
Abstract
Toxicity of pharmaceuticals to aquatic biota is still largely unknown, and no research on the stereoselective toxicity of chiral drugs to these organisms has been undertaken to date. Because of the lack of analytical methods available for this purpose, this manuscript deals, for the first time, with the enantioselective analysis of the non-steroidal anti-inflammatory drugs (NSAIDs) ibuprofen, naproxen and ketoprofen in freshwater fish. The method was based on the microextraction of NSAIDs from fish muscle with a supramolecular liquid made up of inverted hexagonal aggregates of decanoic acid, their enantiomeric separation by liquid chromatography onto a (R)-1-naphthylglycine and 3,5-dinitrobenzoic acid stationary phase and quantification by tandem mass spectrometry. Limits of quantitation (LOQs) for NSAID enantiomers were in the range 1.7–3.3 ng g−1. Absolute recoveries were from 97 to 104 %, which indicated the high extraction efficiency of the supramolecular solvent. Extraction equilibrium conditions were reached after 10 min which permitted fast sample treatment. Relative standard deviations for enantiomers in fish muscle were always below 6 %. Isotopically labelled internal standards were used to compensate for matrix interferences. The method in-house validation was carried out with the Oncorhynchus mykiss species, and it was applied to the determination of NSAID enantiomers in different fortified freshwater fish species (Alburnus alburnus, Lepomis gibbosus, Micropterus salmoides, O. mykiss and Cyprinus carpio).
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Information on the toxicity of pharmaceuticals to biota is still scarce [1] and mostly inaccurate [2]. Lack of toxicological data is partly due to the still unresolved analytical challenges associated with the complexity of quantifying pharmaceuticals in biological matrices, which demand exhaustive sample treatments and highly sensitive and selective detection techniques [1]. Inaccuracy of toxicological data mainly arises from the fact that enantioselective analysis of chiral drugs, which represent around 56 % of the pharmaceuticals currently in use, has been overlooked, which leads to under- or overestimation of environmental risk [2].
Exposure of biota to pharmaceuticals mainly occurs in rivers, lakes and coastal waters that receive discharge from wastewater treatment plants (WWTPs). The concentration of pharmaceuticals in such surface waters is usually in the range from nanograms per litre to micrograms per litre [3], and an increasing body of literature indicates their accumulation in the wild-caught fish population at the concentration of nanograms per gram [4–10]. There is growing concern about the effects of pharmaceuticals to aquatic organisms because of both their inherent biological activity and pseudo-persistence (i.e. environmental dissipation rates are exceeded by introduction rates from effluent loadings).
Non-steroidal anti-inflammatory drugs (NSAIDs) are chiral pharmaceuticals prescribed in high quantities over the world. Human and veterinary consumption rates for NSAIDs follow the order ibuprofen, naproxen and ketoprofen, the former being one of the top-ten drugs sold worldwide http://www.fiercepharma.com/special-reports/top-20-generic-molecules-worldwide. Ibuprofen and ketoprofen are marketed as racemic mixtures, although their therapeutic effect resides almost exclusively in the S-enantiomers, whereas naproxen is only distributed as an S-enantiomer since R-naproxen is hepatotoxic. These NSAIDs are present in WWTP influents at concentrations from the nanogram-per-litre to the microgram-per-litre level, ibuprofen reaching the highest concentrations (up to 603 μg L−1) [11]. Treatments used in WWTPs are inefficient to remove them, and accordingly, ibuprofen, ketoprofen and naproxen are also present in WWTP effluents with concentration values up to 55, 3.92 and 5.09 μg L−1, respectively [11]. The concentrations of these pharmaceuticals in surface waters, including both sea- and freshwaters, usually are in the nanogram-per-litre range [11, 12] and those present in fishes living in contaminated waters in the nanogram-per-gram one [13–15]. The enantiomeric distribution of NSAIDs in the aquatic environment depends on the extent of both the chiral inversion of (R)-enantiomers during human metabolism and the preferential biodegradation of (S)-enantiomers during wastewater treatment. Their enantiomeric fractions, defined as [S-enantiomer]/[S-enantiomer] + [R-enantiomer], are around 0.6–0.7 for ibuprofen and ketoprofen and close to 1 for naproxen [2], which indicates the preferential occurrence of S-enantiomers in the aquatic environment. Previous research has shown that chronic exposure to ibuprofen may alter reproduction [16] and development [17] in fish; however, to the best of our knowledge, no research on the stereoselective toxicity of NSAIDs to aquatic organisms has been undertaken, nor methods for their enantioselective analysis in biota have been reported so far. In fact, animal [18, 19] and human [20, 21] body fluids have been the only biological samples for which methods of determining NSAID enantiomers have been proposed so far. In order to fill this gap, this paper deals with the development of a simple and reliable method for the enantioselective determination of NSAIDs in fish tissues with the aim of fostering research able to improve our understanding on the environmental toxicity of chiral drugs.
