
Abstract
Pyrrolizidine alkaloids (PAs) are a large class of natural compounds amongst which the esterified 1,2-unsaturated necine base is toxic for humans and livestock. In the present study, a method was developed and validated for the screening and quantification of nine PAs and one PA N-oxide in teas (Camellia sinensis (L.) O. Kuntze) and herbal teas (camomile, fennel, linden, mint, rooibos, verbena). Samples were analysed by HPLC on a RP-column, packed with sub-2 μm core-shell particles, and quantified using tandem mass spectrometry operating in the positive electrospray ionisation mode. These PAs and some of their isomers were detected in a majority of the analysed beverages (50/70 samples). In 24 samples, PA concentrations were above the limit of quantification and the sum of the nine targeted PAs was between 0.021 and 0.954 μg per cup of tea. Thus, in some cases, total concentrations exceed the maximum daily intake recommended by the German Federal Institute for Risk Assessment and the UK’s Committee On Toxicity (i.e. 0.007 μg kg−1 bw).

ᅟ
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Pyrrolizidine alkaloids (PAs) are secondary metabolites naturally occurring in botanicals worldwide. Over 6,000 plant species are known to contain PAs, largely distributed in certain genera of the Asteraceae (Senecio and Eupatorium), Boraginaceae (Heliotropium, Echium and Symphytum) and Fabaceae (Crotalaria) [1]. PAs are a large class of heterocyclic compounds formed as esters of a necine base (1-hydroxymethylpyrrolizidine) with mono and dibasic acids (see Fig. 1). The necine bases may have different structures, which divide PAs into several types: crotanecine, heliotridine, otonecine, platynecine, retronecine and supinidine [2, 3]. Platynecine-type PAs are non-toxic and saturated [4]. The other types, which are 1,2-unsaturated PAs, are potentially carcinogenic, mutagenic and hepatotoxic compounds when they have at least one hydroxymethyl esterifying a necine acid [3, 5]. Consumption of PA-contaminated food or beverages may cause acute or chronic disease by inducing hepatic sinusoidal obstruction syndrome [6, 7]. Numerous cases of PA intoxications have been reported either for humans or livestock, and in several cases outcomes were lethal [8–15]. Based on different cases of intoxications and toxicological studies, several risk assessment agencies have proposed various limits for daily PA intake, the most restrictive being established by the Austrian agency, which do not authorize any PAs in final products. In Switzerland, as well as in Germany, the maximum daily PA value recommended in phytopharmaceuticals is 1 μg per person [16]. Furthermore, the German Federal Institute for Risk Assessment (BfR, Bundesinstitut für Risikobewertung) reduced the maximum value to 0.1 μg per day if the period of use exceeds 6 weeks [5]. In 2005, the Dutch National Institute for Public Health and Environment calculated a virtually daily safe dose of up to 0.00043 μg kg−1 body weight (bw) of riddelliine as harmless [1]. In 2008, the UK’s Committee On Toxicity (COT) of Chemicals in Food Consumer Products and the Environment indicated that PA doses of up to 0.007 μg kg−1 bw were unlikely to be of concern for cancer risk [17]. In 2013, the BfR, in its opinion on PAs in herbal teas and teas, conformed to the line of the COT assessment [5].

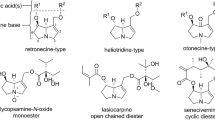
Structures, names and CAS numbers of the targeted PAs, PANO and necine base
There are numerous studies dealing with the occurrence of PAs and PA N-oxides (PANO) in honey and pollen as well as in culinary herbs, traditional Chinese medicines and herbal teas [18–22], as well as a large body which have recently been completed [23–29] as recommended by the European Food Safety Agency (EFSA) [30].
The present study reports on the development and validation of a high-performance liquid chromatography (HPLC) coupled with tandem mass spectrometry (MS/MS) to routinely quantify nine PAs and one PANO (see Fig. 1) in various kinds of teas. The objective of this work was to determine the PA concentrations in the beverages and compare daily exposures with current recommendations. For this, 70 samples of teas, herbal teas and instant teas were purchased from the Swiss market and analysed with the developed method.
