
Abstract
The potential of fluorescence resonance energy transfer (FRET) in a photonic crystal (PC) nanostructured array to enhance the speed and sensitivity of a protein-based immunoassay was tested. Forty-nanometer carboxylated particles conjugated with donor-labeled capture antibodies were trapped by electrophoresis and used as a FRET energy donor. The PC array was able to enhance fluorescent excitation and emission by phase matching. To provide a proof of concept for this FRET-based homogeneous assay on a PC chip, an immunoassay was tested with a simple immunoglobulin G (IgG)-based reaction. A standard curve was generated by testing two different antibody reaction times: 20 min. and 1 min. The results were compared directly to those obtained from a FRET assay that used a modern, high-sensitivity plate reader with a 96-well plate and a reaction time of 1 h. The rabbit-IgG detection limits of the FRET-based homogeneous assay on the PC were 0.001 and 0.1 μg/mL for incubation times of 20 and 1 min, respectively; the sensitivities were 103 and 10 times better than the 96-well plate reader, respectively. Thus, FRET on a PC immunoplatform was shown to be a facile, effective, rapid, and sensitive detection technology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fluorescence resonance energy transfer (FRET) is a spectroscopic method involving nonradiative energy transfer from a fluorescent donor molecule to an acceptor molecule due to a dipole–dipole interaction [1]. The efficiency of energy transfer is dominated by the distance between the donor and acceptor [2]. Because the efficiency of FRET is sensitively dependent on the distance between donor and acceptor, FRET has been widely employed in bioassays that rely on binding between biological molecules, offering high sensitivity and specificity [3]. In addition, given that there is then no need to separate and purify the biological molecules during the assay, FRET has been a favored format for homogeneous immunoassays [4–6], where it decreases the number of false-positive results due to reduced background interference from nonspecific binding of fluorescent labels to extraneous surfaces.
To obtain high sensitivity when using FRET-based immunoassays in biosensors, it is critical to improve the efficiency of energy transfer between the two different fluorescent dyes attached to the biological molecules by increasing spectral overlap and/or increasing the extinction coefficient of the acceptor [7]. However, in general, fluorescent dyes are susceptible to photobleaching and have wide emission spectra and narrow absorption spectral bands [8]. Quantum dots (QDs) [4, 7, 8] or upconversion luminescent nanoparticles [9–11] have been considered as alternative luminescent labels due to their photochemical stability and high quantum yield—properties that could lead to more robust FRET-based biosensors as well as ultrahigh sensitivity once they are incorporated into the photonic crystal.
In the study reported in the present paper, we tested the potential of a photonic crystal nanostructure to enhance FRET. We developed and tested a FRET-based homogeneous immunoassay (HIA) on a photonic crystal (PC) nanostructured array for use in a generic immunoassay to detect immunoglobulin G (IgG). The PC immunoplatform was found to boost the fluorescent signal from the ensuing immunofluorescent complex, leading to a high signal-to-noise ratio [12]. A nanoparticle-based IgG immobilization using an electrophoretic particle entrapment system can minimize the use of expensive biological reagents and improve total assay time compared to other immobilization methods [12–16]. In previous studies, the utilization of PCs in various kinds of fluorescence-based immunoassays and DNA assays led to enhanced assay sensitivity [14–17]. The novel use of FRET on a PC nanostructured array offsets the inherent disadvantages of fluorescent dyes and simultaneously provides a simple, rapid, but sensitive method for the point-of-use detection of disease markers.
Materials and methods
Materials
Forty-nanometer fluorescent carboxylated polystyrene (PS) nanoparticles (F-8789; ex: 660 nm/em: 680 nm) were purchased from Invitrogen (Carlsbad, CA, USA). In our case, the fluorescence of the particles did not play a role in the FRET assay, as discussed later; these particular particles were simply a suitable size and readily available. Goat-anti-rabbit IgG–Alexa 555 (the donor molecule, for which exCitation occurs at 555 nm and emission at 565 nm; A21428) was purchased from Invitrogen. Rabbit IgG–Alexa 647 (the acceptor molecule, for which excitation occurs at 650 nm and emission at 668 nm; SC-24647) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The hapten 3-phenoxybenzoic acid (3-PBA) was synthesized using a method described in detail previously [18]. Alexa 647 was conjugated to 3-PBA using a commercial protein-labeling kit (A-20173, Invitrogen).
