
Abstract
Mercapturic acids (MAs) are metabolic end products formed from conjugates between glutathione and electrophilic compounds. MAs are, therefore, suitable biomarkers of exposure to toxicants, which are either electrophiles by themselves or metabolized to electrophilic intermediates. We developed and validated two LC-MS/MS methods which allow the complementary, rapid, and sensitive determination of MAs derived from acrolein, acrylamide, acrylonitrile, benzene, 1,3-butadiene, crotonaldehyde, N,N-dimethylformamide, ethylene, ethylene oxide, vinyl chloride, propylene oxide, styrene, toluene as well as methylating and ethylating agents. Since separate determinations of single or small groups of MAs are time-consuming and expensive, we multiplexed several individual methods into two LC-MS/MS methods covering 18 individual mercapturic acids. Method validation according to FDA guidelines showed excellent results in terms of robustness, accuracy, and sensitivity of the methods. Moreover, the use of a minimal, simple, and straightforward sample cleanup procedure further accelerated the analytical workflow, which allows a time- and cost-efficient analysis of up to 18 MAs derived from various toxicants in environmental levels. The methods were applied to urine samples derived from a strictly diet-controlled clinical study, including 25 smoking and 25 non-smoking subjects. Significant increase in the urine concentrations in smokers as compared to non-smokers (p < 0.01; Student t test) was observed for 13 individual MAs. Moreover, a dose dependence was obtained for the majority of the analytes. In conclusion, the newly developed assays represent a powerful tool for the fast and reliable quantification of 18 MAs in clinical studies. A first method application suggests several suitable biomarkers for nine relevant toxicants in tobacco smoke.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Volatile organic compounds (VOCs) are numerous, varied, and ubiquitous, including both human-made and naturally occurring chemical compounds. Amongst these are several highly toxic and in part carcinogenic chemicals, including acrylamide, benzene, 1,3-butadiene, and crotonaldehyde. The absorption into the human body mainly occurs via the lung or skin. Tobacco smoke has been described as the major non-occupational source of exposure to many VOCs [1]. Such VOCs can be determined either by directly measuring mainstream and environmental tobacco smoke (ETS) which, however, does not reflect the actual absorbed dose or by exposure assessment of appropriate biomarkers in different biofluids. VOCs are mainly determined in blood either directly as parent compounds [2–4] and DNA/protein adducts [5, 6], or suitable metabolites are determined in urine [7–10].
Most VOCs exhibit their toxic potential due to an initial bioactivation step resulting in reactive metabolites which readily form adducts with, inter alia, DNA, hemoglobin, or other proteins. On the other hand, these electrophilic intermediates usually conjugate with glutathione (GSH) in a well-characterized detoxification pathway, also known as mercapturic acid (MA) pathway [11]. Mercapturic acids (MAs), N-acetyl-l-cysteine-S-conjugates, represent the metabolic end products from GSH conjugation and may serve as biomarkers of exposure (BoE) for various VOCs.
MAs are frequently analyzed in exposure assessment of various toxicants for several reasons. Firstly, as MAs are predominantly excreted in urine [12], samples can be obtained non-invasively in sufficient amounts. Furthermore, the short elimination half-lives of MAs allow the determination of recent exposures. Besides, the short half-lives allow repeated sampling and the correlation between the internal dose and a specific environmental impact [13].
Several LC-MS/MS methods have been reported in the past for the analysis of MAs resulting from exposure to toxicants from various sources [8, 11, 14, 15], including tobacco smoke [1, 7]. However, the vast majority of methods comprise only single analytes and small sets of MAs [7–10, 12, 16–26]. Recently, Alwis et al. published a method covering 16 MAs relevant for assessing the exposure to a total of 28 VOCs [27]. However, in order to quantify several MAs resulting from environmental (background) exposure, there is a need for sensitive methods.
Our goal was to develop a robust and quantitative methodology which allows for a complementary, rapid, and sensitive determination of multiple MAs resulting from different major toxicants, for example those identified as harmful or potentially harmful (HPHC) in tobacco smoke by the FDA [28]. The toxicants covered by our methodology are acrolein, acrylamide, acrylonitrile, benzene, 1,3-butadiene, crotonaldehyde, N,N-dimethylformamide, ethylene oxide, propylene oxide, styrene, toluene, vinyl chloride, and (m)ethylating agents. It emerged that two multiplex-LC-MS/MS methods for the simultaneous determination of 18 MAs in human urine were required. Both methods were applied to urine samples from a confined, diet-controlled clinical study to evaluate the suitability of the MAs as BoE to environmental and tobacco smoke toxicants.