Materials and method
Standards, reagents and samples
Echimidine, lycopsamine, monocrotaline, senecionine N-oxide and seneciphylline were purchased from PhytoLab (Vestenbergsgreuth, Germany); heliotrine and lasiocarpine from Amber (Orléans, France); senecionine and senkirkine from Carl Roth GmbH & Co. KG (Karlsruhe, Germany); and retrorsine as well as zinc powder were obtained from Sigma-Aldrich (Buchs, Switzerland).
Stock solutions were prepared by dissolving accurately weighted amounts of each PA in methanol. These stock solutions were further mixed and diluted to generate calibration curves ranging from 0.5 to 100 μg L−1.
Ethyl acetate was purchased from Biosolve (Valkenswaard, The Netherlands) and ammonium formate from Agilent Technologies (Plan-les-Ouates, Switzerland). All other solvents were purchased from Merck (Darmstadt, Germany). Water was purified with a Milli-Q system from Millipore (Molsheim, France).
All samples of teas, herbal teas and instant teas were obtained in Swiss supermarkets or tea shops and were stored at room temperature (RT).
Sample preparation
Boiled water (200 mL) was poured over a teabag (or 2.0 g of loose sample). After 10 min of brewing under slight agitation at RT, the solid material was removed. An aliquot (1.5 mL) was basified with a 6 M ammonia solution (30 μL) and a standard mixture (15 μL) was added for calibration or quality controls. After vortex mixing, aliquot (1 mL) was loaded on an EXtrelut NT1 column (Merck) and equilibrated for 10 min. Alkaloids were then extracted three times with 2 mL of ethyl acetate. The combined organic solutions were evaporated under nitrogen and solubilised in 100 μL of a mixture of 5 mM ammonium formate-methanol (1:1, V/V) (Fig. 2).

Workflow of sample analysis, including the optional step for the PANO reduction
For PANO analysis, an additional reduction step was necessary before purification using the EXtrelut NT1 column. Zinc powder (100 mg) was added in the infusion aliquot (1.5 mL). After 30 min in an ultrasonic bath at RT, samples were basified and purified by solid-supported liquid-liquid extraction (SLE) as described above (Fig. 2).
Plant extracts
To compare the PA extraction in boiling water (tea preparation) and in acidic water, powdered plant material (200 mg) was extracted in 0.05 M H2SO4 (10 mL) for 30 min in an ultrasonic bath (Branson, Danbury, CT). Basification and purification steps were identical to the procedure described above.
HPLC-MS/MS parameters
HPLC analysis was performed with a 1290 Infinity instrument from Agilent Technologies. For the separation, a Kinetex pentafluorophenyl (PFP) column (50 × 2.1 mm, 1.7 μm) was used (Phenomenex, Torrance, CA). The column temperature was maintained at 40 °C and the flow rate was set at 250 μL min−1. The solvent gradient started with 10 % methanol with 5 mM ammonium formate buffer at pH 4, for 1 min; was then increased linearly to 97 % over 8.5 min; remained constant for 5 min; and was then decreased over 0.1 min back to 10 % (Fig. 3). Column re-equilibration time was set at 3 min. The injection volume was 2 μL. In addition, four other HPLC columns were evaluated, namely a Phenomenex Synergi Polar RP (50 × 2 mm, 2.5 μm), a Phenomenex Kinetex C18 (100 × 2.1 mm, 2.6 μm), a Phenomenex Kinetex Phenyl-hexyl (50 × 2.1 mm, 1.7 μm) and an Agilent Zorbax Eclipse Plus C18 (50 × 2.1 mm, 1.8 μm).