Analytical methods for the non-enantioselective determination of NSAIDs in fish tissue involve the use of rather aggressive sample treatments combined with GC–MS or LC–MS [5–8, 14, 22]. Common sample treatments require the extraction of 0.1–2 g of fish fillet in around 10 mL of solvent, often using repetitive microwave-assisted extraction [6, 22] or sonication [6], followed by solvent evaporation to dryness [7, 8, 13] and SPE cleanup in Oasis HLB [6, 11] or Florisil [14] cartridges. Both GC–MS [14] and LC–MS [5, 7, 8, 22] have been used in studies related to fish exposure to NSAIDs, the latter being preferred because of the need of NSAID derivatization in GC. Method quantification limits (LODs) for NSAIDs are in the range 14–46 ng g−1 using GC–MS or LC–triple quadrupole (QQQ) MS [7, 8, 14], these values increasing for LC–(TOF) MS (e.g. 138–644 ng g−1 [5]) or LC–UV (e.g. 100–410 ng g−1 [6]). Concentrations of NSAIDs in fish depend on the exposure level (e.g. 21.14 and 18.74 ng g−1 after exposure to 250 μg L−1 of ibuprofen for 1 and 7 days, respectively [14]), which demands for techniques giving the lower LODs (e.g. LC–(QQQ) MS) in order to cover a wide range of lab and/or field exposure studies.
Regarding the enantioselective determination of NSAIDs, the above-described sample treatments should be also applicable to their chiral analysis; however, the ability of these approaches to keep unaltered the enantiomeric fraction of NSAIDs throughout the whole process should be proved. So, sample treatments should be simple and using mild conditions in order to avoid enantiomer conversion or degradation [23]. On the other hand, stationary phases for enantiomeric resolution of individual NSAIDs have become commercially available (http://www.sascorp.jp/english/public_html/sumichiral/d0107.pdf, http://www.sascorp.jp/english/public_html/sumichiral/d0814.pdf), so their suitability for the simultaneous analysis of these chiral drugs in fish tissue can be assessed. A key point in chiral chromatography is to prove that detection is not stereoselective (i.e. there is no enantioselective signal suppression or enhancement) which could cause deviations in the measurement of the respective enantiomeric fractions.
This paper presents the development of a method for the determination of the enantiomeric composition of ibuprofen, ketoprofen and naproxen in fish tissue based on supramolecular solvent (SUPRAS) microextraction and chiral LC–MS/MS on a (R)-1-naphthylglycine 3,5-dinitrobenzoic acid stationary phase (http://www.sascorp.jp/english/public_html/sumichiral/d0107.pdf, http://www.sascorp.jp/english/public_html/sumichiral/d0814.pdf). Selection of these NSAIDs was based on their occurrence in surface waters as well as their high human consumption. To the authors’ knowledge, this is the first reported method for the determination of enantiomers of NSAIDs in fish, which constitutes one of the most studied organisms regarding the effect of pharmaceuticals on the environment [1].
The SUPRAS selected consisted of inverted hexagonal aggregates of decanoic acid [24], synthesized by a spontaneous self-assembly process from ternary mixtures of decanoic acid, tetrahydrofuran (THF) and water at the proportions shown in the phase diagram depicted in Fig. 1. The hexagonal nanostructures making up the SUPRAS (viz. aqueous cavities surrounded by the carboxylic groups with the hydrocarbon chains dissolved in THF) are also displayed in that figure. Selection of this SUPRAS was based on its high extraction potential derived from both the possibility to act as a mixed mode extractant (viz. it provides hydrogen bonding and dispersion interactions) and the huge concentration of binding sites it offers (viz. decanoic concentration in the SUPRAS is around 0.8 mg μL−1). On the other hand, the size of the aqueous cavities can be tailored by controlling the THF-to-water ratio in the solution where decanoic acid self-assembles and, consequently, this solvent has the potential to exclude the extraction of macromolecules present in the fish. So this SUPRAS can simplify sample treatment by combining both isolation of NSAIDs and sample cleanup. In the following paragraphs, the main results of this study are described and discussed.

Experimental
Chemicals
All chemicals were of analytical reagent grade and were used as supplied. Decanoic acid (DeA) and ammonium acetate were purchased from Fluka (Buchs, Switzerland), (R)/(S)-ibuprofen (≥98 % purity) and (R)/(S)-ketoprofen (≥98 % purity) were supplied by Sigma (St. Louis, MO, USA) whereas (R)/(S)-naproxen (≤100 % purity) was obtained from European Pharmacopoeia Reference Standard (Strasbourg, France). The isotopically labelled compounds (R)/(S)-ibuprofen (U-Ring-13C6, 99 % purity), (R)/(S)-ketoprofen (13C6, 99 % purity) and (R)/(S)-naproxen (13C, 2H3, 98 % purity), acquired from ALSACHIM (Strasbourg, France), were used as internal standards (ISs). All NSAID standards contained R- and S-enantiomers in a 50:50 ratio, which was confirmed by LC–UV. Both individual stock standard (1 g L−1) and isotopically labelled internal standard (40 mg L−1) solutions of racemic NSAIDs were prepared on a weight basis in methanol and stored under dark conditions at 4 °C. They were stable for at least 2 months. Working solutions containing mixtures of NSAIDs (5 mg L−1 of each enantiomer) or ISs (4.2 mg L−1 of each enantiomer of (R)/(S)-ibuprofen and 0.84 mg L−1 of each enantiomer of (R)/(S)-ketoprofen and (R)/(S)-naproxen) were prepared in methanol and stored at 4 °C. Tetrahydrofuran (THF) and hydrochloric acid were purchased from Panreac (Barcelona, Spain) and LC-grade methanol and acetonitrile from HiPerSolv Chromanorm (Fontenay-Sous-Bois, France). Ultra-high-quality water was obtained from a Milli-Q water purification system (Millipore, Madrid, Spain).