Materials and methods
Chemicals, standards, stock solutions, and quality controls
N-Acetyl-S-(3-hydroxypropyl)cysteine (3-HPMA), N-acetyl-S-(3-hydroxypropyl)cysteine-15N13C3 (3-HPMA-15N13C3), and N-acetyl-S-(2-hydroxypropyl)cysteine (2-HPMA) were purchased from AptoChem (Montreal, Canada). N-Acetyl-S-(2-hydroxypropyl)cysteine-D3 dicyclohexylammonium salt (D3-2-HPMA), (R,S)-N-acetyl-S-[1-(hydroxymethyl)-2-propenyl-l-cysteine + (R,S)-N-acetyl-S-[2-(hydroxymethyl)-3-propenyl-l-cysteine (MHBMA), (R,S)-N-acetyl-S-[1-(hydroxymethyl)-2-propenyl-l-cysteine-D6 + (R,S)-N-acetyl-S-[2-(hydroxymethyl)-3-propenyl-l-cysteine-D6 (D6-MHBMA), N-acetyl-S-(3,4-dihydroxybutyl)-l-cysteine (DHBMA), N-acetyl-S-(3,4-dihydroxybutyl)-l-cysteine-D7 (D7-DHBMA), N-acetyl-S-(N-methylcarbamoyl)-l-cysteine (AMCC), N-acetyl-D3-S-(N-methylcarbamoyl)-l-cysteine (D3-AMCC), N-acetyl-S-(2-hydroxyethyl)-l-cysteine sodium salt (HEMA), N-acetyl-S-(2-hydroxyethyl-D4)-l-cysteine (D4-HEMA), N-acetyl-S-(2-cyanoethyl)-l-cysteine (CEMA), N-acetyl-S-(2-cyanoethyl)-l-cysteine-D3 (D3-CEMA), N-acetyl-S-methyl-l-cysteine (MMA), N-acetyl-S-methyl-l-cysteine-D3 (D3-MMA), N-acetyl-S-ethyl-l-cysteine (EMA), N-acetyl-S-ethyl-l-cysteine-D5 (D5-EMA), N-acetyl-S-(3-hydroxypropyl-1-methyl)-l-cysteine dicyclohexylammonium salt (HMPMA), N-acetyl-D3-S-(3-hydroxypropyl-1-methyl)-l-cysteine dicyclohexylammonium salt (D3-HMPMA), N-acetyl-S-(3-carboxy-2-propyl)-l-cysteine disodium salt (CMEMA), N-acetyl-S-(3-carboxy-2-propyl)-l-cysteine-D3 disodium salt (D3-CMEMA), N-acetyl-S-(2-hydroxy-1-phenylethyl)-l-cysteine + N-acetyl-S-(2-hydroxy-2-phenylethyl)-l-cysteine (mixture of PHEMA 1 and PHEMA 2), N-acetyl-S-(2-hydroxy-1-phenylethyl)-l-cysteine-13C6 + N-acetyl-S-(2-hydroxy-2-phenylethyl)-l-cysteine-13C6 (mixture of 13C6-PHEMA 1 and 13C6-PHEMA 2), N-acetyl-S-phenyl-l-cysteine (SPMA), and N-acetyl-S-benzyl-cysteine (SBMA) were obtained from Toronto Research Chemicals (Ontario, Canada). N-Acetyl-S-(3-amino-3-oxopropyl)-cysteine (AAMA), N-acetyl-D3-S-(3-amino-3-oxopropyl)-cysteine (D3-AAMA), N-acetyl-S-(3-amino-2-hydroxy-3-oxopropyl)-cysteine (GAMA), N-acetyl-D3-S-(3-amino-2-hydroxy-3-oxopropyl)-cysteine (D3-GAMA), N-acetyl-S-(N-methylcarbamoyl)-l-cysteine (AMCC), and N-acetyl-D3-S-(N-methylcarbamoyl)-l-cysteine (D3-AMCC) were supplied by Chemical Laboratories Dr. Mark (Worms, Germany). N-Acetyl-S-Phenyl-D5-l-cysteine (D5-SPMA) was purchased from Chemotrade (Düsseldorf, Germany). N-Acetyl-S-benzyl-cysteine-D7 (D7-SBMA) was obtained from a custom synthesis. The purity of all reference compounds commercially available was ≥98 %. Ammonium acetate (0.1 %) in water was supplied by Fluka (Taufkirchen, Germany). Acetonitrile (LC-MS grade) and methanol (HPLC grade) were purchased from LGC Standards GmbH (Wesel, Germany). Ultrapure water was prepared by a Seralpur Pro 90 C apparatus (Seral, Minden, Germany). Acetic acid, ammonium acetate, ammonium formate, formic acid, and sodium hydroxide were purchased from Sigma-Aldrich (Taufkirchen, Germany). All chemicals were of analytical grade or higher.
Stock solutions were prepared in methanol at a concentration of 1 mg/mL. Primary stock solutions were stored at −20 °C. The mixed working solutions for calibrators of SPMA, SBMA, EMA, MMA, HEMA, CEMA, PHEMA, HMPMA, and CMEMA and for all internal standards (ISs) were prepared from the primary stock solutions in methanol, whereas calibrators for 2-HPMA, 3-HPMA, AAMA, AMCC, GAMA, MHBMA, and DHBMA were prepared from the primary stock solution in water. Working solutions were stored at −20 °C until analysis. Under these conditions, stock and working solutions were found to be stable for more than a year.
A mixture of non-smoker and smoker urine pools was used to generate the low-level quality control (QC) samples. Medium- and high-level QC samples were prepared by pooling smokers’ urine samples. QC samples were aliquoted and stored at −20 °C until use.
Calibration
Calibration was performed with the standard addition method without weighting. Generally, a set of eight calibrators were analyzed, except for SPMA, EMA, SBMA, and CEMA where 10 calibrators were used, with each batch of unknown samples. Calibrators were prepared by spiking non-smoker urine samples (for most of the mercapturic acids, urine samples with no detectable amounts of analytes (<LOD) were available) with increasing amounts of analytes, yielding calibration ranges as indicated in Table 1. The same amount of IS mixture (method 1: 20 μL of 1.25 μg/mL of each D4-HEMA, D3-MMA, D5-EMA, 13C6-PHEMA; 2.5 μg/mL of each D5-SPMA and D7-SBMA; 5 μg/mL D3-CEMA; 15 μg/mL of each D3-HMPMA and D3-CMEMA; method 2: 10 μL of IS mix in methanol (3 μg/mL of each 3-HPMA-15N13C3, D3-AAMA, and D3-2-HPMA; 0.6 μg/mL D3-GAMA; 4 μg/mL D3-AMCC; 1 μg/mL D6-MHBMA; 10 μg/mL D7-DHBMA)) was added to each calibration level.
Study samples
Study samples were obtained from 25 smoking (9–18 cigarettes/day) and 25 non-smoking subjects. Subjects were healthy individuals participating in a diet-controlled smoking study. The study was carried out under the German national guidelines, and the protocol was approved by the ethic commission of the Medical Chamber of Nordrhein-Westfalen, Germany. Urine samples were collected at two different time points (spot urines) and after 24 h; however, only the 24-h urine samples were considered for analysis. Urine samples were kept at −20 °C until analysis. Aliquots of 100 and 500 μL were taken from each tube and used for analysis using methods 1 and 2, respectively.
