
Abstract
The structures and energetics of the binuclear phospholyl vanadium carbonyls (C4H4P)2V2(CO)n (n = 7, 6, 5, 4, 3, 2, 1) have been investigated using density functional theory. The lowest energy heptacarbonyl (C4H4P)2V2(CO)7 structures resemble those of the dimanganese pentacarbonyl (C4H4P)2Mn2(CO)5 with one seven-electron donor bridging η5,η1-C4H4P ring and no direct vanadium–vanadium bond. Similarly, the lowest energy hexacarbonyl (C4H4P)2V2(CO)6 structure resembles that of the dimanganese tetracarbonyl (C4H4P)2Mn2(CO)4 with two seven-electron donor bridging η5,η1-C4H4P rings and no direct vanadium–vanadium bond. The lowest energy pentacarbonyl (C4H4P)2V2(CO)5 structure has terminal η5-C4H4P phospholyl rings and a formal V≡V triple bond of length ~ 2.45 Å similar to the experimentally known and structurally characterized cyclopentadienyl analogue (η5-C5H5)2V2(CO)5. Various combinations of V=V double bonds or V≡V triple bonds, four-electron donor bridging carbonyl groups and seven-electron donor bridging η5,η1-C4H4P rings are found in the more highly unsaturated derivatives (C4H4P)2V2(CO)n (n = 4, 3, 2) to give 17- and 18-electron vanadium configurations for triplet- and singlet-state structures, respectively. Formal V≣V quadruple bonds are found only in the highly unsaturated lowest energy singlet (C4H4P)2V2(CO)n (n = 2, 1) structures, which, however, lie at somewhat higher energies than isomeric triplet structures.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The analogy between a phosphorus atom and a CH unit based on electron count and electronegativity [1] makes the phospholyl ligand (C4H4P) an analogue of the cyclopentadienyl (C5H5 or Cp) ligand (Fig. 1). Thus, both ligands, considered as neutral species, can function as pentahapto ligands toward a metal atom. This analogy is manifest in their manganese tricarbonyl derivatives. Thus, (η5-C4H4P)Mn(CO)3 or its more readily available 3,4-dimethyl derivative (η5-3,4-C4H2Me2P)Mn(CO)3, synthesized by thermal reactions of Mn2(CO)10 with the corresponding P-phenylphosphole [2, 3], is a stable yellow solid analogous to CpMn(CO)3. In such phospholylmetal carbonyl derivatives, the phosphorus lone pair is not involved in the ligand–metal bonding.

Comparison of five-electron donor phospholyl and Cp ligands in mononuclear metal complexes with bridging µ-η5,η1-C4H4P phospholyl ligands in binuclear metal complexes
Binuclear phospholyl metal carbonyl derivatives can contain either terminal or bridging phospholyl ligands (Fig. 1). Neutral terminal phospholyl ligands are five-electron donor pentahapto η5-C4H4P ligands similar to those in mononuclear phospholyl metal carbonyls. However, phospholyl ligands bridging the central M2 unit are seven-electron donor µ-η5,η1-C4H4P ligands in systems which may or may not contain metal–metal bonds.
The limited range of known phospholyl metal carbonyl derivatives as well as challenges in their synthesis suggested theoretical studies by density functional methods. Our initial studies on the phospholyl metal carbonyls of manganese [4], iron [5], and cobalt [6] showed that the lowest energy iron and cobalt (C4H4P)2M2(CO)n (M = Fe, Co) structures have exclusively terminal η5-C4H4P phospholyl ligands indicating the lack of involvement of the phosphorus lone pairs. However, the lowest energy structures of the phospholyl manganese derivatives (C4H4)2Mn2(CO)n (n = 5, 4) were found to have bridging µ-η5,η1-C4H4P ligands and long Mn…Mn distances indicating lack of direct metal–metal bonding (Fig. 2). This initially suggested that seven-electron donor bridging µ-η5,η1-C4H4P ligands were a feature of early-transition metal binuclear phospholyl carbonyl derivatives. In order to explore this point further, we then investigated phospholyl chromium carbonyl derivatives using similar density functional methods [7]. However, we found the energetically preferred (C4H4P)2Cr2(CO)n structures (n = 6, 4, 3) to have exclusively terminal η5-C4H4P phospholyl ligands. Only for the pentacarbonyl (C4H4P)2Cr2(CO)5 was the lowest energy structure found to contain a bridging µ-η5,η1-C4H4P ring. This may be a reflection of the instability of the pentacarbonyl Cp2Cr2(CO)5 in the cyclopentadienyl series relative to disproportionation into Cp2Cr2(CO)6 + Cp2Cr2(CO)4 [8]. In any case, the results on the chromium (C4H4P)2Cr2(CO)n systems showing little involvement of the phospholyl phosphorus lone pairs in most of the lowest energy structures indicated that the energetic preference for structures with bridging µ-η5,η1-C4H4P rings was not an inherent characteristic of early-transition metal structures.

The lowest energy (C4H4P)2Mn2(CO)5 and (C4H4P)2Mn2(CO)4 structures having one and two µ-η5,η1-C4H4P bridging phospholyl ligands, respectively. Neither structure has a manganese-manganese bond
In order to clarify the types of binuclear phospholyl metal carbonyl derivatives for which structures containing bridging µ-η5,η1-C4H4P phospholyl rings are energetically preferred and thus clearly do not have cyclopentadienyl analogues, we have now studied the binuclear phospholyl vanadium carbonyls (C4H4P)2V2(CO)n (n = 7, 6, 5, 4, 3, 2, 1) using density functional theory methods related to those used in earlier studies. The experimentally known related cyclopentadienyl vanadium carbonyl chemistry starts from CpV(CO)4, which is accessible by reaction of Cp2V with carbon monoxide at elevated pressures [9] or from reaction Na(diglyme)2V(CO)6 with CpHgCl at ambient temperature and pressure [10]. Photolysis of CpV(CO)4 [11] or protonation of the anion V(CO) −6 [12] gives the binuclear derivative Cp2V2(CO)5 shown by X-ray crystallography [13, 14] to have a short V≡V distance of 2.459 Å. This suggests the formal triple bond required to give each vanadium atom the favored 18-electron configuration. No evidence was observed for formation of intermediate Cp2V2(CO)7 or Cp2V2(CO)6 expected on the basis of the 18-electron rule to have a V–V single bond or a V=V double bond, respectively. Previous theoretical studies on the complete series of Cp2V2(CO)n (n = 7, 6, 5, 4, 3, 2, 1) derivatives [15, 16] using related density functional methods provide data for comparison with their phospholyl analogues.