Apparatus
The LC–MS system used was a hybrid triple quadrupole/linear ion trap Applied Biosystems MSD Sciex 4000QTRAP (Applied Biosystems, Foster City, CA, USA) coupled to a liquid chromatograph Agilent HP 1200 Series (Agilent Technologies, Palo Alto, CA, USA) with a TurboIonSpray (TIS) interface. All data were acquired and processed using Analyst 1.5.1 Software. The analytical column used for separation of NSAID enantiomers was a Sumichiral OA-2500 (stationary phase: (R)-1-naphthylglycine and 3.5-dinitrobenzoic acid, particle size 5 μm, i.d. 4.6 mm, length 250 mm) from Sumika Chemical Analysis Service (Osaka, Japan). It was preceded by a guard column (Chirex 3005 from Phenomenex, Torrance, CA, 30 mm length) with the same chiral selector, particle size and internal diameter as those of the analytical column. A magnetic stirrer BasicMagMix from Ovan (Barcelona, Spain) and a digitally regulated centrifuge Mixtasel equipped with an angle rotor 4 × 100 mL from J.P. Selecta (Abrera, Spain) were used for SUPRAS production. A homogenizer–disperser Ultra-Turrax T25 Basic from Ika (Werke, Germany), a vortex-shaker REAX Top equipped with an attachment for ten microtubes from Heidolph (Schwabach, Germany) and a high-speed brushless centrifuge MPW-350 R equipped with an angle rotor 36 × 2.2/1.5 mL from MPW Med-Instruments (Warsaw, Poland), were used for sample treatment.
Supramolecular solvent production
The following procedure, which permits to obtain a SUPRAS volume (~8.5 mL) able to treat 25 fish samples, was routinely followed. DeA (6.5 g) was dissolved in THF (4.2 mL) at room temperature in a 100-mL glass centrifuge tube. Then, 80 mL of a 10-mM hydrochloric acid aqueous solution was added. After sealing the tube with parafilm to avoid THF evaporation, the mixture was magnetically stirred for 5 min at 900 rpm, time in which the SUPRAS spontaneously formed into the bulk solution. Then, the suspension was centrifuged at 3.500 rpm for 10 min to accelerate the separation of two liquid phases, namely the SUPRAS and a DeA-poor hydro-organic solution. Next, the SUPRAS, which is less dense than the hydro-organic solution, was withdrawn using a 20-mL glass syringe, transferred to a hermetically closed storage glass vial to avoid THF losses and stored at 4 °C. Under these conditions, the solvent produced was stable for at least 1 month. This procedure is similar to that followed for the synthesis of SUPRAS obtained from other alkyl carboxylic acids (e.g. dodecanoic acid [25]). A scheme of the synthesis procedure is shown in Fig. 1. The volume of solvent obtained can be adjusted at will by varying the amount of DeA, THF and water while keeping constant their above-specified proportion.
Determination of NSAID enantiomers in fish tissue
Sample collection and pretreatment
Different species of freshwater fish, namely rainbow trout (Oncorhynchus mykiss), common bleak (Alburnus alburnus), pumpkinseed sunfish (Lepomis gibbosus), largemouth bass (Micropterus salmoides) and common carp (Cyprinus carpio), were analysed. Rainbow trout were acquired from local markets in Córdoba (south of Spain) while the rest of the fishes were captured in Breña dam, which is not impacted by effluent discharges and has a capability to house water of 100.131 hm3. The Breña dam is located in the centre-west of the province of Córdoba (Spain). The head, fishbone and backbones of fishes were removed and the muscles were filleted. Cleaned fish muscles were stored at −20 °C until analysis. After thawing, about 200 g of sample was chopped and homogenized using a homogenizer–disperser.
SUPRAS-based microextraction
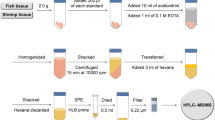
About 200 mg of chopped sample and 340 μL of SUPRAS were mixed in a 2-mL microtube Safe-Lock from Eppendorf Ibérica (Madrid, Spain). A micro Teflon-coated bar (3 × 10 mm) was introduced in the microtube to favour sample dispersion during extraction, which was made by vortex shaking at 2.500 rpm for 10 min. Then, the mixture, thermostated at 20 °C, was centrifuged at 15.000 rpm for 5 min and the extract separated from the sample residue. A volume of 100 μL of the extract was spiked with 5 μL of the IS working solution containing 8.4 mg L−1 of racemic (R)/(S)-ibuprofen and 1.7 mg L−1 of racemic (R)/(S)-ketoprofen and (R)/(S)-naproxen and directly analysed by chiral LC–MS. Figure 2 shows a scheme of the sample treatment and microextraction procedure.