HPLC-ESI-SRM chromatogram of the nine PAs and the PANO at 50 μg L−1, and the corresponding HPLC gradient
Identification and quantification were performed in the positive electrospray ionisation (ESI) mode with a jet stream source coupled to a 6460 triple quadrupole MS from Agilent Technologies. MS acquisition was based on a conventional selected reaction monitoring (SRM) mode by targeting each PA at two specific mass transitions: one for the quantification and one for the confirmation. Source parameters were set as follows: gas temperature at 300 °C, gas flow at 11 L min−1, nebulizer 35 psi, sheath gas heater at 400 °C, sheath gas flow at 12 L min−1, capillary at 3,500 V and V charging set at 0. The dwell times were fixed at 10 ms for each mass transition and the cell accelerator voltage was set at 4 V. Mass transitions, fragmentor voltages and collision energies are detailed in Table 1 for each targeted compound. MassHunter version B.06.00 software from Agilent was used for data processing.
High-resolution mass spectrometry
LC separation was performed on an UltiMate 3000 UHPLC from Dionex (Olten, Switzerland) with the same LC parameters as those described above.
Identification of compounds was performed on a Q Exactive Plus Hybrid Quadrupole-Orbitrap MS from Thermo Scientific (Ecublens, Switzerland) with targeted selected ion monitoring (t-SIM) combined with a data dependent MS2 (DD-MS2) acquisition mode. ESI source was used in positive ionisation mode and automatic parameters were set as follows: spray voltage at 3,500 V, capillary temperature at 253 °C, sheath gas at 46.25 units, auxiliary gas at 2.13 units, max. spray current at 100, probe heater temperature at 406.25 °C, S-lens RF level at 50, in-source collision induced dissociation at 0 eV and default charge state at 1. During t-SIM, exact mass of the 10 compounds was incorporated in a list of inclusion (Table 1). Isolation windows were set at 2.0 m/z and retention times at ± 0.4 min. Resolution was tuned at 70,000, the microscan level was fixed at 1, automatic gain control (AGC) at 5e4, maximum ion time at 100 ms, and loop and MSX counts both at 2. The DD-MS2 parameters were set as follows: microscans at 1, resolution at 35,000, AGC target at 2e4, maximum ion time at 100 ms, loop count at 3, MSX count at 1, topN at 3, isolation window at 2.0 m/z, normalized collision energy at 35 V, underfill ratio at 0 % and intensity threshold at 0.0. Data treatment was generated by the Xcalibur 3.0 software from Thermo Scientific.
Isomer separation
Isomeric compounds were separated on a porous graphite carbon (PGC) column (100 × 2.1 mm, 3 μm, Hypercarb, Thermo Scientific), with a HPLC 1290 Infinity instrument from Agilent Technologies. The column temperature was maintained at 60 °C and the flow rate was set at 250 μL min−1. The solvent gradient started with 30 % methanol with 0.1 % of formic acid, for 1 min; was then increased linearly to 97 % over 8.5 min; and then remained constant for 8.5 min. Column re-equilibration time was 3 min and the injection volume was 2 μL.
Compounds were targeted in SRM mode on a 6460 triple quadrupole MS from Agilent with the same MS parameters as those described in the section “HPLC-MS/MS parameters”.
Validation
To validate the method, four validation standards (VS) (1, 5, 25 and 75 μg L−1) were replicated four times and repeated in three independent series. The spiked concentrations were correlated within the range of calibration (0.5 to 100 μg L−1). PA mixtures were spiked in crude extract before the basification step. For this, a linden infusion (devoid of PAs) was chosen to be representative of all infusions.
Results and discussion
An HPLC-MS/MS method was developed to screen nine PAs and one PANO in green, black, rooibos and various herbal teas, as well as three instant tea preparations. For the quantification of these PAs, the beverage preparations were carried out according to the instructions labelled on packages to mimic home-made brewing. Thus, boiling water (200 mL) was poured on 1 teabag or 2 g of loose sample. For the three instant teas, granules (10 or 12.5 g) were dissolved in boiling water as indicated by the manufacturers.
After basification of an aliquot (1 mL) of the infusions, non-ionised compounds were extracted with SLE on chemically inert EXtrelut NT1 columns [31] as described in Fig. 2. This sample preparation was not adapted for the non-ionisable senecionine N-oxide, and an additional reduction step with zinc powder was necessary before purification by SLE to extract the corresponding PA (i.e. senecionine). Quantification of senecionine N-oxide was thus not possible directly.