Sample preparation
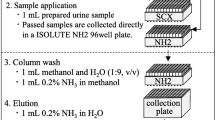
Due to the different physicochemical properties of the analytes, their quantification was split into two different methods. Both methods use a minimal, simple, and straightforward sample cleanup procedure. Urine samples were thawed at room temperature and split into two aliquots. Aliquot 1 (500 μL urine) was used for the analysis of the following analytes: HEMA, CEMA, HMPMA, CMEMA, MMA, EMA, PHEMA 1, PHEMA 2, SPMA, and SBMA (method 1). Twenty microliters of the IS mix in methanol containing the stable isotope-labelled analytes (1.25 μg/mL of each D4-HEMA, D3-MMA, D5-EMA, 13C6-PHEMA; 2.5 μg/mL of each D5-SPMA and D7-SBMA; 5 μg/mL D3-CEMA; 15 μg/mL of each D3-HMPMA and D3-CMEMA) was added to each sample. Previous studies have shown that for the determination of urinary SPMA as a biomarker of exposure to benzene, it is essential to completely convert pre-SPMA to SPMA [23]. In order to allow pre-SPMA conversion, the pH of the urine samples was reduced to approximately 1 by adding 20 μL of 37 % HCl. After 5 min of shaking, ammonium formate buffer (pH 2.5) and 10 μL of 50 % NaOH were added. Samples were centrifuged, and 50 μL of the supernatant was analyzed by a column-switching LC-MS/MS methodology (Fig. 1).

Column-switching procedure and gradient elution for method 1
Aliquot 2 (100 μL urine) was used for the analysis of the following analytes: 3-HPMA, 2-HPMA, AAMA, GAMA, 1-MHBMA, 2-MHBMA, DHBMA, and AMCC (method 2). Urine samples were placed in an Eppendorf cup and mixed with 10 μL of IS mix in methanol (3 μg/mL of each 3-HPMA-15N13C3, D3-AAMA, and D3-2-HPMA; 0.6 μg/mL D3-GAMA; 4 μg/mL D3-AMCC; 1 μg/mL D6-MHBMA; 10 μg/mL D7-DHBMA). The mixture was evaporated in a SpeedVac (Jouan RC10.22, Thermo Scientific, Dreieich, Germany) to dryness and further reconstituted in 100 μL methanol. After thoroughly shaking for approximately 20 min, the supernatant was transferred into microvials and 2 μL was injected into the LC-MS/MS system.
LC-MS/MS analysis
Liquid chromatography was performed with an AT 1200 system including a binary pump and a column oven (Agilent Technologies, Waldbronn, Germany) connected to an HTC Pal autosampler (CTC Analytics, Zwingen, Switzerland). For the column-switching procedure (method 1, Fig. 1) an AT 1100 external isocratic pump (Agilent Technologies, Waldbronn, Germany) was used for additional analyte purification and concentration. A hybrid triple quadrupole mass spectrometer API 5000 equipped with a Turbo V source ion spray, operating in negative ESI mode, was used for detection (AB Sciex, Darmstadt, Germany). High-purity nitrogen was produced by a nitrogen generator NGM 22-LC/MS (cmc Instruments, Eschborn, Germany). The turbo ion spray source parameters were kept the same for both methods using the following settings: ion spray voltage = −4500 V, ion source heater temperature = 600 °C, source gas 1 = 30 psi, source gas 2 = 40 psi, and curtain gas setting = 30 psi. Since numerous mass transitions are required to cover the up to 11 metabolites and their respective IS, analytes were monitored in scheduled multiple reaction monitoring (SMRM) mode, applying a retention time window of 90 s. Target scan time was set to 0.25 s. Quantifier and qualifier (as far as available) mass transitions, MS parameters, and retention times (RT) are shown in Table 2. Quadrupoles Q1 and Q3 were operated at unit resolution.
To simplify the sample preparation process for method 1, an automated online restricted access material (RAM) coupled with a column-switching LC-MS/MS system was developed (Fig. 1). Eluent A and C consisted of 0.1 % formic acid in water, eluent B was composed of 0.1 % formic acid in acetonitrile, and eluent D was a mixture of eluent A and B (40/60, v/v). The procedure included an automated online analyte loading step, which retained the analytes on a LiChrospher reversed-phase C8 (RP-8) (25 μm particle size) RAM column. During that time period, the pump 1 flow was sent via the analytical column into the MS (Fig. 1, position A). Then, at 0.5 min, the six-port valve was switched (Fig. 1, position B) and the pump 1 flow back-flushed the RAM column and the analytes were loaded onto the analytical column (Luna C8 column, 150 mm × 4.6 mm i.d., 3 μm; equipped with a 4-mm × 3-mm pre-column, Phenomenex, Aschaffenburg, Germany). Meanwhile, pump 2 delivered the flow into the waste (Fig. 1, position B). At 1.3 min, the six-port valve was switched back to the initial position (Fig. 1, position A), during which the chromatographic separation of the analytes on the analytical column and detection via the MS/MS were achieved. The column oven was maintained at 30 °C. Chromatographic separation was optimized to obtain good resolution amongst the different mercapturic acids. The gradient elution along with the six-port valve switching cycle is highlighted in the table at the bottom of Fig. 1. The flow rate was set to 500 μL/min for pump 1 and to 300 μL/min for pump 2, respectively.
Chromatographic separation for method 2 was performed on a Waters (Eschborn, Germany) Acquity ultraperformance liquid chromatography (UPLC) BEH C18 column (150 mm × 3 mm, 1.7 μm particle size). The column was maintained at 30 °C and the injection volume was set to 2 μL. Eluent A consisted of 5 mM ammonium acetate buffer in water; eluent B was composed of acetonitrile containing 5 % of ammonium acetate (100 mM). Gradient elution was performed with 1 % A for 0.5 min, a linear increase to 7 % A until 14 min, 50 % A from 14.01 until 16 min, and re-equilibration with 1 % A from 16.01 until 21 min. The flow rate was set to 700 μL/min.
