
Abstract
Davis’ group and Setser’s group using different experimental techniques measured water vibrational distribution for OH/OD + XH → H2O/HOD + X hydrogen abstraction reactions (X ≡ D, Br, NH2, GeH3). Theoretically, this issue has been studied by several groups: different potential energy surfaces (PESs) have been developed for each system, and quasi-classical trajectory (QCT) and quantum mechanics (QM) calculations have been performed. However, important experimental/theoretical controversies still exist. In the present work, we have revisited and performed new kinetics and dynamics calculations, comparing the theoretical and experimental results on the same footing. In general, theoretical results reproduce reasonably well experimental rate constants and total vibrational energy released to water, ~ 50–60%, although they underestimate water bending excitation. In the present work, we analyse different causes of this theory/experiment discrepancy and propose different mechanisms to explain the water bending excitation for diatom–diatom and polyatomic systems. Finally, we reflect on the ability of QCT and QM calculations to provide reasonable (although not quantitative) predictions for polyatomic reactions and observe that even using full-dimensional QM calculations on very accurate PESs, agreement with the experiment is far from that reached in the case of triatomic systems.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Reactions with hydroxyl radicals have attracted experimental and theoretical interest because these free radicals are prominent in the atmosphere and in combustion, producing innocuous water as a product: OH/OD + XH → H2O/HOD + X (X ≡ D, Br, NH2, GeH3). Due to the high dissociation energy of the new bond formed, OH, these reactions are exothermic and so a large amount of energy is available to products as translation, rotation or vibration. This product energy distribution can help with understanding of the reactive process and in the explanation of the reaction mechanism. These hydrogen abstraction reactions correspond to the heavy + light–heavy (HLH) mass combination class of reactions, characterized by early barriers in the reaction path and a high product vibrational energy fraction. In addition, another very interesting dynamics feature is the distribution of this vibrational energy between the vibrational modes in water: ν1, OH symmetric stretching, ν2, HOH bending and ν3, OH asymmetric stretching, in the case of the H2O product; and ν1, OD stretching, ν2, HOD bending and ν3, OH stretching, in the case of the HOD product.
In general, theoretical/experimental agreement worsens as more detailed dynamical magnitudes are measured: from total available energy to fraction of energy as vibration and finally to population in the water vibrational modes, in the latter case related to theoretical and experimental difficulties in its description. Experimentally, these water vibrational populations have been observed by cross-beam experiments [1] or by chemiluminescence infrared [2] techniques. In the first case, at fixed collision energy for the OH + D2 reaction, Davis et al. obtained two sharp peaks associated with two quanta and one quantum in the OD stretching mode of the HOD product. From this time-of-flight (TOF) spectra, Davis et al. obtained the final vibrational distribution from Gaussian convolution simulations. However, this method is not without risk, because although the construction of a final distribution from several Gaussian functions is unique, the reverse way is not unique. In the second case, Setser et al. at fixed room temperature obtained the water vibrational spectra and, from them, the vibrational distributions. Theoretically, while the description of diatomic products is easy and straightforward, the description of triatomic products, as in the water case (and in general polyatomic products), is rather more complicated. Basically, two methods have been developed: fast Fourier transform (FFT) [3, 4] and normal-mode analysis (NMA) [5]. Finally, to make things worse, the theory/experiment comparison has not always been made on the same footing, i.e. different physical properties are measured in each case. As a consequence of this issue, which is usually not considered in the literature, serious theory/experiment discrepancies have been reported. An important question arises at this point: Can theoretical calculations simulate these fine experiments? Is it possible to achieve quantitative accuracy?
Using several hydrogen abstraction reactions yielding water as product, the main objectives of the present paper are: (1) to re-analyse the theory/experiment comparison on the same footing, (2) to propose water bending excitation mechanisms and (3) to think about the capacity and limitations of theoretical methods to simulate experimental water vibrational distributions. The paper is organized as follows: Sect. 2 is devoted to the presentation of the theoretical tools used in the present work, where variational transition-state theory (VTST) for the kinetics analysis and quasi-classical trajectory (QCT) calculations for the dynamics analysis are detailed. In addition, the PESs for each reaction analysed are also presented. Results and discussion are presented in Sect. 3. The VTST and QCT results on different PESs are compared with QM results previously reported and with experimental data. In addition, possible causes for water bending excitation are discussed and different mechanisms are proposed. The main conclusions are presented in Sect. 4. Finally, note that the present paper is not a perspective nor a review about the kinetics and dynamics studies on these reactions and so only the most recent or most interesting literature has been used in the present comparison.
2 Theoretical tools
2.1 Previous considerations
The following hydrogen abstraction reactions have been considered:
where H2O (ν1, ν2, ν3) and HOD (ν1, ν2, ν3), the focus of the theory/experimental comparison performed in the present work, represent water vibrational distributions with ν1 and ν3 being stretching modes and ν2 the bending mode. Experimentally, the assignment of these distributions is complicated due to collisional coupling of populations: in the case of the H2O, due to the proximity of the symmetric and asymmetric stretching modes, ν1: 3756 cm−1 and ν3: 3657 cm−1, only the populations of the (ν1,3, ν2) states can be assigned, where ν1,3 = ν1 + ν3 refers to combined stretch modes. In the case of the HOD product, the coupling is due to 2:1 quasi-degeneracy, ν1 (OD): 2724 cm−1 and 2ν2: 2782 cm−1 (i.e. two quanta in the bending mode) and so only the populations of the (ν1,2, ν3) states are assigned, ν1,2 being the population of the combined ν1 + ν2 modes and ν3 the pure stretch population of the OH mode.
The studied reactions present a wide variety of barrier heights, from 5.8 kcal mol−1 to barrierless, and reaction heats, from − 10.0 to − 32.4 kcal mol−1 [6,7,8,9,10,11,12,13,14,15,16,17,18,19]. Theoretically, the accurate description of the kinetics and dynamics of a reaction depends mainly on two factors: the dynamics method used and the potential energy surface describing the nuclear motion. In dynamics problems such as those dealt with here, QM and QCT calculations have been performed by several groups, and in the largest polyatomic systems, only the QCT method has been used. With regard to PESs, different strategies have been used in their development: valence bond–molecular mechanics (VB-MM)-based surfaces, reflecting bond properties, and molecular orbital (MO)-based surfaces, reflecting atomic properties. Recently, Warshel [20] noted that in the study of chemical reactions it is more physical to calibrate surfaces with the first method, although this statement is not universally accepted. Finally, note that both factors are intimately related, because the quality of the results of the QCT (or QM) calculations depends on the accuracy of the PES.