Compound identification
Compounds were characterised by four identification parameters, namely retention time, two mass transitions and their mass transition ratio. The acceptance limits were set at ± 2 % for the retention time window and ± 30 % for the relative mass ratio. These criteria were established with matrix-matched calibration standards (Table 1). During the development of the method, special attention was paid to the chromatographic resolution to selectively screen and quantify each PA. Indeed, fragments of the 10 analysed compounds were often similar or identical, in particular in the case of senecionine N-oxide and retrorsine which have the same mass transitions. Thus, a baseline separation was necessary and five HPLC columns were tested. Only the Kinetex PFP column permitted full separation of the critical pair of isobars. Two eluent temperatures (40 and 50 °C) and three flow rates (250, 400 and 1,000 μL min−1), using the same gradient profile with the times geometrically scaled, were investigated [32]. The best resolution, especially for the two isobaric compounds, was obtained at 40 °C with a flow rate at 250 μL min−1 (Fig. 3).
High mass resolution analyses
Several times, only three identification criteria were fulfilled, namely retention time and two mass transitions, but their ratio was not within the tolerance of ± 30 %. The 20 problematic samples were thus further analysed by high-resolution mass spectrometry (HRMS) to differentiate possible co-eluting isobaric compounds and compare their MS data. The t-SIM DD-MS2 mode described by Kumar et al., permitted not only measurement of the exact masses of molecular ions but also their fragments produced at 35 eV [33].
Figure 4 shows HRMS results of a spiked sample (A) and two infusions containing unknown compounds co-eluting with echimidine (B and C). In each sample, the same molecular ion was detected with an exact mass of 398.2170 Th within 1 mu, corresponding to the molecular formula of echimidine, as well as a fragment at 120.0810 Th within 1 mu. In sample C, the relative abundance of the fragments produced is different to that of the echimidine standard (A) and sample B. These spectral data suggest that the co-eluting compounds are isomers of echimidine. For the latter, nine PA isomers are described [34]. As reported by Bieri et al. for tropane alkaloids, in the case of an esterified moiety with isomeric C5 acids (tiglic, senecioic and angelic), the same fragmentation pattern may occur but with different intensity ratios between the product ions [35].

HRMS data comparison of a spiked infusion at 10 μg L−1 of standards (A) and two infusions containing unknown compounds co-eluting with echimidine (B, C). Spectra A1, B1 and C1 are the extracted masses of 398.217 Th in SIM mode, with the corresponding exact masses and formulae. Spectra A2, B2, and C2 are the full MS2 spectra of ion 398.217 Th
To specifically separate the putative echimidine isomers, a PGC column was used with 0.1 % formic acid and methanol. Three isomers were detected (retention times 4.64, 5.10 and 6.60 min) (Fig. 5). Proportions of these three isomers were different in samples B and C. It is noteworthy to mention that two peaks were systematically detected in the echimidine solutions (calibration and stock solutions). However, we could not determine if the isomer was ubiquitous or not in the standard powder, which was certified with a purity > 96 %. During method development, none of the five other HPLC columns permitted separation of these echimidine isomers.

Separation of echimidine isomers using the PFP and the PGC columns. Part A, infusion spiked with 10 μg L−1 of echimidine standard. Parts B and C, two samples of herbal teas containing unknown isomers co-eluting with echimidine. SRM1 = 398 → 120; SRM2 = 398 → 83 Th
To avoid isomer problems and the need of expensive reference standards, Cramer et al. hydrolysed the 1,2-unsaturated PAs and PANOs, which allows quantification of the necine base [25]. Nevertheless, this approach has the disadvantage of requiring an approximate conversion factor to express the sum of PAs in milligrams per kilogram from the molar necine base concentration and the loss of structural information of the individual PA present in the sample, which may indicate botanical origins. The purpose of the present study was to investigate the PA profiles and their concentrations in beverages. Thus, the PA isomers were systematically quantified using the corresponding PA standard and are expressed in their PA equivalent.