Phospholyl vanadium carbonyls have not yet been synthesized. However, some of the synthetic methods for analogous cyclopentadienyl vanadium carbonyls are potentially adaptable for their phospholyl analogues. Thus, (η5-C4H4P)V(CO)4 is potentially accessible from Na(diglyme)2V(CO)6 with C4H4PHgX analogous to one of the synthetic methods for CpV(CO)4. Binuclear phospholyl vanadium carbonyl derivatives, including (C4H4P)2V2(CO)5 analogous to the known Cp2V2(CO)5, are potentially accessible by photolysis of C4H4PV(CO)4.
2 Theoretical methods
Electron correlation effects were considered by using density functional theory (DFT) methods, which have evolved as a practical and effective computational tool, especially for organometallic compounds [17,18,19,20,21,22,23]. The reliability of such density functional theory (DFT) methods is affected by the quality of the approximate exchange–correlation (XC) energy functional. Two DFT methods were used for the geometry optimization and vibrational frequency analyses in the present study. The first method used was the M06-L functional of Zhao and Truhlar [24], and the other DFT method used was BP86 [25, 26]. These two functionals were developed using different strategies. We include both methods because when two very different DFT methods agree, confident predictions can be made. Indeed, the M06-L and BP86 results agree with each other fairly well in predicting the structural characteristics of the (C4H4P)2V2(CO)n derivatives. For the vibrational frequencies, the BP86 method is known to give values that are closer to the experimental values without using any scaling factors [27,28,29]. This concurrence may be accidental, since the theoretical vibrational frequencies predicted by BP86 are harmonic frequencies, whereas the experimental fundamental frequencies are anharmonic.
Since the DFT methods have been reported to be less sensitive to the basis set size [30], all computations were performed using the double-ζ plus polarization (DZP) basis sets [31, 32], which is consistent with our previous similar studies. The DZP basis set used for hydrogen is the Huzinaga–Dunning DZ set augmented with a set of p polarization functions αp(H) = 0.75. The DZP sets for carbon, oxygen, phosphorus are the Huzinaga–Dunning DZ sets augmented with one set of d functions with orbital exponents αd(C) = 0.75, αd(O) = 0.85 and αd(P) = 0.60. The loosely contracted DZP basis set for vanadium is the Wachters primitive set [33] augmented by two sets of p functions and a set of d functions, contracted following Hood et al. [34], designated as (14s11p6d/10s8p3d).
All of the computations were carried out with the Gaussian 09 program [35], exercising the default fine grid option (75 radial shells, 302 angular points) for evaluating integrals numerically [36].
3 Results and discussion
3.1 Molecular structures
3.1.1 (C4H4P)2V2(CO)7
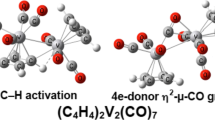
Three (C4H4P)2V2(CO)7 low-energy structures were found (Fig. 3 and Table 1). The lowest energy structure 7S-1 has one (η5-C4H4P)V(CO)4 moiety and one (η5-C4H4P)V(CO)3 moiety linked by a P→V dative bond of length 2.369 Å (BP86) or 2.371 Å (M06-L). A very weakly semibridging carbonyl group in (C4H4P)V(CO)3 bonds to this bridging phosphorus atom with a long C–P distance of ~ 2.7 Å. The predicted V–V distance in 7S-1 is quite long, > 4.5 Å, suggesting no bond between the two vanadium atoms. Thus, the vanadium atoms have the favored 18-electron configurations. The (C4H4P)2V2(CO)7 structure 7S-2, lying only 0.7 kcal/mol (BP86) or 1.5 kcal/mol (M06-L) in energy above 7S-1, has a geometry similar to 7S-1, but with two weakly semibridging CO groups across the P→V dative bond with long P–C distances of ~ 2.7 Å. The (C4H4P)2V2(CO)7 structure 7S-3, lying only 1.2 kcal/mol (BP86) or 1.5 kcal/mol (M06-L) above 7S-1, has geometry close to that 7S-2, except for the relative orientations of the two (C4H4P)V(CO)n moieties.

The low-energy (C4H4P)2V2(CO)7 structures. Distances are given in Å. The upper numbers were obtained from BP86, while the lower numbers were from M06-L
3.1.2 (C4H4P)2V2(CO)6
Three low-energy (C4H4P)2V2(CO)6 structures were found (Fig. 4 and Table 2). The lowest energy structure 6S-1 is a C2h structure with two (η5-C4H4P)V(CO)3 moieties connected by two P→V dative bonds of lengths 2.389 Å (BP86) or 2.392 Å (M06-L). The predicted V–V distance of ~ 4.0 Å suggests the lack of a direct vanadium–vanadium bond. Each vanadium atom in 6S-1 has the favored 18-electron configuration.

The low-energy structures for (C4H4P)2V2(CO)6. Distances are given in Å. The upper numbers were obtained from BP86, while the lower numbers were from M06-L
The (C4H4P)2V2(CO)6 structure 6S-2, lying 20.5 kcal/mol (BP86) or 7.8 kcal/mol (M06-L) in energy above 6S-1, has two (η5-C4H4P)V(CO)3 moieties like 6S-1 (Fig. 4 and Table 2). However, unlike 6S-1, these two moieties are connected by only one P→V dative bond of length 2.452 Å (BP86) or 2.507 Å (M06-L). The predicted V–V distance of 3.368 Å (BP86) or 3.086 Å (M06-L) suggests the formal single V–V bond required to give each vanadium atom the favored 18-electron configuration. However, this rather long apparent V–V single bond could be very weak since the two strong P→V dative bonds are sufficient to hold together the two halves of the molecule.
