
Abstract
The electrostatic potential at the nucleus of an atom, whether in the free state or in a molecule, is qualitatively a characteristic property of the atom. It changes remarkably little from one molecular environment to another. The energies of atoms and molecules can be expressed both rigorously and approximately in terms of the electrostatic potentials at their nuclei. Molecular energies can be written entirely as summations over atomic contributions, with no explicit interatomic terms. This provides a basis for estimating the energy of an atom in a molecule. Overall, the present study supports the validity of the atoms-in-molecules concept.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 The ambiguous nature of the chemical bond
In 1992, taking care to avoid understatement, Pauling expressed his view that “The concept of the chemical bond is the most valuable concept in chemistry. Its development over the past 150 years has been one of the greatest triumphs of the human intellect.” [1] However, just 15 years later, in a special issue of the Journal of Computational Chemistry entitled “90 Years of Chemical Bonding,” Frenking and Shaik commented that “we are still far from understanding the nature of the chemical bond” [2], which was echoed by Jacobsen, “many questions remain open in the quest for a unified and complete understanding of the true nature of the chemical bond” [3].
But is such a quest even meaningful? Jacobsen goes on to say that “it has become clear that the chemical bond per se does not exist. Each bond possesses its very own specific character,” [3]. Foroutan-Nejad et al. seem to share this view: “there is no general theoretical Scheme that may claim to encompass the description of all known types of chemical bonds.” [4]. Gillespie and Robinson go further: “a bond has no physical reality.” [5] Bader, as always is emphatic: “a ‘bond’ is neither measurable nor susceptible to theoretical definition.” [6] Instead of bonds, he speaks of “bonded interactions.”
Questioning the very concept of a chemical bond is not a recent development. Just a year after Pauling’s famous treatise [7], The Nature of the Chemical Bond, Allen and Shull noted that it is “extremely difficult to see factors significant in stabilizing a particular configuration of electrons and nuclei” [8].
This brings up a yet more fundamental issue of contention. Chemical bonds are typically viewed as linking atoms in molecules. But is it even meaningful to speak of atoms in molecules? Quantum chemistry views a molecule as a collection of electrons and nuclei. As Ivanic et al. pointed out, “the resolution of molecules in terms of atoms is not fundamental to rigorous physical theory.” [9] Parr et al. noted that the atoms-in-molecules concept is ambiguous and arbitrary [10], and Gilbert et al. put it succinctly, “There exists no quantum mechanical definition of an atom within a molecule and, therefore, any attempt to extract atomic identity is arbitrary.” [11]
Bader, developer of the Quantum Theory of Atoms in Molecules (QTAIM) [12, 13] naturally strongly disagrees [14]. However, while QTAIM is mathematically elegant, its relevance to chemical realities is increasingly being questioned [4, 15,16,17,18,19,20,21,22,23,24,25,26,27,28,29].
Can one aspire to explain the nature of bonds between atoms in molecules if it is not even established that there are indeed atoms in molecules? This dilemma has not prevented a variety of explanations of chemical bonding from being put forth: resonance, electronic charge buildup in internuclear regions, kinetic energy lowering, orbital interference, charge transfer, etc. Carbó-Dorca commented that “It might be almost impossible to describe in a short account the different manners by which the chemical bond has been described” [30].
In this brief overview, we have sought to demonstrate some fundamental disagreements that prevail in the area of chemical bonding. Our present objective is not to try to resolve any issues, but simply to provide some additional perspective.
2 Electrostatic potentials at nuclei of atoms and molecules
The electrostatic potential V0,A that is created at any nucleus A of a molecule by its electrons and the other nuclei is given rigorously, following Coulomb’s Law, by Eq. (1):
RA is the position of nucleus A, having charge ZA, and RB and ZB are the position and charge of any other nucleus B; ρ(r) is the molecule’s electronic density. The two terms in Eq. (1) represent, respectively, the positive contribution of the other nuclei and the negative one of the electrons.
If A is the nucleus of just a single free atom, then the summation over other nuclei does not appear. The electrostatic potential V0 at the nucleus of a free atom is,
Now ρ(r) is the electronic density of the atom, and r is relative to its nucleus.
An important feature of the electrostatic potential is that it is a real physical property, an observable. It can be determined experimentally, by diffraction methods [31,32,33], as well as computationally.
It has been pointed out in the past that the electrostatic potential at the nucleus of a given atom is relatively insensitive to the atom’s molecular environment [34, 35]. We will now investigate this more extensively and in greater detail.
In Table 1 are the computed electrostatic potentials at the nuclei of the indicated atoms in a variety of molecules as well as in the free atoms. The calculations were carried out at the B3P86/6–311 + G(3df,2p) level, using Gaussian 09 [36].
