
Abstract
Here for the first time, we applied approach developed earlier for higher fullerenes to investigate the features of molecular structures of non-IPR isomers 270 (D3) and 271 (D5h) of small fullerene C50. The bond distributions are presented as structural formulas. The instability of the studied isomers is caused by a significant local overstrain due to the excessive folding of pentagons in pentalene fragments, which typically are planar molecules. It is found that the chains of π-bonds are passing through some cycles like in the previously studied higher non-IPR fullerenes C66 and C68. Chemical shifts of the centers of the pentagons and hexagons (NICS(0)) are reported. It is shown that chlorine atoms in the exohedral derivative C50Cl10 of isomer 271 (D5h) are attached to the indacene-like substructures confirming the preference of addition to π-delocalized hexagons in radical reactions. The identified features in the structures of smaller fullerene molecules can be predictive of the ability to be synthesized as derivatives and will assist in determination of their reactivity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
An empirical rule of isolated pentagons (IPR), that appeared after the discovery of the molecular structures of first most stable fullerenes C60, C70, etc., suggested that fullerene molecule in which all 12 pentagons are separated by hexagons has to be stable and conversely the molecule with some abutting pentagons is unstable [1, 2]. In fact, all fullerenes that were obtained, isolated and characterized during the first period after their discovery in 1985 were obeying IPR. Nevertheless, sometimes some of non-IPR fullerene Cn (n > 20) can be obtained in the form of endohedral or exohedral derivatives; in other words, molecules of such fullerenes can be stabilized to study them. However, basically, this is a random unexpected luck. The unused potential of a huge amount of non-IPR fullerenes and the scanty proportion of IPR fullerenes obtained and used in practice is a paradox of the current situation in the field of research of fullerenes. It is obvious that only theoretical studies can (and not always) shed light on the molecular features of non-IPR fullerenes so far in connection with the possibility of their stabilization and, accordingly, production.
Earlier, we developed and used a semiempirical approach for analyzing the structure of higher fullerenes Cn (n > 60) [3,4,5], mainly IPR ones, among which there were several non-IPR molecules. It has been shown that pentalene substructures with two paired pentagons have a different bonds distribution in molecules of various non-IPR fullerenes (Fig. 1) [6,7,8], i.e., pentalene does not have a common structure in fullerene molecule, in contrast to the previously considered substructures in higher IPR fullerenes. Nevertheless, the partial delocalization of bonds and the presence of chains of conjugated delocalized bonds passing through cycles are common to them. It means that the electronic structure of pentalene varies significantly depending on its environment.

In contrast to IPR fullerenes C60, C70 and higher ones, the structures and stabilities of small fullerenes Cn (n < 60), all of them being the non-IPR ones, were studied significantly less. Among them the fullerene C50 is interesting due to an unexpected good luck of its obtaining as decachlorofullerene C50Cl10, which molecular structure was unambiguously identified as isomer 271 (D5h) [9, 10]. The series of theoretical works, some before this event [11,12,13,14] and most of them after it [15,16,17,18,19,20,21,22,23,24,25,26,27], were devoted to different aspects of structures of isomers of C50.
The non-IPR C50 fullerene has 271 isomers [28]. Selected isomers of C20–C70 were first studied by tight-binding molecular-dynamics total energy optimization [11] and MNDO calculations [12, 13]. Two of 271 possible isomers [270 (D3) and 271 (D5h)] which were considered in singlet states are the most energetically favorable cages according to several theoretical studies [14,15,16, 18,19,20,21,22, 25, 29, 30]. The presence of paired fused pentagons, i.e., pentalene fragments, leads to structural distortions and enhanced strains of the cage that were estimated as energy per atom [20] or in terms of the pyramidalization angles [10]. It was shown that isomer 270 (D3) containing six pairs of adjacent pentagons is energetically more favorable than isomer 271 (D5h) containing five pentalene substructures due to values of the sphericity parameter according to which isomer 270 (D3) is more spherical than isomer 271 (D5h) [19]. However, non-IPR fullerenes (including small fullerenes) can be isolated as less reactive adducts at the pentagon–pentagon fusions [31]. Nonetheless, there was no detailed structural evidence of instability of small fullerene molecules. In our report, for the first time we applied our approach of theoretical study of molecular structure developed for higher fullerenes [3,4,5, 32] to small fullerenes. We investigate the molecular structures of non-IPR isomers 270 (D3) and 271 (D5h) and show that the reason of their instability is the local molecular overstrain due to the excessive folding of pentagons in pentalene substructures of fullerene molecules.