Scheme of the procedure used for sample treatment
LC–MS/MS analysis
Enantiomers of NSAIDs were separated and quantified by using chiral LC coupled with a turbo ion spray (TIS) source operating in the negative ion mode and a hybrid triple quadrupole/linear ion trap analyser operating in the selected reaction monitoring (SRM) mode. The mobile phase consisted of 90 % tetrahydrofuran and 10 % ammonium acetate (50 mM) in methanol working at a variable flow: 0.5 mL min−1 from 0 to 18 min and then 1.2 mL min−1. The injection volume used was 10 μL. The temperature of the analytical and guard column was kept at 25 °C. The eluates from the analytical column were diverted by the switching valve to waste from 0 to 14 min in order to protect the mass spectrometer from the entrance of matrix components or decanoic acid. The TIS source and analyser conditions were as follows: curtain gas 30 psi; nebulizer gas 65 psi; turbo gas 30 psi; temperature of the turbo gas 425 °C; ion spray voltage −4.500 V; entrance potential −5 V; and collision gas 3.0 × 10−5 Torr. Unit resolution was used for both first and third quadrupoles. Figure 3 shows typical mass spectra for the target analytes and Table 1 the quantifier and qualifier ions used for each native NSAID and ISs. Only one SRM transition could be recorded for ibuprofen because of its poor fragmentation. Declustering potential, collision energy and collision cell exit potential parameters were optimized for each analyte (Table 1). Calibration curves were constructed from standard solutions in methanol containing the target enantiomers in the ranges 0.4–4.000, 1.0–4.000 and 1.2–4.000 μg L−1 for (R)/(S)-ibuprofen, (R)/(S)-ketoprofen and (R)/(S)-naproxen, respectively, and constant concentrations of ISs [200 μg L−1 of each enantiomer of (R)/(S)-ibuprofen and 40 μg L−1 of each enantiomer of (R)/(S)-ketoprofen and (R)/(S)-naproxen]. The concentration of the target analytes in the extract was calculated from calibration curves obtained by plotting peak area ratios (A/A IS; A = peak area of individual enantiomers and A IS = peak area of the corresponding IS) versus the concentration of analytes injected.

MS2 product ion spectra for A (R)/(S)-ibuprofen, B (R)/(S)-ketoprofen and C (R)/(S)-naproxen obtained from a standard solution containing 100 ng L−1 of each enantiomer
Results and discussion
Enantioselective separation/detection of NSAIDs in fish
LC–MS was the preferred choice for the enantioselective separation/detection of NSAIDs in fish because it far surpasses the sensitivity and selectivity of LC/UV and the sample throughput and experimental convenience of GC–MS. The chiral stationary phase (R)-1-naphthylglycine and 3.5-dinitrobenzoic acid and the mobile phase 30–80 mM ammonium acetate in methanol, which provide good resolution for the enantiomeric separation of individual NSAIDs in chiral LC–UV (http://www.sascorp.jp/english/public_html/sumichiral/d0107.pdf, http://www.sascorp.jp/english/public_html/sumichiral/d0814.pdf), were used for this purpose. Ammonium acetate was required for the enantioselective separation of NSAIDs and highly influenced retention times. Unfortunately, this additive caused suppression of NSAID ionization in LC–(ESI) MS so the concentration of ammonium acetate in the mobile phase should be kept as low as possible (e.g. 5 mM) in order to overcome signal suppression. At such low concentration, retention times became unacceptably long and, consequently, a stronger mobile phase had to be used (e.g. mixtures of THF and methanol). Figure 4A shows a representative chromatogram obtained from a standard solution of NSAIDs run under the experimental conditions proposed for their determination in fish muscle (i.e. 90 % THF and 10 % methanolic 50 mM ammonium acetate). Resolution values for the R/S-enantiomers were in the range 1.4–2.8.

LC–MS/MS selected ion chromatograms obtained from A a standard solution containing 250 μg L−1 of each enantiomer of (R)/(S)-ibuprofen (IBP), (R)/(S)-ketoprofen (KTP) and (R)/(S)-naproxen (NPX) and B, C a rainbow trout sample spiked with B 400 ng g−1 and C 15 ng g−1 of each enantiomer
Selection of SUPRAS composition
Figure 1 shows a scheme of the self-assembly process that leads to SUPRAS formation. Addition of aqueous 10 mM hydrochloric acid to a solution of decanoic acid (DeA) in THF causes the spontaneous self-assembly of the amphiphiles in oily droplets, named coacervate droplets, which associate and finally flocculate as a new liquid phase (i.e. coacervate or supramolecular solvent). Acidification of the coacervating agent (i.e. water) is required to ensure protonation of DeA (pKa = 4.8 ± 0.1).