The lowest energy triplet (C4H4P)2V2(CO)6 structure 6T-1, lying 25.9 kcal/mol (BP86) or 13.8 kcal/mol (M06-L) above 6S-1, is a C2 symmetry structure with two (η5-C4H4P)V(CO)2 moieties connected by two semibridging µ-CO groups as well as a vanadium–vanadium bond (Fig. 4 and Table 2). These semibridging µ-CO groups in 6T-1 are predicted to exhibit ν(CO) frequencies at 1807 and 1816 cm−1 (BP86) which lie significantly below the terminal ν(CO) frequencies ranging from 1918 to 1983 cm−1. These predicted bridging ν(CO) frequencies for 6T-1 are close to the 1799 and 1810 cm−1 bridging ν(CO) frequencies predicted for the triplet spin state cyclopentadienyl analogue (η5-C5H5)2V2(CO)6 using a similar method. The V=V distance of length 2.948 Å (BP86) or 2.976 Å (M06-L) in 6T-1 is significantly shorter than that in 6S-2 suggesting the formal double bond required to give each vanadium atom the favored 18-electron configuration. The unpaired spins for the triplet spin state of 6T-1 can reside in the V=V double bond, which then is of the σ + 2⁄2π type similar to the O=O double bond in triplet dioxygen rather than the σ + π type C=C double bond in ethylene. A related organometallic (η5-Me5C5)2Fe2(µ-CO)3, which has been synthesized and characterized structurally by X-ray crystallography, is a triplet spin state species with a similar Fe = Fe σ + 2⁄2π double bond similar to that suggested for 6T-1 [37,38,39,40].
3.1.3 (C4H4P)2V2(CO)5
Six (C4H4P)2V2(CO)5 low-energy isomers (three triplets and three singlets) were found (Fig. 5 and Table 3). The lowest energy (C4H4P)2V2(CO)5 structure 5S-1 has a terminal η5-C4H4P ring bonded to each vanadium atom and two bridging carbonyl groups. One vanadium atom in 5S-1 bears two terminal CO groups, whereas the other vanadium atom bears only one terminal CO group. The predicted ν(CO) frequencies of 1836 and 1865 cm−1 (BP86) are very close to those of 1849 and 1877 cm−1 for the experimentally known cyclopentadienyl analogue (η5-C5H5)2V2(CO)5 with an analogous structure [16]. The predicted V≡V distance of 2.455 Å (BP86) or 2.453 Å (M06-L) is close to the experimental distance of 2.459 Å in the cyclopentadienyl analogue and suggests the formal triple bond required to give each vanadium atom in 5S-1 the favored 18-electron configuration. Structures 5S-2 and 5S-3, lying 4.6 and 10.2 kcal/mol (BP86) or 3.5 and 8.9 kcal/mol (M06-L), respectively, above 5S-1, are conformers of 5S-1 with similar overall geometries differing only in the orientations of the C4H4P and CO ligands.

The low-energy structures for (C4H4P)2V2(CO)5. Distances are given in Å. The upper numbers were obtained from BP86, while the lower numbers were from M06-L
The lowest energy triplet (C4H4P)2V2(CO)5 structure 5T-1, lying 13.9 kcal/mol (BP86) or 12.6 kcal/mol (M06-L) in energy above 5S-1, has a terminal η5-C4H4P ring bonded to each vanadium atom (Fig. 5 and Table 3). The two vanadium atoms in 5T-1 are bridged by a four-electron donor η2-µ-CO group exhibiting the expected very low predicted ν(CO) frequency of 1654 cm−1 and a short V–O distance of ~ 2.23 Å as well as a two-electron donor semibridging µ-CO group exhibiting a predicted ν(CO) frequency of 1880 cm−1. The predicted V–V distance of 3.008 Å (BP86) or 2.961 Å (M06-L) suggests a formal single bond thereby giving each vanadium atom a 17-electron configuration consistent with a binuclear triplet. Structure 5T-2, has a similar geometry to 5T-1 except for the orientation of one of the C4H4P ring phosphorus atoms.
The triplet (C4H4P)2V2(CO)5 structure 5T-3, lying 17.7 kcal/mol (BP86) or 14.9 kcal/mol (M06-L) in energy above 5S-1, has a terminal η5-C4H4P ring bonded to each vanadium atom (Fig. 5 and Table 3). The central V2 unit in 5T-3 is bridged by two of the five carbonyl groups as two-electron donors. One of these bridging carbonyl groups is a normal bridging µ-CO group with a predicted ν(CO) frequency of 1771 cm.−1 The other bridging carbonyl group in 5T-3 is only weakly semibridging with a long V–C distance of ~ 2.7 Å and exhibits a significantly higher ν(CO) frequency of 1877 cm−1, only 32 cm−1 below the lowest terminal ν(CO) frequency. The predicted V=V distance of 2.653 Å (BP86) or 2.653 Å (M06-L) in 5T-3, which is intermediate between the V–V single bond distances in 5T-1 and 5T-2 and the V≡V single bond distances in 5S-1, 5S-2, and 5S-3, suggests a V=V double bond, thereby giving each vanadium atom the favored 18-electron configuration.
3.1.4 (C4H4P)2V2(CO)4
Sixteen (C4H4P)2V2(CO)4 structures were found within 12 kcal/mol of the lowest energy structure. However, some of the 16 structures are conformers with similar geometries and energies from which eight representative structures (Fig. 6 and Table 4) are selected for discussion.