FRET immunoassay using a 96-well plate
A 96-well plate (Nunc Maxisorp, Thermo Fisher Scientific, Waltham, MA, USA) was coated with goat-anti-rabbit IgG (RIgG)–Alexa 555 at 128 μg/mL in phosphate-buffered saline (PBS) during 4 h of incubation at 37 °C. The wells were then washed five times with PBS. Other nonspecific sites in the wells were blocked using 300 μL of 2 % bovine serum albumin (BSA) for 1 h at room temperature. After the blocking solution was removed, 100 μL of rabbit IgG (RIgG)–Alexa 647 at 10−5, 10−4, 10−3, 0.01, 0.1, 1, and 10 μg/mL were added to the wells. After 1 h of incubation at room temperature, the fluorescent intensities were obtained at 670 nm using a plate reader (Infinite M1000 Pro, Tecan, San Jose, CA, USA). To obtain a negative control, hapten–Alexa 647 was added as a target to anti-rabbit IgG–Alexa 555-coated wells. 3-PBA was chosen as the hapten because this reagent had already been conjugated with Alexa 647 in our lab and was readily available to us.
FRET immunoassay using a photonic crystal nanostructured array
One milliliter of 0.05 % fluorescent carboxylated PS particles was coated with goat-anti-RIgG–Alexa 555 in deionized (DI) water by passive adsorption for 2 h at room temperature with gentle shaking. The amount of antibody used was estimated to provide full coverage of the particle surface, following a protocol provided by the vendor (TechNote 205, Bangs Laboratories, Fishers, IN, USA). Particles with anti-RIgG–Alexa 555 were trapped using an electrophoretic particle entrapment system (EPES) followed by removal of the solution [19]. After particle trapping was completed, 10 μL of RIgG–Alexa 647 were dropped onto the photonic crystal array and then incubated for either 20 or 1 min, followed by removal of the solution. Both the particle and IgG solution was removed from the surface of the array using Couette flow, as described previously [19]. The array was excited by a 532-nm laser and the signal was collected at 670 nm using a single-photon-counting detection system. The materials and detailed procedures used to fabricate the PC nanostructured array with EPES for nanoparticle-in-well assembly and the single-photon-counting detection system were described previously [14, 17].
Results and discussion
FRET assay using a 96-well plate
We performed a FRET-based immunoassay using a plate reader on a 96-well plate, which is a conventional laboratory-based method. Seven different concentrations of RIgG–Alexa 647 were applied to the wells of the plate to which anti-RIgG–Alexa 555 had been already coated: 10−5, 10−4, 10−3, 0.01, 0.1, 1, and 10 μg/mL. Three replicates were performed to obtain each datum on the standard curve (Fig. 1). PBS buffer without RIgG was used to determine the blank noise. The LOD was defined as the concentration of RIgG that produced a signal that was three standard deviations greater than the background noise (dashed line), leading to 1 μg/mL. 3-PBA–Alexa 647 was used to test the crossreactivity of anti-RIgG–Alexa 555 with the other target molecule. The resulting signal was within the interval of the background noise.

Detection of RIgG on a 96-well plate using a plate reader. Seven different concentrations of RIgG–Alexa 647 in PBS were detected using FRET: 10−5, 10−4, 10−3, 0.01, 0.1, 1, and 10 μg/mL. LODs were determined from the mean plus three standard deviations of the background noise (dashed line). Error bars are based on the standard deviation of three replicates
FRET assay on a PC nanostructured array
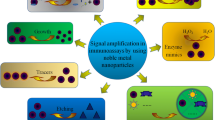
To perform FRET immunoassays on a PC nanostructured array (Fig. 2), carboxylated fluorescent PS particles conjugated with anti-RIgG–Alexa 555 were trapped using an EPES. Five different concentrations of RIgG–Alexa 647 were used: 10−3, 0.01, 0.1, 1, and 10 μg/mL. There were six arrays patterned with a separation of 5 mm on a 37.5 × 25 mm PC chip. These arrays could be used independently to measure the signals from all five concentrations of the target on a chip. Figure 3 shows the standard curve used to quantify the concentration of RIgG–Alexa 647. Background noise (primarily backscattered laser light that was not rejected by a notch filter) from the 532-nm laser was measured by shining the laser onto the arrays in the absence of particles and immunoassay reagents. The signal was 114 ± 10 photons/s. Nonspecific binding of a different target molecule to the PC system was measured using the same target molecule as that employed for the 96-well plate-reader detection. The signal arising from the test was 137 ± 29 photons/s, which was not significant in comparison to the background signal.