2.2 Potential energy surfaces
Although several surfaces have been developed [6,7,8,9,10,11] for the OH + H2 reaction, we have considered two PESs that use different strategies in their construction. One was developed by Ochoa and Clary [7] in 1998 and is a potential function expressed as a many-process expansion of rotation bond order (ROBO) potential, the LAGROBO method, and referred to here as the OC surface. The other is the most recent surface describing the reactive system developed by Zhang et al. [11] in 2016, using the fundamental invariant neural network (FI-NN) method, which is an MO surface fitted to ~ 17,000 high-level ab initio points, referred to here as the FI-NN surface. Both the OC and FI-NN surfaces have a similar energetic description, with barriers of 5.8 and 5.4 kcal mol−1 and reaction heats of − 14.9 and − 16.0 kcalmol−1, respectively; so the FI-NN surface has a slightly lower barrier and a larger available energy. With respect to the geometry of the transition state, especially the bending angle which will influence the final dynamics description of the system, both surfaces give a similar picture, with HOH´ bending angles ~ 96° and imaginary frequencies of 1102 i and 1208 i cm−1, respectively.
For the OH + HBr reaction, the first analytical PES was developed by Clary et al. [12] in 1994 and since then several surfaces have been made [13,14,15]. Here, we use the most recent PES developed by Bowman et al. [15] in 2014, which is a permutationally invariant polynomial (PIP) fitted to ~ 26,000 high-level ab initio points and referred to here as PIP. This surface presents a negative classical barrier, − 0.52 kcal mol−1, and a reaction heat of − 32.4 kcal mol−1, with the presence of intermediate complexes in the entrance and exit channels. The HOH´ bending angle in the transition state is 105.7°, with an imaginary frequency of 631 i cm−1.
In the case of the larger molecular size systems, OH + NH3 and OH + GeH4, we used PESs developed by our own group, PES-2012 [16, 17] and PES-2019 [18, 19], respectively. Basically, they are VB-MM surfaces fitted to a reduced number of high-level ab initio calculations. The barrier heights are, respectively, 3.3 and 0.1 kcal mol−1, while the heats of reaction are − 10.0 and − 32.4 kcal mol−1. Both systems show intermediate complexes in the entrance and exit channels. In the transition state, the HOH´ bending angles are ~ 105° in both reactions, with imaginary frequencies of 1716 i and 404 i cm−1.
2.3 QCT dynamics details
QCT calculations were performed on these five surfaces using the same strategy. Trajectories were begun and stopped when the reactants and products, respectively, were separated 10 Å. For each reaction, 1,000,000 trajectories were run, where the maximum value of the impact parameter, bmax, was determined by increasing the value of b until no reactive trajectories were obtained. The following bmax values were obtained: OH + D2 (OC): 1.6 Å; OH + D2 (FI-NN): 1.1 Å; OH + HBr (PIP): 7.7 Å; OH + NH3 (PES-2012): 2.9 Å; and OH + GeH4 (PES-2019): 10.4 Å. To simulate the corresponding experimental conditions, for the OH + D2 reaction the collision energy was fixed at 6.6 kcal mol−1, where the OH and D2 reactants were in their vibrational and rotational ground states; the other reactions were simulated at fixed room temperature, the reactants’ vibrational and rotational energies being selected by thermal sampling at 298 K. It is well known that QCT calculations are classical, and to deal with the zero-point energy (ZPE) violation problem, i.e. the artificial flow of energy between products, in addition to considering all trajectories in the analysis, we considered only trajectories in which both products had a vibrational energy above their ZPEs [21, 22], named here the double ZPE approach, DZPE. All QCT calculations were performed using the VENUS 96 code [23, 24].
To obtain the vibrational state distributions from the QCT calculations, once the trajectories finished, the normal-mode analysis (NMA) [5] algorithm was used to calculate the vibrational actions of the triatomic water product, H2O or HOD, which yields a triplet of non-integer vibrational actions (a1, a2, a3), which are rounded to the nearest integer values (n1, n2, n3). To deal with quantized vibrational distributions, two methods were used: the standard histogram binning (SB), which attributes the same statistical weight at each trajectory, i.e. the rounded actions are directly used; and the energy-based Gaussian binning (1 GB) [25,26,27], which assigns at each trajectory a weight for each product given by
where E(a), E(n) and Eo are, respectively, classical vibrational energy, quantum energy and zero-point energy (ZPE), and ε is kept at a small value, usually 0.05, to obtain full width at half maximum of 10%. In polyatomic reactions, two non-atomic products are formed, and so the weight assigned at each trajectory is
In the same way that an artificial flow of energy between products is possible, a flow of energy between vibrational modes is also possible, so in QCT calculations, due to their classical nature, the conservation of the ZPE per mode, even in an isolated molecule, is not guaranteed. Thus, in a previous paper [28], on studying the water bending excitation in the OH + GeH4 reaction, we found that many reactive trajectories finished with bending energy below its ZPE, even though the overall water vibrational energy was above the water ZPE.
When many degrees of freedom are involved, coupling between vibrational modes is favoured [21, 29]. To analyse this issue, we calculated the Bi,F(s) and Bi,i´(s) coupling terms, where s is the reaction coordinate. Within the Hamiltonian reaction path [30] implemented in the POLYRATE code [31], these coupling terms are given by
where the first one measures the coupling between the mode i and the reaction coordinate, F, and from them the reaction path curvature, κ(s), can be obtained:
The second coupling term measures the coupling between modes i and i´. In these expressions, \( c_{l\gamma }^{i} \left( s \right) \) is the lγ component of the eigenvector for mode i and νlγ(s) is the lγ component of the normalized gradient vector.
Finally, taking into account that the average total energy available to products is
where Ecoll, \( E_{\text{rot}}^{\text{react}} \) and ΔHr°(0 K) are, respectively, collision energy (or sampled translational energy at 298 K), reactant rotational energy and the 0 K enthalpy of reaction, the corresponding fractions of products’ energy in vibration (v), rotation (r) and translation (t) are given by
where the subindex i represents v, r or t.