Validation
Based on protocol V5 of the SFSTP’s (Société Française des Sciences et Techniques Pharmaceutiques) proposal, the validation design consisted of three independent series of one blank and six matrix-matched calibration points, replicated twice, and four VS replicated four times [36]. Response functions of all PAs were linear, within the selected range (0.5 to 100 μg L−1). Back-calculated concentrations of the VS were estimated by linear regression using different mathematical transformations (e.g. weighted factors: 1/X and 1/X 2, square-root transformation and log transformation) without correction factor. Accuracy profiles were built to visualise the most appropriate regression for each PA. For the nine PAs, confidence intervals were between 7 and 40 %, and trueness were between 91 and 114 % for the four VS (1, 5, 25 and 75 μg L−1) (see Electronic Supplementary Material (ESM) Table 4). For lycopsamine and retrorsine, the limit of quantification (LOQ) was 0.1 μg per cup of 200 mL, and 0.02 μg per cup of tea for the seven remaining compounds. LOQs were calculated as the lowest concentrations at which, confidence intervals are <40 % while, LODs correspond to a signal to noise ratio of at least three for both mass transitions (i.e. target and qualifier).
PA concentrations in beverages
A majority of the samples contained PAs. At least one PA was detected in 50 of the 70 teas analysed (see ESM Table 5). Amongst these samples, several PAs were detected in 31 beverages, and concentrations were higher than LOQs in 24 herbal teas as shown in Table 2. The sum of PAs ranged from 0.021 to 0.954 μg per cup of beverages (Table 2). Referring to the evaluation of the COT (0.007 μg per kg bw), the sum of PAs was higher than the recommended maximum dose of 0.42 μg per cup in 10 tea samples [17].
Considering all samples, the measured amounts correspond to a Margin of Exposure (MOE) lying between 200,000 and 4,400 for a 60 kg bw person consuming one cup of infusion daily. This MOE is used for carcinogenic and genotoxic substances and corresponds to the ratio between the dose response curve for the adverse effect of lasiocarpine (i.e. benchmark dose lower confidence limit for a 10 % excess cancer risk (BMDL10) of 70 μg per kg bw per day) and the level of exposure (sum of PAs in μg per cup) [37]. According to EFSA, a MOE of 10,000 or higher poses little health risk which is the case for samples 1–14 amongst the 24 (Table 2).
Botanical composition of 70 samples
Seventy samples were analysed and classified in nine groups according to their composition. Fifty-five teas were composed of a single plant only: camomile, fennel, linden, mint, rooibos, verbena and black and green teas. Conversely, 15 preparations were mixtures of three to eight plants. No PA was quantified in fennel (n = 5), black (n = 10) and green (n = 6) teas. In linden (n = 6) and plant mixtures (n = 15), some samples contained low concentrations of PAs with a maximum of 0.05 and 0.03 μg per 200 mL, respectively. For verbena, only one sample was analysed and overall PA concentration was 0.11 μg per cup. In the majority of camomile (n = 10) and mint (n = 8) samples, PAs were quantified and mean concentrations were 0.38 and 0.20 μg per cup of tea, respectively. All samples of rooibos (n = 9) contained PAs, with a mean concentration of 0.29 μg per cup of beverage (Table 3).
In rooibos, we can assume that small amounts of PAs are naturally occurring as they were systematically detected in the nine samples. To the best of our knowledge, rooibos is considered as devoid of PAs and this is the first report dealing with their presence in this species. In the other botanicals (linden, mint, camomile), PAs were not always detected. As far as we know, these species were not reported as containing PAs.
Possible explanations for the presence of PAs in herbal teas could be due to cross-contaminations with botanicals containing PAs during harvesting, storage and/or transport. Additionally, we noticed that PA concentrations in teabag samples were statistically higher than those in loose teas (Mann-Whitney U test: α 5 %, P value 1.51 %) [38].