Schematic of the FRET homogeneous immunoassay on a photonic crystal array

Detection of RIgG using a photonic crystal array. Incubation times of 20 min (diamonds) and 1 min (circles) were used for the immune reaction of the RIgG and anti-RIgG. Concentrations of 10−3, 0.01, 0.1, 1, 10 μg/mL were measured. LODs were 10−3 and 0.1 μg/mL, respectively, for incubation times of 20 and 1 min. Error bars represent the standard deviation based on three replicates
The fluorescent particles were chosen to match the particle size with the well size (diameter of the well: 60 nm, depth: 240 nm), which was engineered to form intensified electromagnetic waves in the PC. The fluorescence of the particles was not used in this assay, but we did assess the potential for particle fluorescence to play an extrinsic role. The background FRET signal from the fluorescent particles conjugated to anti-RIgG–Alexa 555 was measured by exciting the conjugate with the 532-nm laser in the absence of RIgG–Alexa 647. The measured signal due to energy transfer from anti-RigG–Alexa 555 to the fluorescent particle was insignificant compared to the background noise.
To demonstrate the performance of FRET-based PC detection for rapid point of care, two different incubation times (20 and 1 min) were used for antibody–antibody binding (Fig. 3). The limit of detection with 20 min of incubation was 0.001 μg/mL, which was a hundred times better than the LOD obtained with 1 min of incubation. However, the sensitivity obtained from 1 min of incubation was still ten times better than the detection limit obtained using a plate reader (1 μg/mL, Fig. 2). In comparison to the plate reader, the PC array showed superior sensitivity. As a result of the enhanced sensitivity of the PC immunoplatform, a 1-min incubation could be enough to obtain the same sensitivity as afforded by the plate reader system, leading to a dramatic reduction in assay time. However, there may be some limitations on the applicability of the PC array, as the options available in terms of luminescent dyes and sources of incident light are limited by the phase-matching requirement of the array.
Conclusions
A FRET-based homogeneous immunoassay obviates the need for washing and simplifies the immunoassay. In a conventional FRET-based assay, signal strength may limit the sensitivity of the method. Migration of the FRET format to a photonic crystal array offers greatly improved sensitivity compared to a conventional plate reader, with no loss of specificity. In addition, the FRET PC array system can achieve detection levels commensurate with much more expensive and complex laboratory instruments in a time as short as 1 min.