2.4 Rate constant calculations
The measure of the rate constants at different temperatures gives a macroscopic view of the reactive system and represents the first test of quality of the PES used. The rate constants were calculated using the canonical variational transition-state theory (CVT) [32, 33],
where σ is the number of equivalent reactions paths (symmetry factor), kB, h and Ko are, respectively, Boltzmann’s constant, Planck’s constant and the reciprocal of the standard-state concentration in molecule cm−3. ΔG is the maximum of the free energy of activation located in the reaction coordinate, s. In this calculation, the electronic partition function of the OH reactant includes the 2Π1/2 excited state of OH (excitation energy of ΔE = 140 cm−1) [34], while the spin–orbit coupling in the transition state is assumed to be quenched (electronic partition function of 2). Note that the OC and FI-NN surfaces for the OH + H2 reaction and the PIP surface describing the OH + HBr reaction were constructed from high-level ab initio calculations data without including spin–orbit coupling and so the reactant OH electronic partition function can be included in two ways:
In the first case, we assume that the zero of energy is set to the energy of the lowest electronic state of OH, 2Π3/2, while in the latter we assume that the true ground state is ΔE/2 lower (obviously the spin–orbit excited state is ΔE/2 higher than the given state) [35]. As these surfaces do not include s–o splitting, the second way (Eq. 10) is the logical consistent alternative to consider s–o coupling and is the one we used in this work. For the polyatomic reactions, OH + NH3 and OH + GeH4, developed by our research group, this s–o coupling was indirectly considered in the PES construction (increasing the barrier height, which is equivalent to lowering the non-relativistic ground state by 1/3 ΔE), so in the kinetics calculations the first way (Eq. 9) was used. As has been shown, the spin–orbit treatment is a delicate and non-resolved problem in non-relativistic kinetics studies [36]. It represents a source of error when accurate theoretical kinetics results are compared with experimental measures, and doubtless it deserves future theoretical analysis.
The OH + HBr and OH + GeH4 very exothermic systems are barrierless reactions, and here the tunnelling effect is negligible. However, in reaction with the barrier this quantum mechanics effect is taken into account by a multidimensional tunnelling correction, κtun(T), which is here calculated using the microcanonical optimized multidimensional tunnelling approach, μOMT [37]. Therefore, the rate constants are obtained as a product of two factors:
Note that although the variational transition-state theory with multidimensional tunnelling (VTST/MT) is an approximate theory, it has been validated to describe quantum mechanics effects, such as ZPE, recrossing and tunnelling [38]. In addition, in the kinetics study of several polyatomic reactions, we tested the quality of the VTST/MT approach against the very accurate ring polymer molecular dynamics (RPMD) method [39,40,41], finding a good agreement between both theories, although the computational cost is very different. All kinetics calculations were performed using the POLYRATE code [31].
Finally, note that in the present study reactions with and without classical barrier have been considered, where the quantum mechanics tunnelling effect must be taken into account in the former. As the QCT calculations are classical methods, this quantum effect is not considered and, therefore, in the present case, QCT calculations were not performed for rate constant calculations.
3 Results and discussion
In the analysis of the results, we will go from the “macroscopic” (averaged measures) to the “microscopic” (state-to-state measures) point of view. In this voyage, we will see the description that each PES gives the reactive system changes.
3.1 Main features of the surfaces used: energy, geometry and vibrational frequencies
Obviously, dynamics properties of the reactive system, such as product vibrational distribution, are related to some features of the surface used. Therefore, we began by analysing certain features, such as energetic profile, geometry of the transition state and vibrational frequencies.
Figure 1 shows the main energetic features for the five surfaces. The classical barrier heights range from − 0.5 kcal mol−1 for the OH + HBr reaction to +5.8 kcal mol−1 for the OH + H2 reaction, while all reactions are very exothermic, from − 10.0 kcal mol−1 for the OH + H2 reaction to − 32.4 kcal mol−1 for the OH + HBr. All reactions present in the entrance channel van der Waals complexes stabilized in the range − 0.6 to − 3.2 kcal mol−1 with respect to the reactants, and they also stabilized in the exit channel (except for the OH + H2 reaction) with respect to the products from − 3.0 to − 5.6 kcal mol−1. These wells in the entrance and exit channels can influence the dynamics of the reaction. Wells in the entrance channel can have stereodynamics effects, and so the reactants are reoriented by driving the system away or towards the transition-state zone, modifying even mechanisms of reaction. In addition, they can also induce quantum resonances. Wells in the exit channels can influence products’ rotational and scattering distributions [42]. From a classical point of view, the presence of wells in the entrance and exit channels favours collisions between reactants or products, which can include energy transfer between translational, rotational and vibrational motions, thus modifying the dynamics of the reaction. A recent example of the influence of the reactant well in the dynamics was studied for the F + CHD3 reaction, which presents two channels: HF + CD3 and DF + CHD2. In this case, the DF/HF ratio upon CH vibrational excitation of the reactants presents a controversy between experiments [43,44,45] and theory [25, 46,47,48,49,50,51]. Theoretically, three surfaces were used, which yield reactant wells of 45, 249 and 363 cm−1 stabilized with respect to the reactants. The three surfaces presented different reaction cross sections and different DF/HF ratios, but none explained satisfactorily the experiments at low energies.

Energy profile for the OH + XH → H2O + X reactions, in kcal mol−1. RC and PC mean, respectively, the reactant and the product complex. For the OH + H2 reaction, the most recent surface, FI-NN, was represented, although the picture using the OC surface is similar
The transition-state geometries are plotted in Fig. 2. In these exothermic reactions, the transition state appears soon in the reactant path, and so they present “reactant-like” structures. In all reactions, the bond broken, X–H´, increases slightly in length with respect to the reactants by values between 4% (OH + GeH4) and 12% (OH + NH3). In general, all transition states present geometries close to linear, X…H´…O, except the OH + HBr reaction, which presents an angle of 143.7°, while the ν2 HOH´ bending angle is close to that of the water product. The corresponding vibrational frequencies of the imaginary normal modes are 1518 i, 1208 i, 631 i, 1716 i and 404 i cm−1, while the X–H´–O bending mode frequencies are 1120, 1046, 629, 1476 and 1056 cm−1, for the OC, FI-NN, PIP, PES-2012 and PES-2019 surfaces. The former frequencies are related to barrier height, while the latter bending frequencies show a wide range of values, from 629 to 1476. A priori, one could expect that different bending motions give different bending vibrational distributions in the water product. However, as will be shown later (Sects. 3.3 and 3.4) this does not happen, and therefore we can rule out this effect as the source of the theoretical/experimental discrepancy analysed in the present work. And what is the geometry description of the water product with these surfaces? All reactions have in common the formation of water as a product. Do the different PESs give a similar picture of this molecule? A priori, one should expect a positive answer, but this does not happen. Figure 3 plots the results for the five surfaces, together with the harmonic vibrational frequencies and experimental values [34] for comparison. All surfaces give length and angle close to experiments, with the largest difference in the HOH angle with the PIP surface as 107.3°. With respect to the vibrational frequencies, taking into account that theoretically only harmonic vibrational values are reported, agreement with experiments is reasonable, while the difference between the five PESs is about 100 cm−1.