Plant extraction
To compare our results, expressed in micrograms of PAs per cup of tea, with those published elsewhere and expressed in milligrams per kilogram of botanicals, powdered plant materials were also extracted with 0.05 M sulphuric acid as reported by Dubecke et al. [39]. Since our results showed that PA concentrations were not different between boiling water and conventional acidic extractions, PA concentrations have been converted from micrograms per cup to milligrams per kilogram (Table 5, in supplementary data) and results are in line with those published by the BfR [5]. This also means that an aqueous extraction (tea preparation) is as efficient as the standard acidic extraction procedure usually used for PA quantification in botanicals.
Conclusion
The consumption of PAs at daily doses higher than the suggested limits recommended by the Austrian, Swiss, German and British risk assessment agencies can be a problem of public health. In the present study, detection of PAs in various herbal teas is probably due to cross-contaminations with botanicals containing high levels of PAs (e.g. Senecio, Heliotropium sp.). Indeed, such species may contain PAs in the milligram per kilogram range, and thus, have the potential to taint products at the μg/kg level even with a bulk contamination as low as 0.1 %. In comparison, current industry practices are based upon the recommendations of the European Herbal Infusions Association which tolerates 2 % of foreign plants [40].
PA concentrations in herbal teas are not negligible and can induce hepatotoxicity for tea drinkers, particularly for long-term consumption. Supplementary evaluations of the toxicity of the most frequently encountered 1,2-unsaturated PAs might be necessary to define an accurate tolerable daily intake for humans and more particularly for pregnant women, toddlers and children. As several of these potent hepatotoxic alkaloids are considered as genotoxic and carcinogenic, the margin of exposure approach is preferable for the risk assessment of such substances. Yet, it is noteworthy that there is no epidemiological data suggesting a link of PAs exposure and cancer in humans to date.
Results of this survey showed that if considering a long-term daily consumption of two cups of tea of the most highly contaminated herbal tea sample (worst case scenario with product loyalty) the margin of exposure is significantly below 10,000 (i.e. 2,200 for a typical 60-kg bw person), which marks the reference level of improbable health impairment.
Overall, to secure an acceptable margin of exposure that poses little health risk, further efforts should be made to minimize PA contents in teas and herbal teas.
References
Codex Alimentarius Commission (2011) Discussion paper on pyrrolizidine alkaloids. CX/CF 11/5/14
Zhang F, Wang C-H, Xiong A-Z, Wang W, Yang L, Branford-White CJ, Wang Z-T, Bligh SWA (2007) Quantitative analysis of total retronecine esters-type pyrrolizidine alkaloids in plant by high performance liquid chromatography. Anal Chim Acta 605:94–101. doi:10.1016/j.aca.2007.10.021
Fu PP, Xia Q, Lin G, Chou MW (2004) Pyrrolizidine alkaloids: genotoxicity, metabolism enzymes, metabolic activation, and mechanisms. Drug Metab Rev 36:1–55. doi:10.1081/dmr-120028426
Bartkowski J-PB, Wiedenfeld H, Roeder E (1997) Quantitative photometric determination of senkirkine in Farfarae Folium. Phytochem Anal 8:1–4. doi:10.1002/(SICI)1099-1565(199701)8:1<1::AID-PCA328>3.0.CO;2-H
BfR (2013) Pyrrolizidine alkaloids in herbal teas and teas