One of the major properties of SUPRASs is that they are environment responsive. This means that tailoring of the environment or dynamically changing it provides an important means of controlling the self-assembling system. In the case of the SUPRAS here selected, the THF-to-water ratio in the bulk solution determines the relative proportion of DeA, THF and water in the solvent [24, 26]. Thus, there is a gradual incorporation of both THF and water into the SUPRAS, and consequently, it becomes increasingly diluted with respect to DeA, by increasing the THF/water percent ratio in the bulk solution. Interestingly, once formed, the composition of the SUPRAS can be reversed by modifying the THF-to-water ratio in the solution, so SUPRASs are highly adaptive.
In order to select a suitable solvent for extraction of enantiomers of NSAIDs from fish, SUPRASs of different compositions were prepared from a constant amount of DeA (6.5 g) and variable THF/water ratios (5/95–30/60, v/v, total volume = 85 mL) according to the procedure specified in ‘Experimental’. The concentration of DeA in the SUPRAS produced in this range of THF/water ratios varied from 0.76 to 0.21 mg μL−1, respectively. The volume of SUPRAS produced in each of these syntheses can be accurately predicted from the following, previously derived, equation [26]:
where y is the volume (mL) of SUPRAS, a the amount (g) of DeA and b the THF percentage (v/v).
The influence of SUPRAS composition on the extraction efficiency of NSAID enantiomers from fish was investigated by extracting around 300 mg of muscle fillet of rainbow trout spiked with 10 μL of a standard solution containing 6 mg L−1 of each native racemic (R)/(S)-ibuprofen, (R)/(S)-ketoprofen and (R)/(S)-naproxen. So, fortification for each enantiomer was 100 ng g−1. They were allowed to stand at room temperature between 0.5 and 24 h before analysis, which was made in triplicate. Extractions were carried out according to the procedure described in ‘Experimental’. The volume of SUPRAS used was 500 μL. Isotopically labelled internal standards (200 μg L−1 of each enantiomer of (R)/(S)-ibuprofen and 40 μg L−1 of each enantiomer of (R)/(S)-ketoprofen and (R)/(S)-naproxen) were added just prior to injection in LC–MS in order to correct any potential matrix effects and ensure accurate quantitation during optimization.
The recoveries found for the target enantiomers were in the range 97–102 % for all the SUPRAS compositions investigated, which indicated their suitability for the intended purpose. A SUPRAS synthesized from a solution containing the minimum percentage of THF (5 %) was selected for the extraction of the enantiomers of NSAIDs in order to consume the minimal volume of organic solvent in the treatment process (i.e. 170 μL per sample). The time of contact between NSAIDs and the matrix in fortified samples had no effect on recoveries in the interval investigated (0.5–24 h), so further optimization studies involving fortified samples were carried out by extracting them after 30 min of adding the analytes.
Optimization of SUPRAS-based microextraction
The influence of variables such as the volume of SUPRAS required per unit of sample, pH used in the synthesis of the SUPRAS and vortex-shaking and centrifugation time, on the extraction of NSAIDs, was investigated. Optimization was carried out by extracting a muscle fillet of rainbow trout, fortified with native NSAIDs (100 ng g−1 of each enantiomer), under the same conditions above described for the choice of SUPRAS composition. Selection of optimal extraction conditions was based on recoveries.
Table 2 shows the results obtained for SUPRAS volume/sample amount ratios within the range 0.7–2 μL mg−1. Results were given for (R)/(S) NSAIDs instead of individual enantiomers because of their equal behaviour regarding both recoveries and standard deviations. Quantitative recoveries were obtained for SUPRAS volume/sample amount ratios equal to or above 1.7 μL mg−1, independently of the amount of sample used. All the subsamples analysed in the interval tested (200–600 mg) were representative, so the minimum amount (i.e. 200 mg) was selected for further studies. Standard deviations below 6 % were obtained at the SUPRAS volume/sample amount ratio chosen (i.e. 1.7 μL mg−1).
The pH of the aqueous solution used for the synthesis of the SUPRAS did not influence recoveries of NSAIDs in the interval 1–4. Self-assembly was carried out at pH 2 in order to ensure maximal production of SUPRAS, since, as it has been previously mentioned, only the protonated form of DeA (pKa = 4.8 ± 0.1) is able to coacervate.
The time for vortex shaking (vibration motion = 2500 rpm) was investigated in the interval 2–30 min. Extraction equilibrium conditions were reached from 10 min, which was selected as optimal. Recoveries at 2 and 5 min were in the interval 85–88 and 91–94 % for the three NSAIDs. Effective separation of the SUPRAS extract from the sample residue was reached after centrifugation at 15.000 rpm for 5 min.
Analytical performance
Recoveries and matrix effects
Method recoveries (R), absence or presence of matrix effects (ME) and overall process efficiency (PE) were evaluated from three sets of calibration [27]. The first set (A) consisted of eight standards in methanol at concentrations of each enantiomer in the ranges 0.4–4.000, 1.0–4.000 and 1.2–4.000 μg L−1 for (R)/(S)-ibuprofen, (R)/(S)-ketoprofen and (R)/(S)-naproxen, respectively. The second set (B) was prepared by spiking the NSAID enantiomers (same concentration range as used for set 1) in SUPRAS aliquots obtained after rainbow trout extraction according to the procedure described in the section ‘SUPRAS-based microextraction’ in ‘Experimental’. In set 3 (C), rainbow trout samples were fortified and subjected to the whole procedure. Matrix effects, absolute recoveries and process efficiency were evaluated by comparing the slopes of the calibration curves obtained by sets 1 and 2, sets 2 and 3 and sets 1 and 3, respectively, according to
The ME calculated in this manner may be referred to as an absolute matrix effect; percentages higher than 100 indicate ion enhancement, while percentages lower than 100 are indicative of ion suppression.