References
Ko S, Grant SA (2006) A novel FRET-based optical fiber biosensor for rapid detection of Salmonella typhimurium. Biosens Bioelectron 21:1283–1290
Lakowicz JR (2006) Principles of fluorescence spectroscopy, 3rd edn. Springer, New York
Pulli T, Hoyhtya M, Soderlund H, Takkinen K (2005) One-step homogeneous immunoassay for small analytes. Anal Chem 77:2637–2642
Zeng Q, Zhang Y, Liu X, Tu L, Kong X, Zhang H (2012) Multiple homogeneous immunoassays based on a quantum dots–gold nanorods FRET nanoplatform. Chem Commun 48:1781–1783
Qian J, Wang C, Pan X, Liu S (2013) A high-throughput homogeneous immunoassay based on Förster resonance energy transfer between quantum dots and gold nanoparticles. Anal Chim Acta 763:43–49. doi:10.1016/j.aca.2012.12.011
Bu D, Zhuang H, Yang G, Ping X (2014) An immunosensor designed for polybrominated biphenyl detection based on fluorescence resonance energy transfer (FRET) between carbon dots and gold nanoparticles. Sensors Actuators B Chem 195:540–548. doi:10.1016/j.snb.2014.01.079
Dennis AM, Bao G (2008) Quantum dot-fluorescent protein pairs as novel fluorescence resonance energy transfer probes. Nano Lett 8(5):1439–1445
Medintz IL, Clapp AR, Mattoussi H, Goldman ER, Fisher B, Mauro JM (2003) Self-assembled nanoscale biosensors based on quantum dot FRET donors. Nat Mater 2:630–638
Wang L, Yan R, Huo Z, Wang L, Zeng J, Bao J, Wang X, Peng Q, Li Y (2005) Fluorescence resonant energy transfer biosensor based on upconversion-luminescent nanoparticles. Biosensors 44:6054–6057
Chatterjee DK, Gnanasammandhan MK, Zhang Y (2010) Small upconverting fluorescent nanoparticles for biomedical applications. Small 6(24):2781–2795. doi:10.1002/smll.201000418
Wang Y, Liu K, Liu X, Dohnalová K, Gregorkiewicz T, Kong X, Aalders MCG, Buma WJ, Zhang H (2011) Critical shell thickness of core/shell upconversion luminescence nanoplatform for FRET application. J Phys Chem Lett 2(17):2083–2088. doi:10.1021/jz200922f
Han J-H, Sudheendra L, Kim H-J, Gee SJ, Hammock BD, Kennedy IM (2012) Ultrasensitive on-chip immunoassays with a nanoparticle-assembled photonic crystal. ACS Nano 6(10):8570–8582
Han J-H, Kim H-J, Sudheendra L, Hass EA, Gee SJ, Hammock BD, Kennedy IM (2013) Electrophoretic build up of multi nanoparticle array for a highly sensitive immunoassay. Biosens Bioelectron 41:302–308
Han J-H, Kim H-J, Sudheendra L, Gee SJ, Hammock BD, Kennedy IM (2013) Photonic crystal lab-on-a-chip for detecting staphylococcal enterotoxin B at low attomolar concentration. Anal Chem 85:3104–3109
Han J-H, Wang MS, Das J, Sudheendra L, Vonasek E, Nitin N, Kennedy IM (2014) Capture and detection of T7 bacteriophages on a nanostructured interface. ACS Appl Mater Interfaces 6:4758–4765
Han J-H, Tan M, Sudheendra L, Weiss RH, Kennedy IM (2014) On-chip detection of a single nucleotide polymorphism without polymerase amplification. Nano Res 7:1302–1310. doi:10.1007/s12274-014-0494-z
Han J-H, Sudheendra L, Kim H-J, Gee SJ, Hammock BD, Kennedy IM (2012) Ultrasensitive on-chip immunoassays with a nanoparticle-assembled photonic crystal. ACS Nano 6:8570–8582. doi:10.1021/nn301656c
Shan G, Huang H, Stoutamire DW, Gee SJ, Leng G, Hammock BD (2004) A sensitive class specific immunoassay for the detection of pyrethroid metabolites in human urine. Chem Res Toxicol 17:218–225
Han J-H, Kim H-J, Sudheendra L, Hass EA, Gee SJ, Hammock BD, Kennedy IM (2012) Electrophoretic build up of multi nanoparticle array for a highly sensitive immunoassay. Biosens Bioelectron 41:302–308. doi:10.1016/j.bios.2012.08.042
Acknowledgments
The authors thank Professor B. Hammock for permission to use a plate reader system. We thank A. Guillaumin for her help in performing FRET-based immunoassays. This work was supported by the National Institute of Environmental Health Sciences (NIEHS) Superfund Basic Research Program (no. P42ES004699).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Han, JH., Sudheendra, L. & Kennedy, I.M. FRET-based homogeneous immunoassay on a nanoparticle-based photonic crystal. Anal Bioanal Chem 407, 5243–5247 (2015). https://doi.org/10.1007/s00216-015-8708-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-015-8708-0