Transition-state geometries for the five PESs. Distances in Å and angles in degrees

Water product geometry and vibrational frequencies for the five PESs: OC, FI-NN, PIP, PES-2012 and PES-2019. Values in parentheses, experimental values.31 Distances in Å, angles in degrees and vibrational frequencies in cm−1
3.2 Rate constants
Table 1 and Fig. 4 present the VTST/MT rate constants for the different systems in the temperature range 200–800 K, together with experimental values for comparison. This range was chosen as a common range for all systems, because it considers low and high temperatures. In the case of the OH + HBr reaction, very cold temperatures were experimentally determined, 20–300 K, and they will also be analysed.

VTST/MT rate constants for all reactions studied, together with experimental values (from Ref. [55]) and other theoretical results for comparison, in the common temperature range 200–800 K. For the OH + H2 reaction, VTST results from the present work; NN1/QM results from Ref. [35]; and NN1/RPMD from Ref. [53]. For the OH + HBr reaction, PIP/VTST from the present work; PIP/QCT from Ref. [15]; and PIP/QM from Ref. [54]. For the OH + NH3 and OH + GeH4 reactions, from the present work
For the OH + H2 reaction, the VTST/MT results on the OC surface present errors of about a factor 2 in the whole temperature range, 200–800 K, this being a factor of about 20 at 200 K, while on the FI-NN surface agreement with experiments (errors of ~ 10–60%) noticeably improves. This reaction presents a classical barrier, the tunnelling factor plays an important role, and these differences can be due also to the treatment of this quantum mechanical effect, although limitations of the FI-NN surface cannot be ruled out; for this reason, Zhang et al. [52] concluded that in order to achieve a complete agreement with experiments the FI-NN PES must be improved. In addition, based on a previous version, named NN1, fitted to the same ab initio points as the more recent FI-NN surface, two high-level sophisticated quantum mechanical kinetics studies were recently performed [35, 52] (Fig. 4). The theoretical rate constants are in good agreement among themselves, but they all slightly underestimate experimental values (15–20%). Based on this NN1 version, RPMD rate constants have also been reported [53] and are included in Fig. 4 for comparison. They reasonably simulate the experimental evidence, but at the lowest temperature, 200 K, the error is about a factor 3. Finally, note that the CVT/μOMT rate constants reasonably simulate the experimental data in the temperature range, and they present small deviations from the QM calculations (< 50%), which indicates that the VTST/MT method is a good tool to study the kinetics of this reaction.
OH + HBr is a barrierless reaction and here the tunnelling effect is negligible. In the common temperature range, 200–400 K (Fig. 4), the experimental rate constants are practically independent of temperature. The CVT rate constants on the PIP surface reasonably simulate the experimental values and the independence with temperature experimentally reported in this temperature range. In the transition state, the five orthogonal motions to the reaction coordinate have values of 3715, 1503, 629, 348 and 117 cm−1. The harmonic oscillator approximation used to describe the partition function for the two lowest anharmonic frequencies may be responsible for this discrepancy, although QCT/PIP calculations performed by Olivera-Filho et al. [15] using a different treatment of the reactant partition function also found a similar overestimation. These same authors concluded that the discrepancies may be due to inaccuracies of the PES and/or limitations of the dynamics tools used. Finally, Wang et al. [54] performed full-dimensional time-dependent quantum mechanics calculations. They concluded that the QM rate constants present better agreement with experiments than the previous QCT results, although the QM rate constants also overestimated the experimental values. The kinetics study at very cold temperatures (Fig. 5) merits special attention. Experimentally, rate constants in the range 20–400 K have been reported [55]. These values indicate that above 150 K the rate constants are independent of temperature, while at lower temperatures (T < 150 K), the rate constants drastically increase. The CVT (present work), QCT [15] and QM [54] simulate this behaviour, although they present different slopes. Temperatures lower than 20 K have not been analysed because there are no experimental data for comparison.

The CVT/µOMT rate constants for the OH + NH3 reaction were already reported by our group in 2013 [16]. In general, they present good agreement with experiments in the whole 230–2000 K temperature range, with errors of ~ 30%, which represents a test of quality of the PES used. With the original surface, PES-2012, we reported a classical barrier of 3.2 kcal mol−1 obtained at the CCSD(T)/aug-cc-pVTZ ab initio level used in the fitting of the PES. However, after using a higher ab initio level, i.e. CCSD(T)-F12a/aug-cc-pVTZ explicitly correlated calculations to obtain energies near the basis set limit, a value of 3.8 kcal mol−1 was obtained (results not previously published). This finding, which is within chemical accuracy, < 1 kcal mol−1, highlights the need for continuous improvement in the description of the potential energy surface describing any reactive system. In our original paper, we also studied the influence of the complex in the entrance channel on the kinetics. Using the same CVT/µOMT approach, we calculated the rate constants at room temperature taking into account this complex (two-step mechanism) as opposed to the direct mechanism. The rate constant is 1.32.10−13 cm3 molecule−1s−1, as compared to 1.60.10−13 cm3 molecule−1s−1 for the direct mechanism, indicating that at low temperatures multiple encounters in the reactant channel due to the presence of this complex diminish the rate constant (in this case, 19%). It is interesting to note that the better ab initio level used later does not modify the stability of the complex, − 1.54 kcal mol−1, in both cases.
Finally, in the barrierless OH + GeH4 polyatomic reaction the tunnelling effect is again negligible. The CVT rate constants on the modified PES-2019 surface are also shown in Table 1 and Fig. 4 and are compared with the only experimental data at room temperature. The theoretical results reproduce this experimental information, indicating that the PES-2019 is reasonably able to simulate the kinetics of this reactive system. To the best of our knowledge, no other theoretical studies (neither kinetics nor dynamics) have been published on this polyatomic reaction.
In summary, when one compares theoretical and experimental kinetics information two issues are considered: accuracy of the PES describing the nuclear motion and the quality of the dynamics tools used (with the approximations included: harmonicity, tunnelling effect, zero-point energy, recrossing, spin–orbit coupling, etc.), without forgetting the uncertainties associated with experiments. In general, taking into account all these issues and the experimental uncertainties, the kinetics of these reactions are reasonably simulated with theory: VTST/MT and PES.
3.3 Product energy distribution
Product energy distribution is an interesting average dynamics property to know as the available energy is partitioned between different motions: fractions of vibrational and rotational energy in water, fv(H2O), fr(H2O); internal energy in the co-product, fint(X) (which is zero when the co-product is an atom); and as energy in translation, ft. QCT calculations at collision energy of 6.6 kcal mol−1 for the OH + D2 reaction and at T = 298 K for the other reactions are listed in Table 2, together with the available experimental data under the same conditions for comparison.