Data analysis
Analyst software (version 1.5.2, Applied Biosystems, Foster City, CA, USA) was used for peak integration, calibration, and quantification. Processed data were transferred to Excel (Microsoft 2007, Redmond, USA) to perform further statistical analysis. SPSS (IBM, Armonk, NY, USA) was used to perform the bivariate Pearson correlation analysis of the smoking subjects (N = 25).
Method validation
Urine samples for validation experiments were obtained by spiking analyte-free non-smoker urine or pooled samples from smoker urine was used. Validation procedure was performed according to FDA guidelines [29].
Specificity
The specificity of the method was analyzed by comparing retention times of non-smoker urine with reference materials in blank samples. For each compound, two MRMs (as far as available) were monitored to ensure that no interference at the RTs of the corresponding analytes is present.
Precision
In order to assess the precision of the analytical workflow, urine samples at three different concentration levels (low, medium, and high) were analyzed six times in a row and on six consecutive days to determine intra-assay and inter-assay variations, respectively. The low concentration level was a non-smoker urine pool. Medium and high concentration levels were taken from smoker urine pools at the middle and the upper end of the calibration range.
Accuracy
Assay accuracy was calculated using non-smoker urine samples spiked at three different concentration levels, covering the entire calibration range. Each level was analyzed five times in a row.
LOD/LOQ
The limit of detection (LOD) in urine, defined as a signal-to-noise ratio (S/N) of 3, was determined by spiking three analyte-free matrix samples with a low concentration of analyte (S/N < 30). The lower limit of quantification (LLOQ, S/N = 9) was confirmed by analyzing three different urine samples in five replicates, containing analytes close to the LLOQ.
Recovery
Extraction efficiencies were determined by spiking non-smoker urine samples at three different concentration levels (low, medium, high) prior to and after extraction. Samples were analyzed as triplicates.
Matrix effect
The matrix effect was evaluated by comparing urine samples spiked with low and high analyte concentration post-extraction with reference standards at the same concentration in water. Each set of samples was analyzed in triplicates.
Carryover
A high-level smoker urine pool was injected five times in a row followed by the analysis of one blank sample. This procedure was repeated three times. There would be no significant carryover if no peaks were detected above LOD at the RT of the analytes in the blank samples.
Accuracy after sample dilution
A test of sample dilution during the validation procedure was performed. For this purpose, three matrix samples with analyte concentrations higher than the upper limit of quantification (ULOQ) were used for dilution experiments. The samples were diluted with water by factors of 2, 5, and 10 to shift back the concentration into the calibration range.
Sample stability
Several types of stability were tested during the validation procedure:
-
Short-term stability: Triplicate samples in matrix at two different concentration levels (low and high) were analyzed by keeping them at ambient temperature for 4 to 24 h.
-
Post-preparative stability: This stability was determined for ∼24 h to cover the anticipated run time for the analytical batch and to allow for delayed injection. The extracted urine samples (ready to inject) are kept at autosampler temperature (10 °C) for the established time and analyzed with fresh samples serving as reference.
-
Freeze-thaw stability: Urine samples at two concentration levels (low and high) were frozen overnight at storage temperature (−20 °C) and thawed unassisted at room temperature. When completely thawed, the samples were frozen again at −20 °C for 12–24 h. This freeze-thaw cycle was repeated six times in a row before the samples were analyzed. The observed concentrations were compared with their reference value derived from freshly analyzed samples.
-
Post-validation long-term stability: For some analytes, long-term stability data are already available from previous experiments. Upon completion of the validation, long-term stability in matrix for the remaining analytes will be determined by storing a sufficient number of QC samples at the required long-term storage temperature and analyzing them in at least triplicates at a minimum of two QC concentrations (low and high). The long-term stability will be determined at several time points (e.g., 1, 3, 6, 9, and 12 months) depending on the stability time required.
Results
LC-MS/MS analysis
We developed a combination of two methods using LC-MS/MS to simultaneously quantify 18 MAs in human urine samples. Each compound was optimized individually by making use of the automated Compound optimization algorithm of the Analyst 1.5.2 software. De-protonated precursor ions were dominated in the target compounds and, therefore, selected as precursor ions in Q1. As far as possible, a minimum of two fragment ions were taken into account, using the MRM transition with the highest signal-to-noise ratio as quantifier and the MRM transition with the second highest signal-to-noise ratio as qualifier. MS conditions, IS assignment, and mass transitions for individual analytes and ISs are summarized in Table 2.
Optimized chromatographic conditions led to sufficient resolution and thus baseline separation of the individual analytes (Fig. 2; cf. Table 2 for RT of analytes). Furthermore, matrix components which might lead to interferences and would hamper absolute quantitation were successfully separated from analytes (Fig. 2). In order to cope with the relatively huge amount of mass transitions for method 1 (in total 21; see Table 2), necessary to cover the entire set of analytes (quantifier, qualifier) along with their respective ISs, the MS program was split into four periods. This was not required for method 2.

a Multiple reaction monitoring chromatogram of a representative human smoker urine sample, spiked with 20 μL of an internal standard mix (1.25 μg/mL of D4-HEMA, D3-MMA, D5-EMA, 13C6-PHEMA; 2.5 μg/mL of D5-SPMA, D7-SBMA; 5 μg/mL D3-CEMA; 15 μg/mL of D3-HMPMA, D3-CMEMA). (Method 1). b Multiple reaction monitoring chromatogram of a representative human smoker urine sample, spiked with 10 μL of an internal standard mix (3 μg/mL of 3-HPMA-15N13C3, D3-AAMA, D3-2-HPMA; 0.6 μg/mL D3-GAMA; 4 μg/mL D3-AMCC; 1 μg/mL D6-MHBMA; 10 μg/mL D7-DHBMA). (Method 2)
In a first step, various extraction procedures were tested with the primary goal of a simple, fast, and straightforward sample cleanup procedure. Thus, method 1 applies an online column-switching procedure (Fig. 1) with a minimum of sample pre-treatment (see “Material and methods” section). Method 2 was also designed to meet the purpose of high sample throughput. Tests revealed that it is sufficient to evaporate the urine samples, reconstitute in methanol, and directly inject into the LC-MS/MS system.