Eight representative low-energy (C4H4P)2V2(CO)4 structures. Distances are given in Å. The upper numbers were obtained from BP86, while the lower numbers were from M06-L
The lowest energy (C4H4P)2V2(CO)4 structure 4S-1 has two (η5-C4H4P)V(CO) moieties connected through a vanadium–vanadium bond bridged by two four-electron donor η2-µ-CO groups with short V–O distances of 2.395 Å (BP86) or 2.369 Å (M06-L) (Fig. 6 and Table 4) and exhibiting very low ν(CO) frequencies of 1706 and 1720 cm−1. The predicted V=V distance of 2.739 Å (BP86) or 2.705 Å (M06-L) suggests the formal double bond required to give each vanadium atom the 18-electron configuration.
The second (C4H4P)2V2(CO)4 structure 4S-2 is essentially energetically degenerate with 4S-1, lying only 0.8 kcal/mol (BP86) or 0.1 kcal/mol (M06-L) above 4S-1 (Fig. 6 and Table 4). Structure 4S-2 has two (η5-C4H4P)V(CO) moieties like 4S-1. However, unlike 4S-1, these two moieties in 4S-2 are bridged by only one four-electron donor bridging η2-µ-CO group as well as a two-electron donor weakly semibridging µ-CO group with a long V–C distance of ~ 2.56 Å. The η2-µ-CO group exhibits a very low ν(CO) frequency of 1677 cm−1, whereas the semibridging µ-CO group exhibits a much higher ν(CO) frequency of 1799 cm−1, which is still ~ 130 cm−1 below the lowest terminal ν(CO) frequency. The predicted V≡V distance of 2.588 Å (BP86) or 2.540 Å (M06-L) can be interpreted as a formal triple bond to give each vanadium atom the favored 18-electron configuration.
The third (C4H4P)2V2(CO)4 structure 4S-3, lying 1.8 kcal/mol (BP86) or 0.8 kcal/mol (M06-L) above 4S-1, has the central V2 unit bridged by a four-electron donor η2-µ-CO group with a short V–O distance of 2.365 Å (BP86) or 2.347 Å (M06-L) and a low ν(CO) frequency of 1699 cm−1 (Fig. 6 and Table 4). The remaining three CO groups are terminal groups with two bonded to one vanadium atom and the third bonded to the other vanadium atom. One of the phospholyl rings is a terminal ligand but the other phospholyl ring is a seven-electron donor η5, η1-C4H4P bridging ligand forming a P→V dative bond to one vanadium atom and a pentahapto ring-metal bond to the other vanadium atom. The V=V distance of 2.748 Å (BP86) or 2.706 Å (M06-L), suggests a V=V double bond, thereby giving each vanadium atom the 18-electron configuration.
The (C4H4P)2V2(CO)4 structure 4S-4, lying 6.7 kcal/mol (BP86) or 5.1 kcal/mol (M06-L) in energy above 4S-1, has four weakly semibridging µ-CO groups with long V–C distances ranging from 2.30 to 2.58 Å (Fig. 6 and Table 4). Interpreting the V–V distance of 2.302 Å (BP86) or 2.301 Å (M06-L) as a formal triple bond gives one vanadium atom an 18-electron configuration but the other vanadium atom only a 16-electron configuration.
The (C4H4P)2V2(CO)4 structure 4S-5, lying 8.1 kcal/mol (BP86) or 5.8 kcal/mol (M06-L) in energy above 4S-1, has similar geometry to 4S-3, but, instead of a 4-electron donor η2-µ-CO group, structure 4S-5 has a normal two-electron donor bridging µ-CO group and a bridging seven-electron donor η5,η1-C4H4P ring (Fig. 6 and Table 4). The predicted V≡V distance of 2.431 Å (BP86) or 2.425 Å (M06-L) in 4S-5 is close to that in 4S-1, thereby suggesting the formal triple bond to give one vanadium atom an 18-electron configuration but the other vanadium atom only a 16-electron configuration.
The (C4H4P)2V2(CO)4 structure 4S-6, lying 10.1 kcal/mol (BP86) or 7.5 kcal/mol (M06-L) in energy above 4S-1, has four terminal CO groups and two bridging seven-electron donor η5,η1-C4H4P rings (Fig. 6 and Table 7). The predicted V=V distance of 2.692 Å (BP86) or 2.661 Å (M06-L) suggests the V=V double bond required to give each vanadium atom the favored 18-electron configuration.
The lowest energy triplet (C4H4P)2V2(CO)4 structure 4T-1, lying 6.6 kcal/mol (BP86) or 4.1 kcal/mol (M06-L) in energy above 4S-1, has two terminal CO groups and two two-electron donor bridging µ-CO groups (Fig. 6 and Table 4). The bridging µ-CO groups exhibit ν(CO) frequencies of 1804 and 1835 cm−1. The predicted V≡V distance of 2.431 Å (BP86) or 2.430 Å (M06-L) in 4T-1, similar to that in 5S-1, suggests a similar triple bond, thereby giving each vanadium atom a 17-electron configuration, consistent with a binuclear triplet.
The triplet (C4H4P)2V2(CO)4 structure 4T-2, lying 11.7 kcal/mol (BP86) or 5.5 kcal/mol (M06-L) above 4S-1, has two terminal CO groups, two two-electron donor bridging µ-CO groups, one seven-electron donor bridging η5,η1-C4H4P ring and one terminal η5-C4H4P ring (Fig. 6 and Table 7). The predicted V≡V distance of 2.474 Å (BP86) or 2.495 Å (M06-L) in 4T-2 is similar to that in the other (C4H4P)2V2(CO)n (n = 5, 4) structures formulated with triple bonds and thus is also formulated as a triple bond.