Firstly, in general the QCT results underestimate the average total energy available to products, by amounts from 2 to 5 kcal mol−1, and this difference increases with molecular size. However, in the end this limitation is not so important in the theory/experiment comparison because of the use of average fractions. In order to analyse the effect of total available energy on this dynamics property, as an example, we report new QCT calculations for the OH + NH3 reaction at 400 K. This increases the average energy available to 15.6 kcal mol−1, thus reproducing the experimental value (Table 2). The 〈fv〉H2O practically does not change, 0.55, and neither does the fraction of energy as bending, 〈fv2〉H2O, 0.09. Therefore, we rule out the differences in total available energy as the source of the theory/experiment discrepancy in the bending population.
Secondly, in all reactions the largest energy fraction is deposited as water vibrations, about 50–60% of the available energy, in excellent agreement with the experimental consensus [2] and consistent with the release of energy in early barrier reactions associated with the heavy–light–heavy mass combination. In addition, the energy deposited as water rotation, 〈fr〉water, is small, < 10%, in accordance with the experimental evidence [1], and this contribution increases, as expected, with the molecular size, Br and Ge.
Thirdly, when we pass to state-to-state measures, < fv2 > H2O and < fv3 > HOD, the theory/experimental discrepancies increase. In H2O, < fv2 > H2O measures the fraction of the vibrational energy deposited in the bending mode. The QCT results underestimate noticeably this contribution bending. In HOD, < fv3 > HOD measures the fraction of the vibrational energy deposited in the new OH bond formed, because experimentally due to the 2:1 quasi-degeneracy, the contribution from the OD stretching mode and the bending mode is coupled. QCT results overestimate the < fv3 > HOD contribution, which is equivalent to saying that they underestimate the bending contribution. A priori, due to the strong bent character of the transition state for the OH + HBr reaction (Fig. 2), a large bending excitation could be expected. However, as we have seen in this case, this does not happen. In summary, very different reactive systems, described with PESs developed using very different strategies, give a similar picture of the water product, and in all cases underestimating the water bending contribution, which represents a serious discrepancy with regard to the experimental evidence.
3.4 Water product vibrational distribution
Next, we analysed state-to-state water vibrational distribution, which represents the finest dynamics property analysed. In the case of the OH + D2 → HOD (ν1, ν2, ν3) + D reaction (R1), the QCT calculations are compared with Davis et al.’s experiments [1]. These authors employed crossed molecular beams with vibrational state resolution at collision energy of 6.6 kcal mol−1 and found a state-resolved structure in product translational energy distribution, with four peaks, associated with the (0,0), (0,1), (1,1) and (0,2) vibrational population states, where (m,n) refers in the HOD product to excitation in the bending and local OD stretching mode, respectively. The reported vibrational populations are 3%, 30%, 11% and 56%. Note that OH local stretching (ν3) remains as a spectator mode and is not included in the analysis, while the bending motion (1,1) presents a noticeable population, 11%. Figure 6 shows the vibrational populations for these four states available for HOD using QCT calculations on the OC and FI-NN surfaces, together with QM calculations [56, 57] on the OC and NN1 surfaces and experimental data for comparison [1]. All theoretical methods and surfaces reproduce the bimodal structure experimentally reported, with the population (0, 2) > (0, 1). However, all theoretical results underestimate the bending contribution, 3% versus 11%, as was anticipated and explained in the previous section. Recently, QM calculations on the NN1 surface [57] slightly improved the bending population, ~ 8%, but they overestimated and underestimated, respectively, the (0, 1) and (0, 2) populations. Therefore, the fact that full-dimensional QM calculations performed on very accurately based ab initio surfaces in diatom–diatom reactions do not match experimental results made us somewhat pessimistic with respect to the accuracy that we might expect in the study of reactivity in larger polyatomic systems.

For the remainder of the hydrogen abstraction reactions, OH/OD + HBr, NH3 and GeH4, a comparison is made with Setser and Butkovskaya’s experimental results [2, 18]. These authors measured the nascent vibrational distributions of the water product (H2O or HOD) by analysis of the infrared chemiluminescence from the water molecule at 298 K. In these studies, assignation of water vibrational distributions is difficult due to collisional coupling of populations in the case of the H2O and 2:1 quasi-degeneracy in the case of the HOD. In the first case, therefore only the (ν1,3, ν2) populations were assigned, with ν1,3 = ν1 + ν3, while in the second case, only the (ν1,2, ν3) populations were assigned, with ν1,2 = ν1 + ν2. So for a theoretical/experimental comparison on the same footing, the theoretical results in Tables 3, 4 and 5 present these populations for the OH/OD + HBr, OH/OD + NH3 and OH/OD + GeH4 reactions, respectively. We began by analysing the OH + HBr → H2O (ν1, ν2, ν3) + Br reaction (R2) (Table 3). The theoretical P2(0-5) pure bending populations (i.e. summing over the ν1 + ν3 stretching levels) are 49:28:12:7:2:1 as compared to the experimental values 37:26:17:10:6:2, while the theoretical P1,3(0-3) populations (i.e. summing over the ν2 bending levels) are 8:40:49:3 versus the experimental values 12:42:35:10. Given the theoretical limitations previously analysed, the agreement with experiments is reasonable. So, the theory simulates the non-inverted distribution of the P2 populations and the inverted distribution of the P1,3 populations. With respect to the isotopic OD + HBr → HOD (ν1, ν2, ν3) + Br reaction (R3) (also in Table 3), the P3(0–3) pure OH stretching distribution and P1,2(0–5) are, respectively, inverted and non-inverted, thus simulating the experimental evidence. To go further into this analysis, we also tested other counting methods to solve the ZPE violation problem in QCT calculations (Tables 3, 4 and 5). Thus, if only trajectories with bending energy above its ZPE are considered, or the 1 GB approach is tested, the theoretical results do not improve.
In the case of the polyatomic OH/OD + NH3 reactions (R4 and R5), theoretical results reasonably simulate the experimental data (Table 4). So, the P1,3(0, 1) and P3(0, 1) populations for H2O and HOD, respectively, present inverted distributions, while the P2(0–2) and P1,2(0–3) populations show non-inverted distributions. However, as in the previous reactions, agreement is not quantitative. Note that the 1 GB approach worsens agreement with experiment, underestimating the populations in the stretching ground state and the bending modes. Finally, for the OH/OD + GeH4 reactions (R6 and R7) the theoretical picture is similar to that found in the previous reactions (Table 5). It is interesting to note that for the H2O product, agreement with experiments is better than for the HOD product, although the reasons for this are unclear.