LC conditions were selected after evaluating the performance of several different mobile phases and column chemistries for the simultaneous detection of 10 analytes in method 1 and 8 analytes in method 2. Optimal separation with well-shaped peaks and good sensitivity across all analytes was achieved for method 1 with a reversed-phase C8 RAM phase in conjunction to a Luna reversed-phase C8 analytical column. The mobile phase consisted of 0.1 % formic acid in water (Fig. 1(A)), 0.1 % formic acid in acetonitrile (Fig. 1(B)), 0.1 % formic acid in water (Fig. 1(C)), and a combination of 40 % A and 60 % B (Fig. 1(D)).
Method 2 used an Acquity BEH HILIC column with a sub-2-μm particle size. Solvents consisted of 5 mM ammonium acetate in water (A) and acetonitrile containing 5 % ammonium acetate (100 mM) (B). As shown in Fig. 2, the method resolves all individual analytes and potential interferences. Additionally, the analytical methods allow us to resolve the primary isomers of monohydroxylated human metabolites of 1,3-butadiene (1-MHBMA and 2-MHBMA) and styrene (PHEMA 1 and PHEMA 2).
Plasma cotinine concentrations were determined in analogy to a previously developed method with minor modifications [30].
Method validation
Validation of the analytical workflow was performed according to FDA guidelines [29]. Therefore, the performance of the method was evaluated by assessing the following parameters:
The specificity of the method was ensured by the fact that no interfering signals at the expected RT in analyte-free matrix samples were detectable for both the quantifier and the qualifier mass transitions.
Intra- and inter-day precisions were evaluated in smokers’ and non-smokers’ urine samples. Precision across all analytes was at 6 % CV (Table 3). We observed slightly higher variability in samples containing lower analyte concentrations, since analytical methods tend to be less precise with lower analyte concentration.
Method accuracy was verified by analyzing spiked non-smoker urine samples with a defined amount of analytes. Accuracy rates were within the range of 80–119 % at the LOQ and 85–110 % at medium and high analyte levels, respectively (Table 3).
The LODs and LOQs for all analytes are given in Table 1 and were defined as S/N ratios of 3:1 for the LOD. LOQs were defined as the lowest concentration which could be quantified in three different urine samples with a precision of CV < 20 % and an accuracy of 80–120 %. Analysis was performed in five replicates per urine sample. The two methods demonstrated excellent sensitivities as compared to previously reported methods [19–22, 27, 31–33]. The lowest LODs were found for SPMA at 5 pg/mL and EMA at 8 pg/mL, respectively (Table 1).
Despite the fact that the current multi-analyte methods cover a wide polarity range, optimal sample preparation with regard to sample throughput along with high extraction yields for all analytes was achieved. Considering the minimal sample cleanup procedure for both methods, not surprisingly, all analytes were almost completely recovered.
Matrix effects were quantitatively assessed by analyzing standard compounds in water and spiked at the same amount into non-smoker urine matrix after extraction. We found a decline/increase in signal intensity due to matrix interferences ±50 %, depending on the analyte and matrix investigated. However, we could demonstrate that ion suppression/enhancement could be fully compensated by using the stable isotope-labelled analogues as ISs (accuracy with IS correction yielding 88–114 %). Matrix effects did not vary with the different concentration levels and were stable across the replicates within each series of analysis.
No significant carryover was observed after analyzing urine samples with high analyte concentrations.
Calibration was achieved by addition of increasing amounts of authentic reference compounds into urine matrix. Calibration curves were calculated by linear regression without weighting. Calibration parameters are summarized in Table 1.
We characterized the stability of analytes in urine stored at different temperature and freeze-thaw cycles. All analytes were found to be stable when stored for 24 h at room temperature. Similarly, post-preparative stability of the extracted urine samples was proven for 24 h at autosampler temperature (10 °C). We also tested freeze-thaw stability of analytes in urine samples and found that all analytes were stable through six freeze/thaw cycles.
Method application
The two newly developed LC-MS/MS methods were applied to samples from a diet-controlled smoking study comprising 25 smoking as well as 25 non-smoking subjects. In order to evaluate the applicability of the methods to determine tobacco-related biomarkers and their suitability as dose-response markers, the 50 subjects were categorized into three different groups based on their plasma cotinine concentration: non-smoker sub-group (NS, N = 25) with a cotinine concentration of <LOD (0.3 ng/mL), light smoking sub-group (LS, N = 12) with a plasma cotinine level between 0.3 and <215 ng/mL, and a smoking sub-group (S, N = 13) with a plasma cotinine level ≥215 ng/mL. The urinary concentrations along with the levels of significance amongst the three different groups are shown in Table 4 and Fig. 3 (boxplots), respectively. SPMA, CEMA, PHEMA 1, 1-MHBMA, and 2-MHBMA showed very low background levels close to the LOQ for the control group (NS), whereas a significant, dose-dependent increase (Student t test) was observed for the smoking groups. In contrast, the urinary concentrations of HEMA, EMA, MMA, CMEMA, and SBMA did not differ significantly with regard to the smoking habits. The most specific correlation for both the smoking status and the smoking dose was observed for SPMA, HMPMA, CEMA, PHEMA 1, 2-HPMA, 3-HPMA, AAMA, 1-MHBMA, 2-MHBMA, and AMCC (Fig. 3; Table 5, A + B).

Boxplot diagrams for the 18 quantified mercapturic acid species in a confined, diet-controlled clinical study
Discussion
Mercapturic acids can be considered as the end products of the metabolic detoxification of various environmental toxicants, which are either electrophiles by themselves (such as acrolein or ethylene oxide) or are metabolically converted to electrophilic intermediates (such as benzene, 1,3-butadiene, and many others). Since electrophiles are involved in a number of toxic processes, including genotoxicity, the quantitative analysis of human urinary MAs is a suitable approach for obtaining crucial information about the relation between exposure to toxicants and potential detrimental health effects, including cancer.