The most striking feature of Table 1 is how little the electrostatic potential at an atom’s nucleus changes even in quite different environments, not only neutral molecules but ions as well. For example, the V0,A of the nitrogen atoms in Table 1 are all within 1.7% of the free atom value; this includes the nitrogen in the N3¯ and NO3¯ anions, in a series of neutral molecules of differing polarities, and in the NH4+ cation. For the chlorine atoms, the V0,A are within 0.6% of the free atom V0 in a series that includes the Cl¯ anion, the highly polar LiCl and several neutral covalent molecules, among them being Cl2.
This relative insensitivity to environment is particularly noteworthy because the two contributions to V0,A, the nuclear and the electronic, can change very considerably from one molecule to another. Consider, for instance, the carbon atoms in H2C=O and (H2N)2C=O. The nuclear and electronic contributions to the electrostatic potentials at the carbon nuclei are quite different in the two molecules:+ 4.496 and– 19.179 hartrees in H2C = O and + 9.916 and– 24.567 hartrees in (H2N)2C=O. Yet the net potentials at the carbon nuclei are nearly the same in both molecules, − 14.683 and − 14.651 hartrees, respectively (Table 1).
As another example, take the phosphorus in PF3 and Cl3P=O. The nuclear and electronic contributions to the potentials at the phosphorus nuclei in these molecules differ considerably: + 9.065 and − 63.101 hartrees in the former and + 16.414 and − 70.422 hartrees in the latter. However, the resulting net potentials at the phosphorus nuclei are very similar,− 54.036 and − 54.007 hartrees.
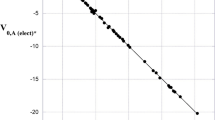
Table 1 and these examples can be interpreted as indicating that the electrostatic potential at the nucleus of an atom A in a molecule reflects almost entirely its own electronic density; the contributions of the other electrons and nuclei largely cancel. The electronic density of the atom undergoes some degree of polarization in forming the molecule and this has a minor effect upon its V0,A, which becomes more (less) negative as electronic charge is polarized toward (away from) the nucleus of A. V0,A is most negative when the atom A is part of an anion, least negative when part of a cation (Table 1). However, V0.A is in all cases very characteristic of the atom A, as can be seen in Table 1 and in the excellent correlation of the free atom V0 with their nuclear charges, Fig. 1.

Relationship between computed electrostatic potential at nucleus of free atom and nuclear charge, for atoms in Table 1 plus hydrogen atom. R2 = 0.9996
Hydrogen is an exception to the generalization that V0,A values are relatively insensitive to molecular environment; its V0,H sometimes differs very considerably from that of the free atom, as shown in Table 2. This can be understood by recognizing that hydrogen has no inner electrons that can provide the basis for a relatively stable V0,H; its only electron is involved in bonding. The absence of inner electrons makes both the hydrogen atom and the hydrogen molecule atypical, as has been pointed out a number of times [29, 37,38,39,40]. (It is ironical that H2 has traditionally been used as the model for the covalent bond.)
It is tempting to try to rationalize the V0,A values in Table 1 in terms of electronegativities. On that basis, the electrostatic potential at the nucleus of an atom would be less (more) negative as the remainder of the molecule is more (less) electron attracting. In some instances, this does account for the observed V0.A. Thus, the carbon in CF4 has a less negative V0,C than does the carbon in CCl4. The nitrogen in pyrrole has a more negative V0,N than the one in NO2.
However, electronegativity does not explain all of the trends and patterns in Table 1. For example, why are the V0,A of many of the homonuclear diatomics less negative than for the free atoms? Why is it that in F2O, which combines the two most electronegative atoms, both V0,F and V0,O are less negative than for the free atoms? The lithium in LiF, LiH and LiCl is expected to be positive in character, yet in both LiF and LiH the potential at the lithium nucleus is more negative than in the free lithium atom.
3 Atomic and molecular energies from electrostatic potentials at nuclei
In 1962, Wilson used the Hellman–Feynman theorem [41, 42] to derive an exact formula for the energy of a molecule in terms of its electronic density [43]. This was two years prior to the Hohenberg–Kohn theorem [44], which established the electronic density as the fundamental determinant of all molecular properties.
It has subsequently been demonstrated that Wilson’s result can be re-formulated to give the energy of an N-electron molecule rigorously as a function of the electrostatic potentials at its nuclei [45, 46]:
In Eq. (3), λ is a scaling parameter such that the charge on any nucleus Zi is λzi, where λ can vary between zero and one. In the actual molecule, λ = 1 and zi = Zi for each nucleus. The effect of λ is to make all of the nuclear charges increase in a concerted manner from zero to their true values.