In sum, the level-to-level comparison with experiments shows that the theoretical tools (dynamics method and potential energy surface) reasonably simulate this fine dynamics property, although agreement is far from quantitative.
3.5 Proposed mechanism of water bending excitation
In order to understand the microscopic mechanism for water bending mode excitation, Nizamov et al. [13] proposed a series of possible causes: energy release due to geometry changes in the transition-state zone, correlation with some initial states of reactants or coupling between bending and stretching vibrational modes along the reaction path. The combined study presented in the present paper affords us an excellent opportunity to analyse these factors. With respect to the first factor, in Fig. 2 we show the transition-state geometries for all reactions and PESs. We find X…H´…O approaches from collinear (OH + GeH4) to strongly bend (OH + HBr) and H–O–H´ water bending angles larger (OH + NH3) and smaller (OH + D2) than the final water bending angle, 104.5°. However, in spite of these geometry differences, all QCT calculations yield average energy in the bending mode of water lower than experiments.
Next, the second factor is analysed, beginning with the diatom–diatom reactions. Figure 7 plots the evolution of the vibrational frequencies along the reaction path for the OH + D2 and OH + HBr diatom–diatom reactions. The OH stretch mode (3805 cm−1) does not change along the path, and it is called spectator mode (i.e. it is related to motions which are not directly involved in the process, black line). The XH stretching mode which is broken transforms in the stretching mode of the new bond formed and is named reactive mode (red line). Finally, the lowest frequencies in reactants are named transitional modes and correspond to free rotations and translations that evolve to vibrations. In these diatom–diatom reactions, the water bending mode (blue line) proceeds from relative rotational motions in the reactants asymptote. In general, in these reactions the analysis is favoured (it is clearer) by the small number of degrees of freedom, 6 in total, 2 stretching, 1 bending and 3 transitional modes (rotations and translations). Therefore, it seems logical that the rotational excitation of the reactants, OH (0, j) + XH (0, j’), will favour the bending excitation of water. And, indeed, this is the behaviour that we have observed in different new QCT calculations increasing the rotation of the reactants (Table 6). 1,000,000 new trajectories were run for each reaction using the same QCT initial conditions as presented in Sect. 2.3, but changing the rotational number of each reactant.

Generalized normal-mode vibrational frequencies (in cm−1) as a function of the reaction coordinate, s (bohr). Values s < 0 correspond to reactants and values s > 0 to products. Upper plot for the OH + D2 reaction and lower plot for the OH + HBr reaction
In both reactions, as the rotation of the reactants increases, OH (0, j) + XH (0, j’), the energy deposited in the rotation is transferred to the water bending mode, which appears more excited. Therefore, the excitation mechanism of the water bending is due to the transfer of energy from the rotational mode in reactants to the water bending mode. In addition, for the OH(0, j) + D2(0, j´) as an example, we also analysed the role of the individual rotational excitation in the reactants, while for the OH(0,2) + D2(0,2) simultaneous combination, the water product (1,1) population is 6% (Table 6), the rotational excitation of the OH reactant [OH(0,2) + D2(0,0)] yields 4%, and the rotational excitation of the D2 reactant [OH(0,0) + D2(0,2)] yields 3%. These results show a synergic behaviour of both reactants.
For a direct comparison with the experiments, the population of each rotational level in reactants and the reactivity of each rotational state must be taken into account. Figure 8 shows the populations of the rotational levels for the reactants, OH + D2 (upper) and OH + HBr (lower) at room temperature. For the OH + D2 reaction, the most populated rotational states correspond to OH (v = 0, j = 2) + D2 (v = 0, j´ = 1), although the distributions extend to j, j´ = 7. In the case of OH + HBr, the most populated states are OH (v = 0, j = 2) + HBr (v = 0, j´ = 3), the distribution extending to j, j´ = 11. From new QCT calculations at different rotational states, the average values (Table 6) present reasonable agreement with the experimental data, although the agreement is not yet quantitative. The reasons for the theoretical/experimental discrepancy are due to the classic nature of the QCT calculations, as will later be analysed.

Reactants (OH, D2 and HBr) rotational distributions at room temperature. Upper plot for the OH + D2 reaction and lower plot for the OH + HBr reaction
Is this mechanism also valid for polyatomic reactions? As was noted above, the analysis is now more complicated due to the large number of degrees of freedom, 18 and 21 for the OH + NH3 and OH + GeH4, respectively (Fig. 9). The OH stretch mode (3805 cm−1) follows as a spectator mode (black line) and now also the two largest NH (and three largest Ge–H) stretching modes, respectively. The reactive mode (red line) corresponds to the NH (GeH) stretching mode broken in reactants which correlates with the new OH stretching mode formed. However, now the water bending mode (blue line) comes from a bending mode of reactants and not from transitional modes, which practically do not change along the reaction path. Therefore, unlike the diatom–diatom reactions, it is not expected that the rotational excitation of the reactants, OH (0, j) + XH (0, j ‘), will favour the bending excitation of water.

Generalized normal-mode vibrational frequencies (in cm−1) as a function of the reaction coordinate, s (bohr). Values s < 0 correspond to reactants and values s > 0 to products. Upper plot for the OH + NH3 reaction and lower plot for the OH + GeH4 reaction
In order to analyse this hypothesis, we performed new QCT calculations for the OH(0, 4) + NH3 and OH(0, 4) + GeH4 reactions, i.e. with the OH reactant rotationally excited, comparing the results with experiments (Table 7). Again, 1,000,000 new trajectories were run for each reaction, changing only the OH(0, j) rotational number with respect to the QCT initial conditions previously presented. In this case, we found that the energy deposited in the rotation of the reactant OH(0, 4) is not transferred to the water bending mode and so does not appear more excited. Therefore, the excitation mechanism of the water bending in polyatomic systems is different.
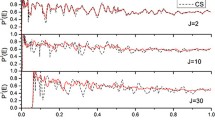
Finally, to analyse the third factor suggested by Nyzamov et al. [13] we used the coupling terms Bi,F(s) and Bi,i´(s) in the Hamiltonian reaction path frame (Eqs. 3, 4), which were independently obtained for each PES. The Bi,F(s) coupling terms measure the coupling of each mode with the reaction coordinate and can be represented by the reaction path curvature, κ(s), Eq. 5. Figure 10 shows this term for the five PESs. All systems show a sharp peak in the exit channel, from 0.5 to 1.0 bohr, associated with coupling of the stretching and bending modes with the reaction coordinate. This coupling is especially important for the OH + NH3 and OH +GeH4 polyatomic reactions, which present κ(s) values larger than for diatom–diatom reactions. Therefore, the bending mode can transfer energy with the reaction coordinate. The Bi,i´(s) coupling terms measure coupling between vibrational modes along the reaction path. Figure 11 presents these couplings for the five PESs. All systems show coupling of the bending mode with other vibrational modes, favouring the transfer of energy among them. So, in the OH + NH3 reaction the water bending mode (mode 5) transfers energy with the reactive mode and the NH2 bending modes, while in the OH + GeH4 reaction, the water bending mode (mode 6) transfers energy with the reactive mode, GeH3 bending modes and GeH stretch mode. Therefore, for polyatomic reactions, OH + NH3 and OH + GeH4, the water bending excitation mechanism is due to strong coupling of this mode with other modes along the reactive process in the exit channel.