Table 3 shows the results obtained for the different enantiomers investigated. Absolute recoveries varied in the range 97–103 %, which confirmed both the similar behaviour of enantiomers regarding extraction and the independence of recoveries of enantiomer concentration in the range evaluated.
Matrix effects were observed for all the NSAIDs (Table 3), although signal suppression progressively decreased with increasing retention time that indicated that most of the matrix components were eluted in the first minutes due to the strength of the mobile phase required for enantiomeric separation. The overall process efficiency (PE) was in the range 43–93 %.
The use of isotopically labelled standards to compensate for matrix effects was investigated by comparing the slopes of calibration curves obtained from standards (n = 8) of native NSAID enantiomers (0.4–4.000, 1.0–4.000 and 1.2–4.000 μg L−1 for (R)/(S)-ibuprofen, (R)/(S)-ketoprofen and (R)/(S)-naproxen) in methanol and fortified SUPRAS extracts obtained from a rainbow trout sample. Both methanol and extracts were also spiked with a constant concentration of IS (200 μg L−1 of each enantiomer of (R)/(S)-ibuprofen and 40 μg L−1of each enantiomer of (R)/(S)-ketoprofen and (R)/(S)-naproxen). Calibration curves were obtained by plotting peak area ratios (A/A IS; A = peak area of individual enantiomers and A IS = peak area of the corresponding IS) versus the concentration of enantiomers injected.
No statistically significant difference between both slopes was found by applying Student’s test. The experimental t values obtained for R-ibuprofen, S-ibuprofen, R-ketoprofen, S-ketoprofen, R-naproxen and S-naproxen were 0.61, 0.40, 0.78, 0.48, 0.85 and 0.88, respectively. These values were below the critical t value (3.05, significant level = 0.01). So, the isotopically labelled standards used were able to compensate for effects produced by matrix components in fish samples.
Sensitivity and linearity
Limits of detection (LODs) were calculated from six independent complete analyses of blank rainbow trout samples, according to the procedure detailed in ‘Experimental’, by using a signal-to-noise ratio of 3. They were 0.6, 0.5, 0.7, 0.8, 1.0 and 0.9 ng g−1 for R-ibuprofen, S-ibuprofen, R-ketoprofen, S-ketoprofen, R-naproxen and S-naproxen, respectively. Limits of quantitation (LOQs) were calculated alike with a signal-to-noise ratio of 10. They were in the range 1.7–3.3 ng g−1. Both LODs and LOQs were practically independent of the type of rainbow trout analysed.
Calibration curves were run from standards of NSAID enantiomers in methanol (n = 8) using the internal standard approach. The range of linearity was confirmed by visual inspection of the plot residuals versus analyte concentration; the residuals were randomly scattered within a horizontal band, and a random sequence of positive and negative residuals was obtained. Correlation between peak areas and enantiomer concentrations was determined by linear regression and 1/x weighted calibration. Correlation coefficients were in the range 0.9995–0.99998 for all enantiomers indicating good fits.
Precision
Precision was studied in terms of repeatability and expressed as relative standard deviation (RSD). For this purpose, 12 independent blank rainbow trout samples, spiked with (R)/(S)-ibuprofen, (R)/(S)-ketoprofen and (R)/(S)-naproxen at two concentrations (i.e. 15 and 400 ng g−1 of each enantiomer), were subjected to the whole analytical process. The values obtained for RSD (n = 6) were 4.8/4.2 % for (R)/(S)-ibuprofen, 4.4/4.6 % for (R)/(S)-ketoprofen and 4.6/5.3 % for (R)/(S)-naproxen for samples spiked with 15 ng g−1 of enantiomers, and they decreased at 1.4/1.7 % for (R)/(S)-ibuprofen, 1.4/1.6 % for (R)/(S)-ketoprofen and 1.3/1.7 % for (R)/(S)-naproxen as the concentration of enantiomers was 400 ng g−1. The precision was satisfactory at both low and high levels of enantiomer concentrations.
Analysis of freshwater fish
The suitability of the method for application in studies involving exposure of freshwater fish to NSAIDs was investigated. Fish individuals belonging to five different species, namely O. mykiss, A. alburnus, L. gibbosus, M. salmoides and C. carpio, were selected for this purpose. Unfortified and fortified samples were analysed according to the procedure specified in ‘Experimental’. Fortification of fish fillets for each NSAID enantiomer was performed at two concentrations (i.e. 15 and 400 ng g−1) in order to prove the suitability of the method for both low- and high-exposure studies. Samples were analysed in triplicate and both recoveries and enantiomeric fractions were measured.