Wiedenfeld H, Edgar J (2011) Toxicity of pyrrolizidine alkaloids to humans and ruminants. Phytochem Rev 10:137–151. doi:10.1007/s11101-010-9174-0
Li YH, Kan WLT, Li N, Lin G (2013) Assessment of pyrrolizidine alkaloid-induced toxicity in an in vitro screening model. J Ethnopharmacol 150:560–567. doi:10.1016/j.jep.2013.09.010
Bach N, Thung SN, Schaffner F (1989) Comfrey herb tea-induced hepatic veno-occlusive disease. Am J Med 87:97–99
Gyorik S, Stricker H (2009) Severe pulmonary hypertension possibly due to pyrrolizidine alkaloids in polyphytotherapy. Swiss Med Wkly 139:210–211
Kumana CR, Ng M, Lin HJ, Ko W, Wu PC, Todd D (1985) Herbal tea induced hepatic veno-occlusive disease: quantification of toxic alkaloid exposure in adults. Gut 26:101–104
McGee J, Patrick RS, Wood CB, Blumgart LH (1976) A case of veno-occlusive disease of the liver in Britain associated with herbal tea consumption. J Clin Pathol 29:788–794
Ridker PN, McDermont WV (1989) Hepatotoxicity due to comfrey herb tea. Am J Med 87:701
Roulet M, Laurini R, Rivier L, Calame A (1988) Hepatic veno-occlusive disease in newborn infant of a woman drinking herbal tea. J Pediatr 112:433–436
Spang R (1989) Toxicity of tea containing pyrrolizidine alkaloids. J Pediatr 115:1025
Sperl W, Stuppner H, Gassner I, Judmaier W, Dietze O, Vogel W (1995) Reversible hepatic veno-occlusive disease in an infant after consumption of pyrrolizidine-containing herbal tea. Eur J Pediatr 154:112–116
Conseil de l’Institut suisse des produits thérapeutiques (2006) Ordinance on complementary and herbal medicinal products
Committee on toxicity of chemicals in food consumer products and the environment (2008) Statement on pyrrolizidine alkaloids in food
Boppre M, Colegate SM, Edgar JA, Fischer OW (2008) Hepatotoxic pyrrolizidine alkaloids in pollen and drying-related implications for commercial processing of bee pollen. J Agric Food Chem 56:5662–5672. doi:10.1021/jf800568u
Coulombe Jr RA (2003) Pyrrolizidine alkaloids in foods. In: Advances in food and nutrition research, 45. Academic, 61–99. doi:http://dx.doi.org/10.1016/S1043-4526(03)45003-1
Deinzer ML, Arbogast BL, Buhler DR, Cheeke PR (1982) Gas chromatographic determination of pyrrolizidine alkaloids in goat’s milk. Anal Chem (Washington, DC, U S) 54:1811–1814. doi:10.1021/ac00248a035
Edgar JA, Roeder E, Molyneux RJ (2002) Honey from plants containing pyrrolizidine alkaloids: a potential threat to health. J Agric Food Chem 50:2719–2730. doi:10.1021/jf0114482
Kempf M, Reinhard A, Beuerle T (2010) Pyrrolizidine alkaloids (PAs) in honey and pollen-legal regulation of PA levels in food and animal feed required. Mol Nutr Food Res 54:158–168. doi:10.1002/mnfr.200900529
Bosi CF, Rosa DW, Grougnet R, Lemonakis N, Halabalaki M, Skaltsounis AL, Biavatti MW (2013) Pyrrolizidine alkaloids in medicinal tea of Ageratum conyzoides. Rev Bras Farmacogn 23:425–432. doi:10.1590/S0102-695X2013005000028
Cao Y, Colegate SM, Edgar JA (2013) Persistence of echimidine, a hepatotoxic pyrrolizidine alkaloid, from honey into mead. J Food Compos Anal 29:106–109. doi:10.1016/j.jfca.2012.11.005
Cramer L, Schiebel H-M, Ernst L, Beuerle T (2013) Pyrrolizidine alkaloids in the food chain: development, validation, and application of a new HPLC-ESI-MS/MS sum parameter method. J Agric Food Chem. doi:10.1021/jf403647u
Griffin CT, Danaher M, Elliott CT, Kennedy DG, Furey A (2013) Detection of pyrrolizidine alkaloids in commercial honey using liquid chromatography-ion trap mass spectrometry. Food Chem 136:1577–1583. doi:10.1016/j.foodchem.2012.02.112