Reaction path curvature, kappa (a.u.), along the reaction coordinate (bohr), for the five PESs analysed

Coriolis-like Bi,i´(s) coupling terms (a.u.) along the reaction path for the normal modes in the product valley. The last modes represent the lowest vibrational modes at s, which eventually become rotational or translational excitation at the product asymptote. They present small or negligible coupling, and so they are not represented. The normal modes are numbered from the highest to lowest vibrational frequencies in both X and Y axes, and the colours show the intensity of the coupling
However, as has been noted throughout the text, the theory/experiment agreement is only qualitative. A reason for this discrepancy is the limitation of QCT calculations due to their classical nature (the ZPE violation problem). As a consequence, an artificial flux of energy from the bending mode to the stretching modes is observed, which explains the low vibrational population observed in this mode as compared to experiments. Table 8 lists the percentage of reactive trajectories with bending energy below its ZPE, and Fig. 12 plots this artificial transfer of energy between modes taking, as an example, the OH + NH3 reaction, where the loss of energy of the bending mode is highlighted. Similar behaviour has been observed for the OH + H2 reaction [13] or for the OH + GeH4 reaction [28].

H2O energy in the bending mode versus the energy in the stretching modes from QCT calculations for the OH + NH3 reaction using PES-2012. Colour zones represent ZPE violation for the bending and stretching modes. Energies in kcal mol−1
In summary, it is possible that the energy transferred from the bending mode, associated with coupling between modes right after water formation and/or with the classical nature of the QCT calculations, could be somehow responsible for the lower bending vibrational populations as compared to experiments. However, this does not seem to be the complete picture. In 2011, we performed [56] an exhaustive analysis of the dynamics of the OH + D2 reaction using the OC surface with the main aim of checking the validity of the QCT method and the approaches used. Different counting methods of the QCT trajectories, such as SB, 1 GB or the more expensive action-based Gaussian binning method, GB, describe a similar picture of low bending excitation. On the other hand, even full-dimensional QM calculations on the same OC PES found low excitation. In that previous paper, we concluded that this exhaustive analysis tends to rule out classical description as the main source of the discrepancies with experiments.
4 Conclusions
In the present paper, we performed an exhaustive theoretical/experimental comparison in order to understand bending mode excitation in the water product in different hydrogen abstraction reactions of type: OH/OD + XH (X ≡ D, Br, NH2, GeH3), which presents serious theory/experiment controversies. So, theoretical results underestimate the water bending excitation experimentally observed. Using PESs from our group and from other researchers, VTST/MT and QCT calculations were performed to describe the kinetics and dynamics of these systems. Having analysed a set of kinetics and dynamics properties, from averaged to level-to-level, and having performed the theory/experiment comparison on the same footing, we conclude:
-
1.
It has been argued that a possible cause of bending excitation in the water product is the bent geometry in the transition state [13]. In the analysed reactions, the H–O…H´ angle ranges from 95° to 105.7° (as compared to the bend angle in the water product, 104.5°). However, all surfaces give similar water vibrational distributions. These results rule out this factor as the main cause of the discrepancy.
-
2.
The rate constants at different temperatures represent a macroscopic kinetics property, and in the comparison with experiments they are a first test of accuracy of the theoretical tools used, i.e. PESs and the kinetics approach. The VTST/MT results reasonably simulate the experimental evidence for all studied reactions. Noticeable is the simulation of the very cold temperatures behaviour (20–150 K) in the case of the OH + HB reaction. Therefore, the different PES developments are adequate to reproduce this kinetics property.
-
3.
The largest fraction of available energy is raised as water vibration, ~ 50–60%, with a small fraction as rotation, ~ 10%, in excellent agreement with experiments. This behaviour corresponds to exothermic reactions with an early barrier in the reaction path and the HLH mass combination. In this analysis, from “macroscopic” to “level-to-level” properties, we analysed the distribution of this vibrational energy between bending and stretching motions. We observe that the theory underestimates the water bending contribution, independently of the reaction, PES or dynamics method used.
-
4.
When the level-to-level water product vibrational distribution is analysed, we find that the theory reasonably simulates experimental measures, although the agreement is far from quantitative, with the bending population being underestimated.
-
5.
Finally, Nizamov et al. [13] proposed different mechanisms to explain water bending excitation. We quantitatively analysed these ideas and concluded that while in diatom–diatom reactions the energy transfer from the rotational modes in reactants is responsible for water bending excitation, in polyatomic systems the strong coupling of the bending mode with other modes along the reactive process is responsible for this excitation. In our analysis, the theory/experiment discrepancies are mainly associated with limitations of the QCT calculations, specifically the non-conservation of the ZPE per mode along the trajectory, which implies that a high percentage of reactive trajectories finish with bending energy below its ZPE. However, this seems not to be the complete story, because sophisticated QM calculations on the same surface give a similar dynamics description.
In sum, by analysing from “macroscopic” to “level-to-level” properties in diatom–diatom and polyatomic reactions, we observe that even using PESs developed with thousands of high-level ab initio calculations and full-dimensional QM calculations, the theory/experiment agreement is far from that reached in atom–diatom reactions and that QCT calculations, with “quantum” restrictions, reasonably simulate experimental measures, although obviously, they are far from being quantitative. Finally, at the beginning of the present paper we asked: Can theoretical calculations simulate level-to-level fine experiments? Is it possible to achieve quantitative accuracy? Throughout the text, we have observed that the answer is optimistic (reasonably optimistic) when macroscopic averaged properties are analysed, but when level-to-level properties are tested the answer is pessimistic.