3.1.5 (C4H4P)2V2(CO)3
Six low-energy (C4H4P)2V2(CO)3 structures (two triplets, four singlets) were found (Fig. 7 and Table 5). The lowest energy (C4H4P)2V2(CO)3 structure 3T-1 has a terminal η5-C4H4P ring bonded to each vanadium atom and all three carbonyl groups in bridging positions. One of the bridging carbonyl groups in 3T-1 is a four-electron donor η2-µ-CO group with a short V–O distance of 2.22 Å and a low ν(CO) frequency of 1636 cm−1. The other two bridging carbonyl groups are normal two-electron donor µ-CO groups exhibiting ν(CO) frequencies of 1819 and 1859 cm−1. The predicted V–V distance of 2.532 Å (BP86) or 2.554 Å (M06-L) suggests a formal triple V≡V bond, thereby giving each vanadium atom a 17-electron configuration, consistent with a binuclear triplet structure.

The six lowest energy (C4H4P)2V2(CO)3 structures. Distances are given in Å. The upper numbers were obtained from BP86, while the lower numbers were from M06-L
The next (C4H4P)2V2(CO)3 structure 3T-2, lying 1.8 kcal/mol (BP86) or 0.4 kcal/mol (M06-L) in energy above 3T-1, has two semibridging carbonyl groups and a seven-electron donor bridging η5,η1-C4H4P ring bridging the central V2 unit with a P→V dative bond of ~ 2.65 Å (Fig. 7 and Table 5) The predicted V≡V distance of 2.412 Å (BP86) or 2.459 Å (M06-L) for 3T-2 is in the typical range for a formal triple V≡V bond, thereby giving both vanadium atoms a 17-electron configuration for a binuclear triplet structure.
The lowest energy singlet (C4H4P)2V2(CO)3 structure 3S-1, lying 7.4 kcal/mol (BP86) or 14.7 kcal/mol (M06-L) above 3T-1, has C2 symmetry with the central V2 unit bridged by all three carbonyl groups (Fig. 7 and Table 5). One of these bridging carbonyl groups is a typical two-electron donor µ-CO group exhibiting a ν(CO) frequency of 1764 cm−1. The remaining two bridging carbonyl groups in 3S-1 are four-electron donor η2-µ-CO groups with short V–O distances of 2.335 Å (BP86) or 2.284 Å (M06-L) and exhibiting ν(CO) frequencies of 1651 and 1682 cm−1. The predicted V–V distance of 2.542 Å (BP86) or 2.531 Å (M06-L) in 3S-1 suggests a V≡V triple bond, thereby giving each vanadium atom the favored 18-electron configuration. The singlet (C4H4P)2V2(CO)3 structure 3S-4, lying 11.7 kcal/mol (BP86) or 17.2 kcal/mol (M06-L) in energy above 3T-1, has Cs symmetry and similar geometry to 3S-1 except for the directions of these two four-electron donor CO groups and the locations of the phosphorus atoms in the phospholyl rings.
The singlet (C4H4P)2V2(CO)3 structure 3S-2, lying 9.7 kcal/mol (BP86) or 15.1 kcal/mol (M06-L) above 3T-1, has similar geometry to 3T-2 except that one of the bridging carbonyl groups is a four-electron donor η2-µ-CO group, as indicated by a short V–O distance of 2.402 Å (BP86) or 2.349 Å (M06-L) (Fig. 7 and Table 5). Thus, 3S-2 has one terminal CO group, one two-electron donor bridging µ-CO group, and one four-electron donor bridging η2-µ-CO group exhibiting predicted ν(CO) frequencies of 1944, 1800 and 1634 cm−1, respectively. Structure 3S-2 has one terminal η5-C4H4P ring and one seven-electron donor bridging η5,η1-C4H4P ring. The bridging η5,η1-C4H4P ring forms a pentahapto bond with one vanadium atom and a P→V dative bond of length 2.618 Å (BP86) or 2.592 Å (M06-L) to the other vanadium atom. The V≡V distance of 2.423 Å (BP86) or 2.416 Å (M06-L) suggests a formal triple bond, thereby giving each vanadium atom the favored 18-electron configuration after considering the four-electron donor bridging η2-µ-CO group and seven-electron donor η5,η1-C4H4P ring. Structure 3S-3, lying 10.5 kcal/mol (BP86) or 16.7 kcal/mol (M06-L) above 3T-1, has almost the same geometry as 3S-2, except for the locations of the phosphorus atoms in the phospholyl rings.
3.1.6 (C4H4P)2V2(CO)2
Three low-energy (C4H4P)2V2(CO)2 structures, namely two triplets and one singlet, were found (Fig. 8 and Table 6). The lowest energy (C4H4P)2V2(CO)2 structure 2T-1 has two seven-electron donor bridging η5,η1-C4H4P rings, one terminal CO group, and one four-electron donor bridging η2-µ-CO group. The η2-µ-CO group in 2T-1 exhibits a short V–O distance of 2.164 Å (BP86) or 2.195 Å (M06-L) and a very low predicted ν(CO) frequency of 1531 cm−1 as compared with the 1915 cm−1 frequency for the terminal CO group. Both η5,η1-(C4H4P) rings bridge the two vanadium atoms with a pentahapto bond to one vanadium atom and a P→V dative bond to the other vanadium atom. The V≡V distance of 2.526 Å (BP86) or 2.637 Å (M06-L) in 2T-1 suggests a V≡V triple bond, thereby giving one V atom a 17-electron configuration and the other V atom 19-electron configuration in a triplet spin state structure.

Three (C4H4P)2V2(CO)2 structures. Distances are given in Å. The upper numbers were obtained from BP86, while the lower numbers were from M06-L
The (C4H4P)2V2(CO)2 structure 2T-2, lying 14.3 kcal/mol (BP86) or 14.6 kcal/mol (M06-L) in energy above 2T-1, has a terminal η5-C4H4P ring bonded to each vanadium atom and two carbonyl groups bridging the central V2 unit (Fig. 8 and Table 6). The V≡V distance of 2.458 Å (BP86) or 2.539 Å (M06-L) in 2T-2 suggests a formal triple bond. This gives each vanadium atom a 15-electron configuration consistent with an unsaturated binuclear triplet structure.