Although numerous methods for the quantification of different MAs by LC-MS/MS have been reported in the recent past, the majority of those assays target a rather small set of up to six MAs [8, 17, 20–22, 24–26]. In addition, many of these methods lack the capability of quantifying MAs from environmental exposures in low levels.
The purpose of the current work was to develop a multi-analyte method for the simultaneous quantification of a series of mercapturic acids for application to non-occupationally exposed non-smokers and smokers as target populations. In order to combine reliable, fast, and sensitive analysis of 18 mercapturic acids, our initial goal was to combine all analytes within one sample work-up procedure and one single LC-MS/MS run. One major challenge in method development was to ensure sufficient peak separation and sensitivity while quantifying a large set of MAs as these analytes largely vary with respect to their physicochemical properties such as polarity, solubility, or ionization efficiency. Additionally, a straightforward, simple sample cleanup procedure should be applied to allow high-throughput analysis, which is of particular importance for their application in large clinical and epidemiological studies. Finally, a broad, linear quantification range is required for assessing exposure of both smokers and non-smokers in such studies.
In order to meet the aforementioned criteria, it was inevitable to split the assays into two separate LC-MS/MS methods. Due to the relatively large range of polarities of the MAs of interest, inherent with different chromatographic behaviors (requiring RP-C8 and HILIC as stationary phases for chromatography), LC-MS/MS methodology including sample work-up steps had to be split into two separate methods. Nevertheless, a fast and simple sample cleanup procedure along with relatively short analysis times for both methods still allows for the high-throughput analysis primarily important when analyzing large batches of samples. Additionally, the use of stable isotope-labelled internal standards enables the full compensation of matrix effects and hence a robust, precise, and accurate quantitation across the entire set of analytes (Table 3). Moreover, a full validation was performed according to U.S. Food and Drug Administration guidelines [29]. This extensive validation procedure showed excellent precision, accuracy, and sensitivity for all analyzed MAs (Tables 1 and 3). Furthermore, the calibration was performed in the sample matrix (urine) by addition of authentic reference compounds prior to sample cleanup. Most importantly, this allows for the full compensation of potential matrix effects on the ionization.
We applied these two methods to urine samples derived from a confined, diet-controlled clinical study including 25 smokers and 25 non-smokers. Confinement and diet control are regarded as major issues in search for suitable exposure markers, since several of the MA precursors can also be inhaled with ambient air and ingested from food. In order to evaluate the applicability of the analytes as dose-dependence marker, smokers were subdivided arbitrarily in two groups—light smokers and moderate smokers as categorized on a plasma cotinine cutoff point of 215 ng/mL—yielding an equal distribution of both sub-groups (12 light smokers and 13 moderate smokers). Not surprisingly, our results demonstrate that urine samples from smokers exhibited significantly higher levels of 13 MAs as compared to non-smokers (Fig. 3). Additionally, plasma cotinine levels of S (N = 25) correlated with the majority of the urinary mercapturic acids in the smoking group (N = 25) except for DHBMA, MMA, EMA, CMEMA, SBMA, and HEMA (Table 5, A + B). A possible reason for the lack of correlation could lie in multiple sources of exposure for the aforementioned analytes.
Moreover, our data confirm recent findings showing significant differences between smokers and non-smokers for AAMA [22], GAMA [34], 2-HPMA [22], 3-HPMA [22], AMCC [22], CEMA [21], SPMA [7], HMPMA [33], and 1-/2-MHBMA [24]. In accordance with the literature, for DHBMA, similar concentrations in smokers and non-smokers were observed [21, 27]. On the one hand, DHBMA may be formed from endogenous butadiene-diol [35]; on the other hand, there are other exogenous sources apart from 1,3-butadiene, namely 2-chloroprene and epichlorohydrine, two chemicals mainly used in polymer synthesis [36]. However, to what extent those sources contribute to the overall concentration of DHBMA is unknown so far and there may be even additional sources, e.g., food intake. In our study design, all subjects had a strictly controlled diet reducing the influence of nutritional habits on the data. Notably, we observed significant differences in DHBMA levels even though those differences are less pronounced when compared to MHBMAs which seem to be more suitable as biomarkers for 1,3-butadiene. Reska et al. observed a significantly higher excretion of the sum of PHEMAs in smokers [37]. We were able to separate the isobaric compounds PHEMA 1 and 2 and quantify both analytes with high sensitivity. We found a significant, dose-dependent increase in both PHEMA 1 (p < 0.01) and 2 (p < 0.001), whereby PHEMA 2 appears to be a more significant biomarker for exposure assessment of styrene from tobacco smoke.
It is noteworthy that the current study was not designed in order to identify suitable dose-dependence biomarkers; it rather aimed at a similar smoking status across all smokers. Thus, the separation of the smoking sub-groups showed a rather small difference in smoking dose reflected by the cotinine plasma levels. Nevertheless, we were able to pick up a dose-dependent increase on MA levels for most of the aforementioned analytes as shown in the correlation matrix (Table 5, A + B). It is most likely that a dedicated study design for this purpose with smokers exhibiting a larger range in the smoking dose would probably have shown a stronger dose dependence for most of the MAs.
In contrast to the aforementioned 13 MAs, HEMA, CMEMA, SBMA, MMA, and EMA are present in similar concentrations in smokers and non-smokers. These data are supported by previous studies for CMEMA [33], HEMA [27], SBMA [27], MMA [17], and EMA [17]. CMEMA is present in very high levels across all groups (mean values 394, 398, and 409 ng/mL for non-smokers, light smokers, and moderate smokers, respectively), whereas smokers showed significant higher levels of HMPMA, a biomarker of crotonaldehyde exposure. Moreover, in accordance to the literature [33], urinary HMPMA excretion was found to significantly correlate to plasma cotinine, whereas CMEMA showed no correlation (Table 5, A). The relatively high background levels of both CMEMA and HMPMA could be attributed to exposure to crotonaldehyde from other sources such as food, an endogenous formation (e.g., lipid peroxidation), or metabolism of other toxicants [33]. Yet, only HMPMA is regarded as a suitable BoE for crotonaldehyde in smokers; the reason for that still remains unclear.