2 Methodology
The semiempirical approach supported by the quantum-chemical calculations provides a complete structural formula of fullerene molecule, showing the distribution of single bonds, double bonds and π-delocalized bonds. We were considering the substructures existing in the most stable fullerene molecules, for example, corannulene substructures in C60 or s-indacene substructures in C70, as a factor which does not reduce the molecular stability of any fullerene. On the contrary, large substructures including only hexagons are the reasons of significant local strains in fullerene molecule as well as radical substructures; both are the reasons of instability of higher fullerenes. In fact, the analysis of all IPR molecular structures of higher fullerenes, such as C72, C74, C76, C78, C80, C82, C84, and C86, confirmed this substructure concept (see Refs. [3,4,5, 32] and references herein). The procedure opens an easy way for identification of radical fullerenes, or fullerenes that are unstable due to the overstrained molecules; moreover, they appear to be useful in finding the most reactive positions of fullerene cages in the addition reactions.
The bond distribution in the researched higher fullerenes was carried out in accordance with the following rules: the bond distribution should not lower the fullerene molecular symmetry; corannulene and indacene substructures that are characteristic for the most stable fullerenes C60 and C70 are preferred; pentagons consist of single bonds; there are two types of hexagons, an alternation of single and double bonds for the first ones, and π-bonds delocalization for the second type of hexagons [3, 4]; the electronic parameters of the substructures (bonds lengths, valence angles, dihedral angles, electronic and spin densities) are mainly preserved in any fullerene molecule (for details see the Refs. [5, 32] with supplements).
However, our approach was used only for higher fullerenes. It is obvious that in small fullerenes the pentagon numbers are comparable with hexagon numbers (for example, in fullerene C50 there are 12 pentagons and 15 hexagons). Considering the constant number of pentagons in any fullerene, the ratio of hexagons and pentagons will also decrease with a decrease in the number of carbon atoms: so, in fullerene C60 this ratio is 1.67, whereas in fullerenes C40 and C50 it will be 0.83 and 1.25, respectively. Accordingly, due to many pentagon–pentagon bonds appearances, some of the above-mentioned rules may not be fulfilled.
In this report, an analysis that was previously used only for higher fullerenes was carried out for the small fullerene C50 to estimate its applicability to small fullerenes, as well as to find features of molecular structures of both non-IPR isomers of C50 which are responsible for their instability and that have not been disclosed previously.
The molecular structures of the investigated isomers of C50 fullerene were fully optimized using DFT B3LYP and M06-2X functionals [33,34,35] with 6-31G and 6-31G(d) basis sets. In the first step, geometry optimization was performed without symmetry constraints. The calculations showed that in all cases, except for triplet configuration of isomer 270 (D3), the symmetry of equilibrium geometry corresponds to the topological molecular symmetry of each isomer. Therefore, subsequent optimization was carried out with the corresponding symmetry constraint. To improve energies, geometry optimization was followed by single-point calculation with 6-311 + G** basis. The calculations have shown a good agreement between the results obtained for all used basis sets.
Since the researched isomers of C50 fullerene were treated also like molecules with the open-shell electronic structures, for these configurations the quantum-chemical calculations were carried out both in singlet and triplet configurations using unrestricted Kohn–Sham methodology. To ensure the calculated structures were indeed minima, vibrational analyses were performed using the same methods. The tests of stability of singlet and triplet wave functions were carried out. All calculations were carried out using Gaussian’09 program [36]. The standard keywords in the Gaussian package were used in optimization processes.
Magnetic shieldings were computed with the direct gauge-including atomic orbitals (GIAO)-SCF method as implemented in Gaussian’09 program [36]. Isotropic chemical shifts, reported as the magnetic shieldings with reversed sign, were evaluated for the center of mass of each individual ring (NICS(0)). The quantum-topological analysis of calculated electron densities was performed by means of the AIMAll package (v 19.10.12) [37].
3 Results and discussion
The calculated main energetic parameters of fullerene molecules in question show that both isomers have closed electronic shells with HOMO–LUMO gaps of 2.27 and 1.33 eV (B3LYP/6-311 + G**) for isomers 270 (D3) and 271 (D5h), respectively (Table 1), and this is in good agreement with the published data [18, 19, 25, 27, 29, 38] (Table S1). Application of the hybrid functional M06-2X from Minnesota group of highly parameterized approximate exchange–correlation energy functionals showed that more HF exchange (54%) [35] could favor strong electronic delocalization and artificially favor isomer 271 (D5h) relative to isomer 270 (D3). Nevertheless, the M06-2X computations also show the closed character of their electronic shells (Table 1).
When analyzing molecular structures of higher fullerenes, we showed that the fullerene molecule could be considered as a set of substructures [3, 4]. These substructures are connected with (or separated from) the rest of the molecule by single bonds. It preserves the intrinsic geometric and electronic characteristics of the substructure since single bonds of the fullerene molecule are poor conductors of delocalization in curved fullerene cage [39]. The corannulene and s-indacene substructures that constitute the most stable fullerenes C60 (Ih) and C70 (D5h), respectively, are preferred and easy to find. A distribution of single, double and delocalized π-bonds in the molecules of researched isomers 270 (D3) and 271 (D5h) of fullerene C50 was supported and specified by quantum-chemical calculations and is presented here for the first time (Figs. 2, 3).