Griffin CT, Gosetto F, Danaher M, Sabatini S, Furey A (2014) Investigation of targeted pyrrolizidine alkaloids in traditional Chinese medicines and selected herbal teas sourced in Ireland using LC-ESI-MS/MS. Food Addit Contam Part A 31:940–961. doi:10.1080/19440049.2014.900193
Koleva II, van Beek TA, Soffers AEMF, Dusemund B, Rietjens IMCM (2012) Alkaloids in the human food chain—natural occurrence and possible adverse effects. Mol Nutr Food Res 56:30–52. doi:10.1002/mnfr.201100165
Tang J, Cheng M, Hattori M (2012) Pyrrolizidine alkaloid profile in a traditional Chinese herbal medicine Chuan Zi Wan (Ligulariae Radix et Rhizoma) by liquid chromatography/electrospray ionization ion trap mass spectrometry. Anal Methods 4:2797–2808. doi:10.1039/c2ay25300c
EFSA Panel on Contaminants in the Food Chain (CONTAM) (2011) Scientific opinion on pyrrolizidine alkaloids in food and feed. EFSA J 9:2406
Crews C (2013) Methods for analysis of pyrrolizidine alkaloids. In: Mérillon J-M, Ramawat KG (eds) Natural products. Springer, Berlin, pp 1049–1068. doi:10.1007/978-3-642-22144-6_186
Guillarme D, Nguyen DTT, Rudaz S, Veuthey J-L (2008) Method transfer for fast liquid chromatography in pharmaceutical analysis: application to short columns packed with small particle. Part II: Gradient experiments. Eur J Pharm Biopharm 68:430–440. doi:http://dx.doi.org/10.1016/j.ejpb.2007.06.018
Kumar P, Rubies A, Centrich F, Granados M, Cortes-Francisco N, Caixach J, Companyo R (2013) Targeted analysis with benchtop quadrupole-orbitrap hybrid mass spectrometer: application to determination of synthetic hormones in animal urine. Anal Chim Acta 780:65–73. doi:10.1016/j.aca.2013.04.017
Mroczek T, Ndjoko-Ioset K, Glowniak K, Mietkiewicz-Capala A, Hostettmann K (2006) Investigation of Symphytum cordatum alkaloids by liquid-liquid partitioning, thin-layer chromatography and liquid chromatography-ion-trap mass spectrometry. Anal Chim Acta 566:157–166. doi:10.1016/j.aca.2006.03.016
Bieri S, Varesio E, Muñoz O, Veuthey J-L, Christen P (2006) Use of porous graphitic carbon column for the separation of natural isomeric tropane alkaloids by capillary LC and mass spectrometry. J Pharm Biomed Anal 40:545–551. doi:http://dx.doi.org/10.1016/j.jpba.2005.07.007
Hubert P, Nguyen-Huu JJ, Boulanger B, Chapuzet E, Chiap P, Cohen N, Compagnon PA, Dewé W, Feinberg M, Lallier M, Laurentie M, Mercier N, Muzard G, Nivet C, Valat L, Rozet E (2007) Harmonization of strategies for the validation of quantitative analytical procedures: a SFSTP proposal—part II. J Pharm Biomed Anal 45:70–81. doi:10.1016/j.jpba.2007.06.013
EFSA (2005) Opinion of the scientific committee on a request from EFSA related to a harmonised approach for risk assessment of substances which are both genotoxic and carcinogenic. EFSA J 282:1–31
Social science statistics. http://www.socscistatistics.com/Default.aspx
Dubecke A, Beckh G, Lullmann C (2011) Pyrrolizidine alkaloids in honey and bee pollen. Food Addit Contam Part A 28:348–358. doi:10.1080/19440049.2010.541594
European Herbal Infusions Association (2014) Compendium of Guidelines for Herbal and Fruit Infusions. 5
Acknowledgments
Dr Pierre-Marie Allard and Dr Florence Mehl (School of Pharmaceutical Sciences, EPGL, Switzerland) are gratefully acknowledged for their assistance with Quadrupole-Orbitrap data acquisition and statistical analysis, respectively.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mathon, C., Edder, P., Bieri, S. et al. Survey of pyrrolizidine alkaloids in teas and herbal teas on the Swiss market using HPLC-MS/MS. Anal Bioanal Chem 406, 7345–7354 (2014). https://doi.org/10.1007/s00216-014-8142-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-014-8142-8