Conclusion
In summary, we have developed two simple, fast, and robust methods (methods 1 and 2) for the simultaneous quantification of 10 and 8 mercapturic acids, respectively, as potential biomarkers for various toxicants present in the environment, food, and tobacco smoke (acrolein, acrylamide, acrylonitrile, benzene, 1,3-butadiene, crotonaldehyde, N,N-dimethylformamide, ethylene oxide, propylene oxide, styrene, toluene, vinyl chloride, and (m)ethylating agents). A full method validation according to FDA guidelines proved the suitability of the multiplex-LC-MS/MS methods for determination of the MAs. Due to the high sensitivity and the straightforward and simple sample cleanup procedures, these methods are able to determine these analytes in a time-efficient manner in exposure assessment studies. High sensitivity with LODs in the low nanogram per milliliter or even picrogram per milliliter range and broad linear calibration ranges allow for the quantitative determination in smoking as well as in non-smoking cohorts. Both methods were applied to urine samples from a strictly confined and diet-controlled clinical study with smokers and non-smokers. We found dose-dependent significant increases in smokers as compared to non-smokers for 13 out of 18 MAs. In contrast to previous reports, we found differing levels in both groups for DHBMA, presumably due to the study design, which reduces influences from diet and environmental exposures. Finally, we identified suitable biomarkers of exposure for nine relevant toxicants in tobacco smoke. In conclusion, the multiplexed methods serve as a powerful tool for the sensitive, high-throughput analysis of 18 urinary mercapturic acids.
References
Hecht SS (2002) Human urinary carcinogen metabolites: biomarkers for investigating tobacco and cancer. Carcinogenesis 23:907–922
Blount BC, Kobelski RJ, Mcelprang DO, Ashley DL, Morrow JC, Chambers DM, Cardinali FL (2006) Quantification of 31 volatile organic compounds in whole blood using solid-phase microextraction and gas chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 832:292–301
Chambers DM, Ocariz JM, Mcguirk MF, Blount BC (2011) Impact of cigarette smoking on volatile organic compound (VOC) blood levels in the U.S. population: NHANES, 2003–2004. Environ Int 37:1321–1328
Chambers DM, Mcelprang DO, Waterhouse MG, Blount BC (2006) An improved approach for accurate quantitation of benzene, toluene, ethylbenzene, xylene, and styrene in blood. Anal Chem 78:5375–5383
Urban M, Kavvadias D, Riedel K, Scherer G, Tricker AR (2006) Urinary mercapturic acids and a hemoglobin adduct for the dosimetry of acrylamide exposure in smokers and non-smokers. Inhal Toxicol 18:831–839
Scherer G, Newland K, Papadopoulou E, Minet E (2014) A correlation study applied to biomarkers of internal and effective dose for acrylonitrile and 4-aminobiphenyl in smokers. Biomarkers 19:291–301
Carmella SG, Chen M, Han S, Briggs A, Jensen J, Hatsukami DK, Hecht SS (2009) Effects of smoking cessation on eight urinary tobacco carcinogen and toxicant biomarkers. Chem Res Toxicol 22:734–741
Ding YS, Blount BC, Valentin-Blasini L, Applewhite HS, Xia Y, Watson CH, Ashley DL (2009) Simultaneous determination of six mercapturic acid metabolites of volatile organic compounds in human urine. Chem Res Toxicol 22:1018–1025
Feng S, Roethig HJ, Liang Q, Kinser R, Jin Y, Scherer G, Urban M, Engl J, Riedel K (2006) Evaluation of urinary 1-hydroxypyrene, S-phenylmercapturic acid, trans, trans-muconic acid, 3-methyladenine, 3-ethyladenine, 8-hydroxy-2′-deoxyguanosine and thioethers as biomarkers of exposure to cigarette smoke. Biomarkers 11:28–52
Scherer G, Urban M, Hagedorn HW, Serafin R, Feng S, Kapur S, Muhammad R, Jin Y, Sarkar M, Roethig HJ (2010) Determination of methyl-, 2-hydroxyethyl- and 2-cyanoethylmercapturic acids as biomarkers of exposure to alkylating agents in cigarette smoke. J ChromatogrB Analyt Technol Biomed Life Sci 878:2520–2528
De Rooij BM, Commandeur JN, Vermeulen NP (1998) Mercapturic acids as biomarkers of exposure to electrophilic chemicals: applications to environmental and industrial chemicals. Biomarkers 3:239–303
Commandeur JN, Stijntjes GJ, Vermeulen NP (1995) Enzymes and transport systems involved in the formation and disposition of glutathione S-conjugates. Pharmacol Rev 47:271–330
Van Welie RTH, Van Dijck RGJM, Vermeulen NPE (1992) Mercapturic acids, protein adducts, and DNA adducts as biomarkers of electrophilic chemicals. Crit Rev Toxicol 22:271–306
Gelpi E (1995) Biomedical and biochemical applications of liquid chromatography-mass spectrometry. J Chromatogr A 703:59–80
Slatter JG, Rashed MS, Pearson PG, Han DH, Baillie TA (1991) Biotransformation of methyl isocyanate in the rat. Evidence for glutathione conjugation as a major pathway of metabolism and implications for isocyanate-mediated toxicities. Chem Res Toxicol 4:157–161
Eckert E, Drexler H, Goen T (2010) Determination of six hydroxyalkyl mercapturic acids in human urine using hydrophilic interaction liquid chromatography with tandem mass spectrometry (HILIC-ESI-MS/MS). J Chromatogr B Analyt Technol Biomed Life Sci 878:2506–2514
Eckert E, Goen T (2014) Rapid determination of four short-chain alkyl mercapturic acids in human urine by column-switching liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 965:54–60