Table 4 shows the results obtained. No NSAIDs were found in any of the fish analysed. Absolute recoveries were in the range 97–104 %, with relative standard deviations from 0.2 to 6 %. Recoveries were independent of the fish species analysed. Calculated enantiomeric fractions were near 0.5, which indicated the suitability of the isotopically labelled internal standard to correct for any sample matrix interferences and consequently to allow for accurate quantification of NSAID enantiomers. Figure 4 depicts, as an example, the chromatograms obtained from the analysis of a rainbow trout sample fortified with 400 ng g−1 (Fig. 4B) and 15 ng g−1 (Fig. 4C) of each NSAID enantiomer.
Conclusion
The need for research on the enantioselective toxicity of chiral drugs to aquatic biota is essential for a correct risk assessment of the presence of such contaminants in the environment. The lack of studies in this area is partly due to the lack of reported analytical methods for this purpose. Because of the difficulty to undertake multiresidue analysis with chiral stationary phases, which are relatively exclusive regarding their binding potential [23], the need for developing methods for the determination of the major groups of chiral drugs (e.g. non-steroidal anti-inflammatory drugs, analgesics, central nervous system drugs, etc.) has been highlighted [2].
The method here developed meets the analytical and operational features required to evaluate the enantioselective exposure of aquatic biota to NSAIDs. Major analytical features include (a) quantitative absolute recoveries (97–104 %) for all the enantiomers investigated, (b) method quantification limits that are 5- to 9-fold lower than those previously reported for the non-stereoselective determination of NSAIDs in biota by using GC–MS or LC–(QQQ) MS [7, 8, 14] and (c) accurate quantification of enantiomers and, consequently, accurate calculation of their enantiomeric fractions.
From an operational point of view, the proposed sample treatment is fast (e.g. sample extraction takes about 15 min and several samples can be treated simultaneously), simple (it uses conventional lab equipment such as vortex shakers or centrifuges) and cheap (340 μL of SUPRAS per sample is only required) and has high sample throughput (no evaporation or further cleanup steps are necessary).
LC–(QQQ) MS is necessary to achieve detection limits low enough to evaluate fish exposure to low concentrations of NSAIDs. The use of isotopically labelled internal standards is encouraged in order to correct for matrix ionization suppression.
References
Huerta B, Rodríguez-Mozaz S, Barceló D (2012) Pharmaceuticals in biota in the aquatic environment: analytical methods and environmental implications. Anal Bioanal Chem 404:2611–2624
Kasprzyk-Hordern B (2010) Pharmacologically active compounds in the environment and their chirality. Chem Soc Rev 39:4466–4503
Brown JN, Paxéus N, Förlin L, Larsson DGJ (2007) Variations in bioconcentration of human pharmaceuticals from sewage effluents into fish blood plasma. Environ Toxicol Pharmacol 24:267–274
Brozinski JM, Lahti M, Meierjohann A, Oikari A, Kronberg L (2013) The anti-inflammatory drugs diclofenac, naproxen and ibuprofen are found in the bile of wild fish caught downstream of a wastewater treatment plant. Environ Sci Technol 47:342–348
Peters RJB, Bolck YJC, Stolker AAM, Nielen MWF (2009) Multi-residue screening of veterinary drugs in egg, fish and meat using high-resolution liquid chromatography accurate mass time-of-flight mass spectrometry. J Chromatogr A 1216:8206–8216
Cueva-Mestanza R, Torres-Padrón ME, Sosa-Ferrera Z, Santana-Rodrígez JJ (2008) Microwave-assisted micellar extraction coupled with solid-phase extraction for preconcentration of pharmaceuticals in molluscs prior to determination by HPLC. Biomed Chromatogr 22:1115–22
Ramirez AJ, Mottaleb MA, Brooks BW, Chambliss CK (2007) Analysis of pharmaceuticals in fish using liquid chromatography-tandem mass spectrometry. Anal Chem 79:3155–3163
Ramirez AJ, Brain RA, Usenko S, Mottaleb MA, O’Donnell JG, Stahl LL, Wathen JB, Snyder BD, Pitt JL, Perez-Hurtado P, Dobbins LL, Brook BW, Chambliss CK (2009) Occurrence of pharmaceuticals and personal care products in fish: results of a national pilot study in the United States. Environ Toxicol Chem 28:2587–2597
Huerta B, Jakimska A, Gros M, Rodríguez-Mozaz S, Barceló D (2013) Analysis of multi-class pharmaceuticals in fish tissues by ultra-high-performance liquid chromatography tandem mass spectrometry. J Chormatogr A 1288:63–72
Azzouz A, Souhail B, Ballesteros E (2001) Determination of residual pharmaceuticals in edible animal tissues by continuous solid-phase extraction and gas chromatography–mass spectrometry. Talanta 84:820–828