The atomic version of Eq. (3) had already been introduced by Foldy [47] some years before Wilson’s work:
The actual application of eqs. (3) and (4) is challenging; e.g., the integrals are to be evaluated holding the numbers of electrons constant. Conceptually, however, these equations are quite significant. They show that the energy of an atom or molecule, which is a two-electron property, can be expressed rigorously in terms of the electrostatic potentials at the nuclei; these are one-electron properties but they evidently take account of electron–electron repulsion. This is a direct manifestation of the Hohenberg–Kohn theorem.
Furthermore, the molecular energy formula, Eq. (3), is simply the atomic formula, Eq. (4), summed over all of the constituent atoms. There are no explicit interaction or “mixing” terms. This is certainly consistent with the concept of atoms in molecules.
The difficulty of actually applying eqs. (3) and (4) has stimulated the development of a variety of approximate relationships between energies and electrostatic potentials at nuclei. There have been several reviews of this work [34, 35, 48,49,50,51,52,53].
An interesting aspect of this arises from the fact that approximate solutions of the Schrödinger equation often give V0 and V0,A more accurately than the total energies. For instance, Hartree–Fock atomic and molecular energies are correct through first-order but Hartree–Fock electrostatic potentials at nuclei are correct through second-order [49, 50, 54]. This means that Hartree–Fock V0 or V0,A inserted into an appropriate approximate E–V0 or E–V0,A relationship can produce energies that are better than Hartree–Fock, i.e., contain significant amounts of correlation energy. This has been demonstrated [49, 50, 55].
4 Energies of atoms in molecules
Equation (4) suggests an approximate proportionality between Eatom and \(ZV_{{0}}\), and Eq. (3) similarly suggests one between Emol and \(\sum\limits_{A} {Z_{A} V_{{{0,}A}} }\). We have investigated these possibilities, using the computed data in Tables 1 and 2 and the exact atomic and molecular energies in Tables 3 and 4.
Figures 2 and 3 show that there are indeed excellent correlations between the exact Eatom and ZV0 for the atoms in Table 3 and between the exact Emol and \(\sum\limits_{A} {Z_{A} V_{{{0,}A}} }\) for the molecules in Table 4. R2 is 1.000 in both cases. Equations (5) and (6) describe these relationships.

Relationship between exact atomic energy and product ZV0 for atoms in Table 3. R2 = 1.0000

Relationship between exact molecular energy and \(\sum\limits_{A} {{\text{Z}}_{A} {\text{V}}_{{{0,}A}} }\) for molecules in Table 4. R2 = 1.0000
Note that the slopes of the lines are essentially the same, and they both go nearly through the origins.
Equation (5) is almost identical with an atomic energy formula that comes out of Thomas–Fermi theory [56, 58,59,60], which was a predecessor of Hohenberg–Kohn density functional theory; the Thomas–Fermi formula is,
Fraga showed that when Hartree–Fock V0 are used, Eq. (7) reproduces true atomic energies to within about 2% [61].
It was later demonstrated that Eq. (7) could be extended to molecules [62]:
With Hartree–Fock V0,A, the errors are usually less than 2%.
Significantly better atomic and molecular energies are obtained with eqs. (5) and (6) and using the V0 and V0,A computed at our present B3P86/6–311 + G(3df,2p) level. For the atoms in Table 3, the average deviation from the exact energies is 0.89%; for the molecules in Table 4, it is 0.44%. While these errors are too large to allow reliable calculations of energy differences, e.g., dissociation energies, the results do indicate that eqs. (5) and (6) provide qualitatively meaningful atomic and molecular energies.
Both the rigorous molecular energy formula, Eq. (3), and the correlation in Eq. (6) express molecular energy entirely as a sum of atomic contributions. This suggests, as proposed some time ago [63], that the energy of a molecule can be apportioned between its constituent atoms in accordance with the ratio of each atom’s contribution to the sum of the contributions of all of the atoms. By that reasoning, the energy of atom M in the molecule is,
Using the data in Tables 3 and 4, we have calculated the energies Eatom,M of the atoms in the molecules in Table 4. They are listed in Table 5. We have also found the differences ΔE between the energies of the atoms in the molecules and the free atoms, Eq. (10); these are also in Table 5.
For homonuclear diatomics, each Eatom,M is simply half of Emol. It also follows from eqs. (9) and (10) that the sum of the ΔE for any molecule equals the atomization energy of that molecule, i.e., the energy needed to separate it into its constituent free atoms. For diatomics, this is the dissociation energy. Since we used the exact Eatom and Emol in eqs. (9) and (10), the sums of the ΔE in Table 5 must equal the exact atomization or dissociation energies of the molecules.