Mascher DG, Mascher HJ, Scherer G, Schmid ER (2001) High-performance liquid chromatographic-tandem mass spectrometric determination of 3-hydroxypropylmercapturic acid in human urine. J Chrom B 750:163–169
Paci E, Pigini D, Cialdella AM, Faranda P, Tranfo G (2007) Determination of free and total S-phenylmercapturic acid by HPLC/MS/MS in the biological monitoring of benzene exposure. Biomarkers 12:111–122
Schettgen T, Musiol A, Alt A, Kraus T (2008) Fast determination of urinary S-phenylmercapturic acid (S-PMA) and S-benzylmercapturic acid (S-BMA) by column-switching liquid chromatography-tandem mass spectrometry. J ChromatogrB Analyt Technol Biomed Life Sci 863:283–292
Schettgen T, Musiol A, Alt A, Ochsmann E, Kraus T (2009) A method for the quantification of biomarkers of exposure to acrylonitrile and 1,3-butadiene in human urine by column-switching liquid chromatography-tandem mass spectrometry. Anal Bioanal Chem 393:969–981
Schettgen T, Musiol A, Kraus T (2008) Simultaneous determination of mercapturic acids derived from ethylene oxide (HEMA), propylene oxide (2-HPMA), acrolein (3-HPMA), acrylamide (AAMA) and N, N-dimethylformamide (AMCC) in human urine using liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom 22:2629–2638
Sterz K, Kohler D, Schettgen T, Scherer G (2010) Enrichment and properties of urinary pre-S-phenylmercapturic acid (pre-SPMA). J ChromatogrB Analyt Technol Biomed Life Sci 878:2502–2505
Sterz K, Scherer G, Krumsiek J, Theis FJ, Ecker J (2012) Identification and quantification of 1-hydroxybutene-2-yl mercapturic acid in human urine by UPLC-HILIC-MS/MS as a novel biomarker for 1,3-butadiene exposure. Chem Res Toxicol 25:1565–1567
Urban M, Gilch G, Schepers G, Van Miert E, Scherer G (2003) Determination of the major mercapturic acids of 1,3-butadiene in human and rat urine using liquid chromatography-tandem mass spectrometry. J Chrom B 796:131–140
Zhang X, Xiong W, Shi L, Hou H, Hu Q (2014) Simultaneous determination of five mercapturic acid derived from volatile organic compounds in human urine by LC-MS/MS and its application to relationship study. J Chromatogr B Analyt Technol Biomed Life Sci 967:102–109
Alwis KU, Blount BC, Britt AS, Patel D, Ashley DL (2012) Simultaneous analysis of 28 urinary VOC metabolites using ultra high performance liquid chromatography coupled with electrospray ionization tandem mass spectrometry (UPLC-ESI/MSMS). Anal Chim Acta 750:152–160
Food and Drug Administration (FDA) (2012) Harmful and potentially harmful constituents in tobacco products and tobacco smoke. In: http://federalregister.gov/a/2012-07727 (last Assessed 6 Feb 2014). Food and Drug Administration (FDA)
Food and Drug Administration (Fda) (2001) Guidance for industry—bioanalytical method validation. In: http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf (last Assessed 22 July 2014). Food and Drug Administration (FDA) p1-22
Meger M, Meger-Kossien I, Schuler-Metz A, Janket D, Scherer G (2002) Simultaneous determination of nicotine and eight nicotine metabolites in urine of smokers using liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 778:251–261
Huang YF, Wu KY, Liou SH, Uang SN, Chen CC, Shih WC, Lee SC, Huang CC, Chen ML (2010) Biological monitoring for occupational acrylamide exposure from acrylamide production workers. Int Arch Occup Environ Health 84:303–313
Negri S, Maestri L, Andreoli R, Manini P, Mutti A, Imbriani M (2006) Mercapturic acids of styrene in man: comparability of the results obtained by LC/MS/MS and by HPLC-fluorimeter, and stability of samples under different storage conditions. Toxicol Lett 162:225–233
Scherer G, Urban M, Hagedorn HW, Feng S, Kinser R, Sarkar M, Liang Q, Roethig HJ (2007) Determination of two mercapturic acids related to crotonaldehyde in human urine: influence of smoking. Hum Exp Toxicol 26:37–47
Boettcher MI, Schettgen T, Kutting B, Pischetsrieder M, Angerer J (2005) Mercapturic acids of acrylamide and glycidamide as biomarkers of the internal exposure to acrylamide in the general population. Mutat Res 580:167–176
Albertini RJ, Sram RJ, Vacek PM, Lynch J, Nicklas JA, Van Sittert NJ, Boogaard PJ, Henderson RF, Swenberg JA, Tates AD, Ward JB, Jr., Wright M, Ammenheuser MM, Binkova B, Blackwell W, De Zwart FA, Krako D, Krone J, Megens H, Musilova P, Rajska G, Ranasinghe A, Rosenblatt JI, Rossner P, Rubes J, Sullivan L, Upton P, Zwinderman AH (2003) Biomarkers in Czech workers exposed to 1,3-butadiene: a transitional epidemiologic study. Res.Rep.Health Eff.Inst.:1–141
Eckert E, Leng G, Gries W, Goen T (2012) A method for the simultaneous determination of mercapturic acids as biomarkers of exposure to 2-chloroprene and epichlorohydrin in human urine. J Chromatogr B Analyt Technol Biomed Life Sci 889–890:69–76
Reska M, Ochsmann E, Kraus T, Schettgen T (2010) Accurate quantification of mercapturic acids of styrene (PHEMAs) in human urine with direct sample injection using automated column-switching high-performance liquid chromatography coupled with tandem mass spectrometry. Anal Bioanal Chem 397:3563–3574
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Pluym, N., Gilch, G., Scherer, G. et al. Analysis of 18 urinary mercapturic acids by two high-throughput multiplex-LC-MS/MS methods. Anal Bioanal Chem 407, 5463–5476 (2015). https://doi.org/10.1007/s00216-015-8719-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-015-8719-x