The lowest energy singlet (C4H4P)2V2(CO)2 structure 2S-1, lying 4.2 kcal/mol (BP86) or 12.6 kcal/mol (M06-L) above 2T-1, has one less P→V dative bond than 2T-1 (Fig. 8 and Table 6). Thus, 2S-1 has one terminal η5-C4H4P ring, one bridging η5,η1-(C4H4P) ring, one terminal CO group and one four-electron donor bridging η2-µ-CO group with a short V–O distance of 2.012 Å (BP86) or 2.030 Å (M06-L) and a very low ν(CO) frequency of 1425 cm−1. Interpreting the very short V≣V distance of 2.337 Å (BP86) or 2.415 Å (M06-L) in 2S-1 as a formal quadruple bond gives each vanadium atom the favored 18-electron configuration.
3.1.7 (C4H4P)2V2(CO)
Two low-energy (C4H4P)2V2(CO) structures, namely one triplet and one singlet, were found (Fig. 9 and Table 7). The lowest energy (C4H4P)2V2(CO) structure 1T-1 can be derived from 2T-1 by removal of the terminal CO group and thus retains the two seven-electron donor bridging η5,η1-C4H4P rings. The remaining CO group in 1T-1 is a four-electron donor bridging η2-µ-CO group with a short V–O distance of 2.145 Å (BP86) or 2.173 Å (M06-L) and a very low ν(CO) frequency of 1481 cm−1. The V≡V distance of 2.338 Å (BP86) or 2.524 Å (M06-L) in 1T-1 suggests a formal triple bond, thereby giving each vanadium atom a 17-electron configuration for a binuclear triplet. The singlet (C4H4P)2V2(CO) structure 1S-1, lying 3.0 kcal/mol (BP86) or 9.0 kcal/mol (M06-L) in energy above 1T-1, has the same geometry as 1T-1 except for a shorter V≣V distance of 2.215 Å (BP86) or 2.247 Å (M06-L). This can be interpreted as the quadruple bond required to give each vanadium atom in 1S-1 the favored 18-electron configuration.

Structures found for (C4H4P)2V2(CO). Distances are given in Å. The upper numbers were obtained from BP86, while the lower numbers were from M06-L
3.2 Thermochemistry
Table 8 shows the energies for the carbonyl dissociation reactions (C4H4P)2V2(CO)n→ (C4H4P)2V2(CO)n−1 + CO (n = 7, 6, 5, 4, 3, 2) considering the lowest energy structures. The CO dissociation energies are all sufficiently endothermic ranging from 20 to 50 kcal/mol, thus suggesting that all of the lowest energy (C4H4P)2V2(CO)n (n = 7, 6, 5, 4, 3, 2) structures are viable toward CO loss. For comparison, the experimental dissociation energies for the well-known simple metal carbonyls Ni(CO)4, Fe(CO)5 and V(CO)6 are 27, 41 and 37 kcal/mol, respectively [41].
Table 9 lists the energies for the disproportionation reactions 2(C4H4P)2V2(CO)n → (C4H4P)2V2(CO)n+1 + (C4H4P)2V2(CO)n–1, which can be readily derived from the CO dissociation energies in Table 8. These show that the tricarbonyl (C4H4P)2V2(CO)3 is clearly viable toward such disproportionation. Interestingly, the pentacarbonyl (C4H4P)2V2(CO)5 analogous to the experimentally known stable cyclopentadienyl derivative (η5-C5H5)2V2(CO)5 is only marginally viable toward disproportionation with ∆E values for the reaction 2(C4H4P)2V2(CO)5 (5S-1) → (C4H4P)2V2(CO)6 (6S-1) + (C4H4P)2V2(CO)4 (4S-1) of only 1.5 kcal/mol (BP86) or 13.5 kcal/mol (M06-L). This may relate to increased stability of the (C4H4P)2V2(CO)6 structure 6S-1 because of the energy gained by the two P→V dative bonds in 6S-1 (Fig. 4). Such a structural feature is obviously not possible in the analogous (η5-C5H5)2V2(CO)6 structure because of the absence of phosphorus atoms.
Also of interest is the dissociation of the (C4H4P)2V2(CO)n derivatives into mononuclear (C4H4P)V(CO)m fragments. In order to obtain such energetic data, the structures of the mononuclear (C4H4P)V(CO)m were optimized (Fig. 10) by the same DFT methods. All of these mononuclear structures are genuine minima. Using this information, the dissociation energies of the binuclear metal-carbonyl derivatives into mononuclear fragments were predicted to be substantial, i.e., larger than 28 kcal/mol (Table 10). These data show that the lowest energy (C4H4P)2V2(CO)n structures are viable toward dissociation into mononuclear fragments with such dissociation becoming monotonically more endothermic with decreasing n.

Optimized geometries for the mononuclear (C4H4P)V(CO)m (m = 4, 3, 2, 1) structures, which are the dissociation products in Table 10
3.3 NBO analysis of the Vanadium–Vanadium bonding
Table 11 lists the natural atomic charges for the vanadium atoms and the Wiberg bond indices (WBI) for the V–V bonds from NBO analysis [42] using the BP86 functional. The V–V distances, the formal V–V bond orders and the types of bridging groups are also listed for comparison. Only singlet structures are considered since WBI analyses of higher spin state open shell structures appear to be less reliable.
