefson ME, Johnson HO (2001) Compound-specific, quantitative characterization of organic fluorochemicals in biological matrices. Environ Sci Technol 35(4):766–770. doi:10.1021/es001489z
Picó Y, Farré M, Llorca M, Barceló D (2011) Perfluorinated compounds in food: a global perspective. Critical Reviews in Food Science and Nutrition 51(7):605–625. doi:10.1080/10408391003721727
Ahrens L, Taniyasu S, Yeung LWY, Yamashita N, Lam PKS, Ebinghaus R (2010) Distribution of polyfluoroalkyl compounds in water, suspended particulate matter and sediment from Tokyo Bay, Japan. Chemosphere 79(3):266–272. doi:10.1016/j.chemosphere.2010.01.045
Campo J, Pérez F, Masiá A, Picó Y, Ml F, Barceló D (2015) Perfluoroalkyl substance contamination of the Llobregat River ecosystem (Mediterranean area, NE Spain). Science of The Total Environment 503–504:48–57. doi:10.1016/j.scitotenv.2014.05.094
Thompson J, Roach A, Eaglesham G, Bartkow ME, Edge K, Mueller JF (2011) Perfluorinated alkyl acids in water, sediment and wildlife from Sydney Harbour and surroundings. Marine Pollution Bulletin 62(12):2869–2875. doi:10.1016/j.marpolbul.2011.09.002
Strynar MJ, Lindstrom AB, Nakayama SF, Egeghy PP, Helfant LJ (2012) Pilot scale application of a method for the analysis of perfluorinated compounds in surface soils. Chemosphere 86(3):252–257. doi:10.1016/j.chemosphere.2011.09.036
Acknowledgments
This work has been supported by the Spanish Ministry of Economy and Competitiveness through the projects “Assessing and Predicting Effects on Water Quantity and Quality in Iberian Rivers Caused by Global Change (SCARCE)” (No. CSD2009-00065, http://www.scarceconsolider.es) and “Evaluation of Emerging Contaminants in the Turia River Basins: From Basic Research to the Application of Environmental Forensics (EMERFOR)” (GCL2011-29703-C02-02, http://mefturia.es). ML also acknowledges the Foundation “Tatiana Pérez de Guzmán el Bueno” for the grant to get the PhD and JC the VALi+d postdoctoral contract (APOSTD/2014/010) of the Generalitat Valenciana for funding.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1005 kb)
Rights and permissions
About this article

Cite this article
Lorenzo, M., Campo, J. & Picó, Y. Optimization and comparison of several extraction methods for determining perfluoroalkyl substances in abiotic environmental solid matrices using liquid chromatography-mass spectrometry. Anal Bioanal Chem 407, 5767–5781 (2015). https://doi.org/10.1007/s00216-015-8759-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-015-8759-2