References
Strazisar BR, Lin C, Davis HF (2000) Science 290:958
Butkovskaya NI, Setser DW (2003) Int Rev Phys Chem 22:1
Duchovic R, Schatz GC (1986) J Chem Phy. 84:2239
Schatz GC (1988) Comput Phys Commun 51:135
Corchado JC, Espinosa-Garcia J (2009) Phy. Chem Chem Phys 11:10157
Schatz GC, Elgersma H (1980) Chem Phys Lett 73:21
de Ochoa Aspuru G, Clary DC (1988) J Phys Chem A 102:9631
Yang M, Zhang D, Collins M, Lee S (2001) J Chem Phys 115:174
Wu G, Schatz GC, Lendvay G, Fang D, Harding L (2000) J Chem Phys 113:3150
Chen J, Xu X, Xu X, Zhang DH (2013) J Chem Phys 138:154301
Shao K, Chen J, Zhao Z, Zhang DH (2016) J Chem Phys 145:071101
Clary DC, Nyman G, Hernandez R (1994) J Chem Phys 101:3704
Nizamov B, Setser DW, Wang HB, Peslherbe GH, Hase WL (1996) J Chem Phys 105:9897
Liu JY, Li ZS, Dai ZW, Huang XR, Sun CC (2001) J Phys Chem A 105:7707
Oliveira-Filho AGS, Ornellas FR, Bowman JM (2014) J Phys Chem Lett 5:706
Monge-Palacios M, Rangel C, Espinosa-Garcia J (2013) J Chem Phys 138:084305
Monge-Palacios M, Corchado JC, Espinosa-Garcia J (2013) J Chem Phys 138:214306
Espinosa-Garcia J, Corchado JC, Butkovskaya NI, Setser DW (2019) Theor Chem Acc 138:119
Espinosa-Garcia J, Rangel C, Corchado JC (2016) Phys Chem Chem Phys 18:16941
Olsson MHM, Mavri J, Warshel A (2006) Phil Trans R Soc B 361:1417
Duff JW, Truhlar DG (1975) J Chem Phys 62:2477
Gray JC, Truhlar DG, Clemens L, Duff JW, Chapman FM, Morrell GO, Hayes EF (1978) J Chem Phys 69:240
Hu X, Hase WL, Pirraglia Y (1991) J Comput Chem 2:1014
Hase WL, Duchovic RJ, Hu X, Komornicki A, Lim KF, Lu D-H, Peslherbe GH, Swamy KN, Van de Linde SR, Varandas AJC et al (1996) QCPE Bull 16:43
Czakó G, Bowman JM (2009) J Chem Phys 131:244302
Bonnet L, Espinosa-Garcia J (2010) J Chem Phys 133:64108
Czakó G (2012) J Phys Chem A 116:7467
Espinosa-Garcia J, Corchado JC (2017) Phys Chem Chem Phys 19:1580
Hofacker GL, Levine RD (1971) Chem Phys Lett 9:617
Miller WH, Handy NC, Adams JE (1980) J Chem Phys 72:99
Zheng J, Zhang S, Lynch BJ, Corchado JC, Chuang Y-Y, Fast PL, Hu W-P, Liu Y-P, Lynch GC, Nguyen KA, Truhlar DG (2010) POLYRATE-2010-A. University of Minnesota, Minneapolis
Garrett BC, Truhlar DG (1979) J Am Chem Soc 101:4534
Truhlar DG, Isaacson AD, Garrett BC (1985) Generalized transition state theory. In: Baer M (ed) The theory of chemical reactions. CRC, Boca Raton
JANAF Thermochemical Tables, 3rd ed.; Chase MW Jr, Davies CA, Downey JR, Frurip DJ, McDonald RA, Syverud AN Eds.; (1985) National Bureau of Standards: Washington, Vol. 14
Welsch R (2018) J Chem Phys 148:204304
Mielke SL, Allison TC, Truhlar DG, Shwenke DW (1996) J Phys Chem 100:13588
Liu Y-P, Lu DH, Gonzalez-Lafont A, Truhlar DG, Garrett BC (1993) J Am Chem So. 115:7806
Mielke SL, Garret BC, Fleming DG, Truhlar DG (2015) Mol Phys 113:160
Gonzalez-Lavado E, Corchado JC, Suleimanov YV, Green WH, Espinosa-Garcia J (2014) J Phys Chem A 118:3243
Suleimanov YV, Espinosa-Garcia J (2016) J Phys Chem B 120:1418
Espinosa-Garcia J, Rangel C, Suleimanov YV (2017) Phys Chem Chem Phys 19:19341
Monge-Palacios M, Yang M, Espinosa-Garcıa J (2012) Phys Chem Chem Phys 14:4824
Zhang W, Kawamata H, Liu K (2009) Science 325:303
Yang J, Zhang D, Jiang B, Dai D, Wu G, Zhang D, Yang X (2014) J Phys Chem Lett 5:1790
Yang J, Zhang D, Chen Z, Blauert F, Jiang B, Dai D, Wu G, Zhang D, Yang X (2015) J Chem Phys 143:044316
Espinosa-Garcia J, Bravo JL (2008) J Phys Chem A 112:6059
Espinosa-Garcia J (2008) Chem Phys Lett 454:158
Czakó G, Bowman JM (2009) J Am Chem Soc 131:17534
Czakó G, Bowman JM (2011) Phys Chem Chem Phys 13:8306
Palma J, Manthe U (2015) J Phys Chem A 119:12209
Espinosa-Garcia J (2016) J Phys Chem A 120:5
Sun P, Zhang Z, Chen J, Liu S, Zhang DH (2018) J Chem Phys 149:064303
Castillo JF, Suleimanov YV (2017) Phys Chem Chem Phys 19:29170
Wang Y, Li Y, Wang D (2017) Sci Rep 7:40314
NIST Chemical Kinetics Database web page. Standard references database 17, Version 7.0 (Web Version), Release 1.6.8. data version 2015.9
Bonnet L, Espinosa-García J, Corchado JC, Liu S, Zhang DH (2011) Chem Phys Lett 516:137
Liu S, Xiao Ch, Wang T, Chen J, Yang T, Xu X, Zhang DH, Yang X (2012) Faraday Discuss 157:101
Acknowledgments
This work was partially supported by Junta de Extremadura and European Regional Development Fund, Spain (Projects No. GR18010 and IB16013). The authors thank Dr. de Oliveira-Filho and Prof. Bowman for sending us the PES of the OH + HBr reaction, Dr. Liu and Prof. Zhang for sending us the FI-NN PES of the OH + H2 reaction and Prof. Butkovskaya for sharing the revised experimental results of the OH/OD + HBr reactions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Espinosa-Garcia, J., García-Chamorro, M. & Corchado, J.C. Rethinking the description of water product in polyatomic OH/OD + XH (X ≡ D, Br, NH2 and GeH3) reactions: theory/experimental comparison. Theor Chem Acc 139, 63 (2020). https://doi.org/10.1007/s00214-020-2577-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-020-2577-0