The natural atomic charges on the vanadium atoms (except one in 1S-1) are negative since the vanadium atoms accept electrons from the CO groups and phosphorus lone pairs without sufficient π back-bonding from the vanadium atoms to the antibonding orbitals of the CO groups to remove all of this extra negative charge. The magnitudes of the vanadium natural charges depend on the number of coordinated CO groups and the number of P→V dative bonds from the C4H4P rings. The vanadium atoms in the (C4H4P)2V2(CO)7 structures have the most negative natural charges from − 1.92 to − 1.98, since each vanadium atom bears either three or four CO ligands plus a lone pair of the bridging phosphorus atom. The vanadium atoms in 6S-1 are in a similar situation, and thus they have a similarly high negative natural charge of − 1.87. One of the vanadium atoms in 6S-2 bears three CO ligands but does not receive a P→V dative bond so it has a less negative natural charge of − 1.27. In the pentacarbonyl (C4H4P)2V2(CO)5, the vanadium atoms with three ligands (either CO groups or phosphorus lone pairs) have natural negative charges around − 1.3, while vanadium atoms with two such ligands have less negative natural charges around − 1.1. In this analysis, a bridging CO group is counted as half of a CO group to each vanadium atom. In the tetracarbonyls (C4H4P)2V2(CO)4, most of the vanadium atoms bear 2 CO ligands and have a natural charge around − 0.9. The exception is one vanadium atom in 4S-3, which is the acceptor of a P→V dative bond and thus has a more negative natural charge of − 1.44. In the tricarbonyls (C4H4P)2V2(CO)3, the natural charges for vanadium atoms with 1.5 CO ligands and with one CO ligand are around − 0.6, while those for vanadium atoms with 2 CO ligands are more negative around − 0.9, like similar vanadium atoms in the tetracarbonyls (C4H4P)2V2(CO)4. For 2S-1 and 1S-1, the vanadium atom with 1.5 CO ligands has a natural charge of − 0.6, whereas vanadium atoms with only half of a bridging CO ligand have only slightly negative natural charges around − 0.1 or − 0.2.
Table 11 shows that the WBI values are related to the formal bond orders. Since the WBI values are also affected by the number and nature of bridging groups, the latter are also listed. The WBI for a V–V single bond (in 6S-2) is 0.31, those for V=V double bonds are 0.86 (4S-1) or 0.85 (4S-3), those for V≡V triple bonds range from 0.92 to 1.14 and those for V≣V quadruple bonds (in 2S-1 and 1S-1) are 1.80 and 2.05, respectively. For those structures without V–V bonds in (C4H4P)2V2(CO)7, the corresponding WBIs are less than 0.03.
4 Conclusion
Simple electron counting not considering other factors such as coordination number suggests that the phospholyl vanadium carbonyls (C4H4P)2V2(CO)n should have low-energy structures analogous to the lowest energy (C4H4P)2Mn2(CO)n–2 with one less CO group per metal atom. This is true for the carbonyl richest structures (C4H4P)2V2(CO)n (n = 7, 6). Thus, the lowest energy structure 7S-1 of the heptacarbonyl (C4H4P)2V2(CO)7 (Fig. 3) can be viewed as a (η5-C4H4P)V(CO)4 species with an 18-electron configuration coordinating to a (η5-C4H4P)V(CO)3 unit through a P→V dative bond leading to a structure with one terminal phospholyl ligand and one bridging phospholyl ligand and no direct vanadium–vanadium bond. This is exactly analogous to the lowest energy (C4H4P)2Mn2(CO)5 structure in which a saturated (η5-C4H4P)Mn(CO)3 unit coordinates to an otherwise unsaturated (η5-C4H4P)Mn(CO)2 unit through a similar P → Mn dative bond giving a structure with no direct manganese–manganese (Fig. 11). Similarly, the lowest energy structure 6S-1 of the hexacarbonyl (C4H4P)2V2(CO)6 has two seven-electron donor bridging η5,η1-C4H4P ligands analogous to the lowest energy (C4H4)2Mn2(CO)4 structure (Fig. 12).

Comparison of the lowest energy (C4H4P)2V2(CO)7 and (C4H4P)2Mn2(CO)5 structures

Comparison of the lowest energy (C4H4P)2V2(CO)6 and (C4H4P)2Mn2(CO)6 structures
The lowest energy structures for the pentacarbonyl (C4H4P)2V2(CO)5 have terminal phospholyl ligands and formal V≡V triple bonds without any P→V dative bonds. They are therefore analogous to the experimentally known and structurally characterized cyclopentadienyl derivative (η5-C5H5)2V2(CO)5 with a similar V≡V triple bond distance of ~ 2.45 Å [11,12,13,14]. However, the viability of (C4H4P)2V2(CO)5 is questionable since the disproportionation process 2(C4H4P)2V2(CO)5 → (C4H4P)2V2(CO)6 + (C4H4P)2V2(CO)4 is predicted to be endothermic by only 1.5 kcal/mol by the BP86 method. The relatively low energy required for this disproportionation process may be a consequence of the energy gained in the (C4H4P)2V2(CO)6 disproportionation product 6S-1 by the presence of two P→V dative bonds. This structural feature is not available in the corresponding binuclear cyclopentadienyl vanadium carbonyl system because of the lack of phosphorus atoms.
Formal V≡V triple bonds are found in most of the lowest energy structures of the more highly unsaturated systems (C4H4P)2V2(CO)n (n = 4, 3, 2) supplemented by other structural features such as four-electron donor bridging η2-µ-CO groups forming V–O bonds, seven-electron donor bridging η5,η1-C4H4P bridging phospholyl rings, and preferred triplet rather than singlet spin states. The general preference for formal V≡V triple bonds in most such unsaturated systems is consistent with the isolation and structural characterization of a number of binuclear cyclopentadienyl metal carbonyls of the first row transition metals with formal M≡M triple bonds such as not only (η5-C5H5)2V2(CO)5 directly relevant to the current work but also (η5-C5H5)2Cr2(CO)4 [43,44,45], and (η5-C5H5)2Mn2(µ-CO)3 [46].