When an atom has ΔE < 0 in a particular molecule, its energy in that molecule is more negative than in the free state; ΔE > 0 indicates that the atom’s energy in the molecule is less negative than as a free atom. To some extent, the signs of the ΔE can be linked to the atoms’ electronegativities, the more electronegative having the more negative ΔE, but as with the V0,A values discussed earlier, the relationship to electronegativity is limited. Note, for instance, that in ClO the oxygen has ΔE = 0.000, while the ΔE of the chlorine is − 0.104 (Table 5).
5 Discussion and summary
The electrostatic potential at the nucleus of an atom A, labeled V0 for the free atom and V0,A when the atom is part of a molecule, is a key property of that atom. Its value changes remarkably little in going from one environment to another–free atom to nonpolar covalent to polar covalent to ionic–even though its nuclear and electronic components separately may vary considerably. The changes in these components largely cancel. The electrostatic potential at the nucleus can thus be viewed as qualitatively characterizing the atom, as shown in Table 1 and Fig. 1.
These observations bring to mind the "Quantum Chemical le Chatelier Principle" formulated by Mezey [64,65,66]. This states that when a molecule at equilibrium is subjected to a perturbation, such as a change in conformation, the effects upon the nuclear repulsion and electronic energies may be quite considerable but of similar magnitudes and opposite signs, so that they will approximately cancel. The total energy is relatively little affected.
The energies of atoms and molecules can be formulated, both rigorously and approximately, in terms of the electrostatic potentials at their nuclei. The molecular expressions are simply summations over the atomic contributions; there are no explicit interatomic ones. This provides a seemingly reasonable basis for estimating the energies of atoms in molecules.
We do not claim that the results that have been presented demonstrate conclusively the existence of atoms in molecules. It may be that this will never be definitively proven. However, a concept does not have to be rigorously established in order to be very useful, e.g., the laws of thermodynamics. It seems fair to say that our results provide strong indications that it is meaningful to view molecules and polyatomic ions as composed of atoms that keep their identities.
References
Pauling L (1992) J Chem Educ 69:519–522
Frenking G, Shaik S (2007) J Comput Chem 28:1–3
Jacobsen H (2009) J Comput Chem 30:1093–1102
Foroutan-Nejad C, Shahbazian S, Marek R (2014) Chem Eur J 20:10140–10152
Gillespie RJ, Robinson EA (2007) J Comput Chem 28:87–97
Bader RFW (2009) J Phys Chem A 113:10391–10396
Pauling L (1960) The nature of the chemical bond, 3rd edn. Cornell university press, Ithaca, NY
Allen TL, Shull H (1961) J Chem Phys 35:1644–1651
Ivanic J, Atchity GJ, Ruedenberg K (2008) Theor Chem Acc 120:281–294
Parr RG, Ayers PW, Nalewajski RF (2005) J Phys Chem A 109:3957–3959
Gilbert ATB, Gill PMW, Taylor SW (2004) J Chem Phys 120:7887–7893
Bader RFW (1990) Atoms in molecules: a quantum theory. Oxford university press, Oxford, UK
Bader RFW (1991) Chem Rev 91:893–928
Matta CF, Bader RFW (2006) J Phys Chem A 110:6365–6371
Perrin CL (1991) J Am Chem Soc 113:2865–2868
Saethre LJ, Siggel MRF, Thomas TD (1991) J Am Chem Soc 113:5224–5230
Wiener JJM, Grice ME, Murray JS, Politzer P (1996) J Chem Phys 104:5109–5111
Haaland A, Helgaker TU, Ruud K, Shorokhov DJ (2000) J Chem Educ 77:1076–1080
De Proft F, Van Alsenoy C, Peeters A, Langenaker W, Geerlings P (2002) J Comput Chem 23:1198–1209
Mitzel NW, Vojinović K, Frӧhlich R, Foerster T, Robertson HE, Borisenko KB, Rankin DWH (2005) J Am Chem Soc 127:13705–13713
Poater J, Solà M, Bickelhaupt FM (2006a) Chem Eur J 12:2889–2895