Luo Y, Guoa W, Ngo HH, Nghiem LD, Hai FI, Zhang J, Liang S, Wang XC (2014) A review on the occurrence of micropollutants in the aquatic environment and their fate and removal during wastewater treatment. Sci Total Environ 473–474:619–641
Lolić A, Paíga P, Santos LHMLM, Ramos S, Correia M, Delerue-Matos D (2015) Assessment of non-steroidal anti-inflammatory and analgesic pharmaceuticals in seawaters of north of Portugal: occurrence and environmental risk. Sci Total Environ 508:240–250
Wang S, Oakes KD, Bragg LM, Pawliszyn J, Dixon G, Servos MR (2011) Validation and use of in vivo solid phase micro-extraction (SPME) for the detection of emerging contaminants in fish. Chemosphere 85:1472–1480
Nallani GC, Paulos PM, Constantine LA, Venables BJ, Huggett DB (2011) Bioconcentration of ibuprofen in fathead minnow (Pimephales promelas) and channel catfish (Ictalurus punctatus). Chemosphere 84:1371–1377
Boström ML, Huang C, Engström H, Larsson E, Berglund O, Jönsson Ȧ (2014) A specific, highly enriching and “green” method for hollow fiber liquid phase microextraction of ionizable pharmaceuticals from fish tissue. Anal Methods 6:6031–6037
Flippin JL, Huggett D, Foran CM (2007) Changes in the timing of reproduction following chronic exposure to ibuprofen in Japanese medaka, Oryzias latipes. Aquat Toxicol 81:73–78
David A, Pancharatna K (2009) Developmental anomalies induced by a non-selective COX inhibitor (ibuprofen) in zebrafish (Danio rerio). Environ Toxicol Pharmacol 27:390–395
Serrano-Rodríguez JM, Serrano JM, Morgaz J, Granados MM, Gómez-Villamandos RJ, Navarrete-Calvo (2014) Pharmacokinetics of the individual enantiomer S-(+)-ketoprofen after intravenous and oral administration in dogs at two dose levels. Res Vet Sci 96:523–525
Cavalganti JL, Lanchote VL, Marques MP, Valadares N, Lepera JS (2014) Analysis of ibuprofen enantiomers in rat plasma by liquid chromatography with tandem mass spectrometry. J Sep Sci 37:944–949
Bonato PS (2003) Recent advances in the determination of enantiomeric drugs and their metabolites in biological fluids by capillary electrophoresis-mediated microanalysis. Electrophoresis 24:4078–4094
Sharma P, Guttikar S, Solanki G, Patel DP, Shrivastav PS (2012) Determination of (S)-(+)- and (R)-(−)-ibuprofen enantiomers in human plasma after chiral precolumn derivatization by reversed-phase LC-ESI-MS/MS. Bioanalysis 4:2909–2927
Contardo-Jara V, Lorenz C, Pflugmacher S, Nützmann G, Kloas W, Wiegand C (2011) Exposure to human pharmaceuticals carbamazepine, ibuprofen and bezafibrate causes molecular effects in Dreissena polymorpha. Aquatic Toxicol 105:428–437
Evans SE, Kasprzyk-Hordern B (2014) Applications of chiral chromatography coupled with mass spectrometry in the analysis of chiral pharmaceuticals in the environment. Trends Environ Anal Chem 1:e34–e51
Ruiz FJ, Rubio S, Pérez-Bendito D (2007) Water-induced coacervation of alkyl carboxylic acid reverse micelles: phenomenon description and potential for the extraction of organic compounds. Anal Chem 79:7473–7485
Caballo C, Sicilia D, Rubio S (2014) Fast, simple and efficient supramolecular solvent-based microextraction of mecoprop and dichlorprop in soils prior to their enantioselective determination by liquid chromatography-tandem mass spectrometry. Talanta 119:46–52
Ballesteros-Gomez A, Ruiz FJ, Rubio S, Perez-Bendito D (2007) Determination of bisphenols A and F and their diglycidyl ethers in wastewater and river water by coacervative extraction and liquid chromatography-fluorimetry. Anal Chim Acta 603:51–61
Matuszewski BK, Constanzer ML, Chavez-Eng CM (2003) Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal Chem 75:3019–3030
Lara AB, Linares C, Sicilia D, Rubio S (2012) Enantiomer-specific determination of hexabromocyclododecane in fish by supramolecular solvent-based single-step sample treatment and liquid chromatography-tandem mass spectrometry. J Chromatogr A 752:62–68
López-Jiménez FJ, Rosales-Marcano M, Rubio S (2013) Restricted access property supramolecular solvents for combined microextraction of endocrine disruptors in sediment and sample cleanup prior to their quantification by liquid chromatography–tandem mass spectrometry. Anal Chim Acta 1303:1–8
Acknowledgments
The authors gratefully acknowledge financial support from the Spanish MICINN (Project CTQ2011-23849). Carmen Caballo acknowledges the Andalusian CICyE for a postgraduate fellowship (P09-FQM-5151).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Caballo, C., Sicilia, M.D. & Rubio, S. Enantioselective analysis of non-steroidal anti-inflammatory drugs in freshwater fish based on microextraction with a supramolecular liquid and chiral liquid chromatography–tandem mass spectrometry. Anal Bioanal Chem 407, 4721–4731 (2015). https://doi.org/10.1007/s00216-015-8675-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-015-8675-5