Despite this general preference for formal V≡V triple bonds in the highly unsaturated (C4H4P)2V2(CO)n (n = 4, 3, 2) structures, the lowest energy structure 4S-1 for the tetracarbonyl (C4H4P)2V2(CO)4, albeit by a minuscule margin of < 1 kcal/mol within computational error, has a formal V=V double bond rather than a formal V≡V triple bond. The favored 18-electron configuration for each vanadium atom is attained by the presence of two four-electron η2-µ-CO groups. The very slightly higher energy (C4H4P)2V2(CO)4 structure 4S-3 also has a similar central V=V double bond. However, the 18-electron configuration for each vanadium atom in 4S-1 is attained by only a single four-electron donor η2-µ-CO group supplemented by a seven-electron donor bridging η5,η1-C4H4P phospholyl ring. No evidence was obtained for a low-energy (C4H4P)2V2(CO)4 structure with exclusively two-electron donor carbonyl groups, terminal phospholyl ligands and the formal V≣V quadruple bond required to give each vanadium atom the favored 18-electron configuration. The only structures found in this work with formal V≣V quadruple bonds are the highly unsaturated singlet (C4H4P)2V2(CO)2 structure 2S-1 and singlet (C4H4P)2V2(CO) structure 1S-1.
References
Dillon KB, Mathey F, Nixon JF (1998) Phosphorus: the carbon copy: from organophosphorus to phospha-organic chemistry. Wiley, New York
Mathey F (1976) Tetrahedron Lett. 17:4155
Mathey F, Mitschler A, Weiss R (1978) J Am Chem Soc 100:5748
Chen X, Du Q, Jin R, Feng H, Xie Y, King RB (2011) New J Chem 35:1117
Chen X, Yuan L, Ren G, Xi Q, King R, Du Q, Feng H, Xie Y, King RB (2016) Inorg Chim Acta 445:79
Chen X, Jin R, Du Q, Feng H, Xie Y, King RB (2012) J Organomet Chem 701:1
Wang X, Feng C, Ren G, Chen X, Jin R, Du Q, Feng H, Xie Y, King RB (2019) Inorg Chim Acta 494:194
Fortman GC, Kégl T, Li Q-S, Zhang X, Schaefer HF, Xie Y, King RB, Telser J, Hoff CD (2007) J Am Chem Soc 129:14388
Fischer EO, Vigoureux S (1958) Chem Ber 91:2205
Werner RPM, Filbey AH, Manastryskyj SA (1964) Inorg Chem 3:298
Herrmann WA, Plank J (1979) Chem Ber 112:392
Fischer EO, Schneider RJ (1967) Angew Chem Int Ed 6:569
Cotton FA, Kruczynski L (1978) J Organomet Chem 160:93
Huffman JC, Lewis LN, Caulton KG (1980) Inorg Chem 19:2755
Zhang X, Li Q, Xie Y, King RB, Schaefer HF (2007) Eur J Inorg Chem 153:1599
Li Q, Zhang X, Xie Y, King RB, Schaefer HF (2007) J Am Chem Soc 129:3433
Ziegler T, Autschbach J (2005) Chem Rev 105:2695
Bühl M, Kabrede H (2006) J Chem Theory Comput 2:1282
Brynda M, Gagliardi L, Widmark PO, Power PP, Roos BO (2006) Angew Chem Int Ed 45:3804
Sieffert N, Bühl M (2010) J Am Chem Soc 132:8056
Schyman P, Lai W, Chen H, Wang Y, Shaik S (2011) J Am Chem Soc 133:7977
Adams RD, Pearl WC, Wong YO, Zhang Q, Hall MB, Walensky JR (2011) J Am Chem Soc 133:12994
Lonsdale R, Olah J, Mulholland AJ, Harvey JN (2011) J Am Chem Soc 133:15464
Zhao Y, Truhlar DG (2006) J Chem Phys 125:194101
Becke AD (1988) Phys Rev A 38:3098
Perdew JP (1986) Phys Rev B 33:8822
Jones V, Thiel W (1995) J Phys Chem 102:8474
Silaghi-Dumitrescu I, Bitterwolf TE, King RB (2006) J Am Chem Soc 128:5432
Assef MK, Dever JL, Brathwaite AD, Mosley JD, Duncan MA (2015) Chem Phys Lett 640:175
Narendrapurapu BS, Richardson NA, Copan AV, Estep ML, Yang Z, Schaefer HF (2013) J Chem Theory Comput 9:2930
Dunning TH (1970) J Chem Phys 53:2823
Huzinaga S (1965) J Chem Phys 42:1293
Wachters AJH (1970) J Chem Phys 52:1033
Hood DM, Pitzer RM, Schaefer HF (1979) J Chem Phys 71:705
Frisch MJ et al (2009) Gaussian, Inc., Wallingford CT. Gaussian 09, Revision A.02
Papas BN, Schaefer HF (2006) J. Mol. Struct. (THEOCHEM) 768:175
Caspar JV, Meyer TJ (1980) J Am Chem Soc 102:7794
Hooker RH, Mahmoud KA, Rest AJ (1983) Chem. Commun. 105:1022
Hepp AF, Blaha JP, Lewis C, Wrighton MS (1984) Organometallics 3:174
Blaha JP, Bursten BE, Dewan JC, Frankel RB, Randolph CL, Wilson BA, Wrighton MS (1985) J Am Chem Soc 107:4561
Sunderlin LS, Wang D, Squires PR (1993) J Am Chem Soc 115:12060
Weinhold F, Landis CR (2005) Valency and bonding: a natural bond order donor–acceptor perspective. Cambridge University Press, Cambridge, pp 32–36
Curtis MD, Butler WM (1978) J Organomet Chem 155:131
King RB, Efraty A, Douglas WM (1973) J Organomet Chem 60:125
Potenza J, Giordano P, Mastropaolo D, Efraty A (1974) Inorg Chem 13:2540
Herrmann WA, Serrano R, Weichmann J (1983) J Organomet Chem 246:C57
Acknowledgements
We are indebted to the Scientific Research Fund of the Key Laboratory of the Education Department of Sichuan Province (Grant No. 10ZX012) for the support of this research.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chen, W., Yan, J., Chen, X. et al. The role of the phosphorus lone pair in the low-energy binuclear phospholyl vanadium carbonyl structures: comparison with cyclopentadienyl analogues. Theor Chem Acc 140, 3 (2021). https://doi.org/10.1007/s00214-020-02692-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-020-02692-y