Poater J, Solà M, Bickelhaupt FM (2006b) Chem Eur J 12:2902–2905
Cerpa E, Krapp A, Vela A, Merino G (2008) Chem Eur J 14:10232–10234
Grimme S, Mück-Lichtenfeld C, Erker G, Kehr G, Wang H, Beckers H, Willner H (2009) Angew Chem Int Ed 48:2592–2595
Lane JR, Contreras-Garcia J, Piquemal J-P, Miller BJ, Kjaergaard HG (2013) J Chem Theory Comput 9:3263–3266
Spackman MA (2015) Cryst Growth Des 15:5624–5628
Wick CR, Clark T (2018) J Mol Model 24:142
Clark T, Murray JS, Politzer P (2018) Phys Chem Chem Phys 20:30076–30082
Politzer P, Murray JS (2019) Struct Chem 30:1153–1157
Carbó-Dorca R (2016) J Math Chem 54:1440–1446
Stewart RF (1979) Chem Phys Lett 65:335–342
Politzer P, Truhlar DG (eds) (1981) Chemical applications of atomic and molecular electrostatic potentials. Plenum press, New York
Feil D (1996) In Murray JS, Sen K (eds) Molecular electrostatic potentials: concepts and applications, chap 13. Elsevier, Amsterdam, pp 543–585
Politzer P (1980) Israel J Chem 19:224–232
Politzer P, Lane P, Murray JS (2002) In Sen KD (ed) Reviews of modern quantum chemistry: a celebration of the contributions of Robert G. parr, vol 1. World Scientific, Singapore, pp 63–84
Frisch MJ, Trucks GW, Schlegel HB, Scuseria G, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakaysuji X et al (2009) Gaussian 09, revision A.1, Gaussian Inc, Wallingford, CT
Bader RFW, Beddall PM (1972) J Chem Phys 56:3320–3329
Hirshfeld FL, Rzotkiewicz S (1974) Mol Phys 27:1319–1343
Dunitz JD, Seiler PJ (1983) J Am Chem Soc 105:7056–7058
Batsanov SS (1998) Struct Chem 9:65–68
Hellmann H (1937) Einführung in die Quantenchemie. Deuticke, Leipzig, p 285
Feynman RP (1939) Phys Rev 56:340–343
Wilson EB Jr (1962) J Chem Phys 36:2232–2233
Hohenberg P, Kohn W (1964) Phys Rev B 136:864–871
Politzer P, Parr RG (1974) J Chem Phys 61:4258–4262
Politzer P (2004) Theor Chem Acc 111:395–399
Foldy LL (1951) Phys Rev 83:397–399
Politzer P (1981) In Politzer P, Truhlar DG (eds) Chemical applications of atomic and molecular electrostatic potentials, chap 2. Plenum Press, New York, pp 7–28
Levy M, Clement SC, Tal Y (1981) In Politzer P, Truhlar DG (eds) Chemical applications of atomic and molecular electrostatic potentials, chap 3. Plenum press, New York, pp 29–50
Levy M, Tal Y, Clement SC (1982) J Chem Phys 77:3140–3147
Politzer P (1987) In March NH, Deb BM (eds) Single-particle density in physics and chemistry, chap 3. Academic press, New York, pp 59–72
Politzer P (2003). In: Brandas E, Kryachko E (eds) Fundamental perspectives in quantum chemistry: a tribute volume to the memory of per-olov löwdin. Kluwer, Dordrecht, pp 631–638
Politzer P, Murray JS (2002) Theor Chem Acc 108:134–142
Cohen M (1979) J Phys B 12:L219–L221
Politzer P, Sjoberg P (1983) J Chem Phys 78:7008–7009
March NH (1982) J Phys Chem 86:2262–2267
Politzer P, Lane P, Concha MC (2002) Int J Quantum Chem 90:459–463
Thomas LH (1927) Proc Camb Philos Soc 23:542–548
Fermi E (1928) Z Physik 48:73–79
March NH (1957) Adv Phys 6:1–101
Fraga S (1964) Theoret Chim Acta 2:406–410
Politzer P (1976) J Chem Phys 64:4239–4240
Politzer P, Daiker KC, Trefonas P III (1979) J Chem Phys 70:4400–4404
Mezey PG (1977) Chem Phys Lett 47:70–75
Mezey PG (1984) Internat J Quantum Chem 25:853–861
Torrent-Sucarrat M, Luis JM, Duran M, Toro-Labbé A, Solà M (2003) J Chem Phys 119:9393
Acknowledgement
We are pleased to join in honoring professor Ramon Carbó-Dorca.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Published as part of the special collection of articles “Festschrift in honour of Prof. Ramon Carbó-Dorca”.
Rights and permissions
About this article

Cite this article
Politzer, P., Murray, J.S. Electrostatic potentials at the nuclei of atoms and molecules. Theor Chem Acc 140, 7 (2021). https://doi.org/10.1007/s00214-020-02701-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-020-02701-0