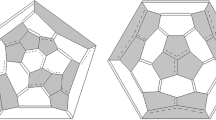
Molecule of isomer 271 (D5h) of fullerene C50: a three-dimensional model, two projections (yellow shows central pentagons of a pair of corannulene substructures; green shows fragments of the equatorial belt); b two-dimensional picture (Schlegel diagram) of bonds distribution with computed NICS(0) values of the individual rings (values not shown are determined by symmetry). Hereinafter: Schlegel diagram marks single and double bonds as single and double lines, respectively; delocalized π-bonds in hexagon are marked by circles, in chains—by dotted lines

Molecule of isomer 270 (D3) of fullerene C50: a three-dimensional model, two projections (yellow shows phenalenyl-like substructures; green shows one of three fragments of the equatorial belt); b two-dimensional picture (Schlegel diagram) of bonds distribution with computed NICS(0) values of the individual rings (values not shown are determined by symmetry); c phenalenyl-radical substructure [44] and d phenalenyl-like substructure
The high symmetry of the isomer 271 (D5h) molecule was preserved in its derivative molecule C50Cl10; its structure was revealed by 13C NMR [9]; a single-crystal X-ray diffraction analysis unambiguously confirmed it [10]. The molecule of fullerene 271 (D5h) has a geodesic shape of a biconvex lens flattened at the poles (Fig. 2a), on which it is easy to detect two corannulene substructures with the fifth-order symmetry axis that passes through the centers of corannulene pentagons, while the equatorial belt of the molecule consists of alternating hexagons with delocalized bonds and pentalene substructures (Fig. 2b).
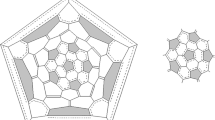
It is interesting to note that the substructures, which form the equatorial belt of molecule 271 (D5h) (Fig. 2b), also occur in the molecule of isomer 270 (D3) (Fig. 3b). The results of quantum-chemical calculations of bond lengths support this substructure with delocalized bonds unlike to substructure with double bonds; all bonds in these hexagons are nearly equal: for isomer 271 (D5h) they are in the range 1.415–1.418 Å (Table S2) and for isomer 270 (D3) they are in the range 1.416–1.431 Å (Table S3). This is also in good agreement with the previously published results showing the preference for benzene-like bonds distribution on the equatorial belt [17, 29]. Another confirmation of the π-delocalization in central hexagons was obtained during our joint analysis of the experimentally obtained data [9, 10] of the addends distribution in exohedral derivative C50Cl10 and the structure of isomer 271 (D5h) molecule. As was shown earlier [9, 10], exohedral derivative C50Cl10 forms a Saturn-like chlorofullerene molecule with 10 chlorine atoms symmetrically bonded to five pairs of two fused pentagons on the equatorial region of the fullerene, thus preserving the D5h point group. As one can see, chlorine atoms are attached at pentagon–pentagon junctions. On the other hand, one can consider chlorine atoms addition as joining to the indacene-like substructures, confirming our recent conclusions about preference of π-delocalized hexagons in radical addition reactions (Fig. 4) [40,41,42], which have been supported by the recent X-ray experimental evidence of radical addition of 12 CF3 groups exclusively to the equatorial belt consisting of six delocalized hexagons of IPR isomer 24 (D6h) of C84 fullerene [43]. In contrast, reactions of radical addition to double bonds are a very rare phenomenon [23, 24].

Besides the three above-mentioned substructures with π-delocalization of central hexagon, the molecule of isomer 270 (D3) also has a pair of equivalent substructures (Fig. 3d) that are similar to the phenalenyl–radical substructure (Fig. 3c). We expected, that such substructure of triple hexagons with a three pentalene environment may be the radical one like phenalenyl–radical, as in the molecule of fullerene C74 (D3h) [44] or some other open-shell higher IPR fullerenes [3,4,5, 32]. However, as was shown earlier [14,15,16, 18,19,20,21,22, 25, 29, 30], and also according to our calculations (Table 1), isomer 270 (D3) and isomer 271 (D5h) have a closed shell. A detailed analysis of the calculated bond lengths revealed a chain of delocalized π-bonds passing through cycles of these substructures (Fig. 3b) similar to previously found in non-IPR higher fullerenes C66 and C68 [6, 7]. This feature seems to be a sign of non-IPR fullerenes, and it was never found in higher IPR fullerene molecules.
Previously, it was stated that the nucleus-independent chemicals shifts (NICS) values can help to identify areas of higher local aromaticity or antiaromaticity (usually associated with hexagons and pentagons, respectively) in each fullerene [45]. Additional analysis of calculated chemical shifts of ring centers showed that the identified regions with delocalized bonds actually have a higher aromatic character (Figs. 2b, 3b).
Comparing Mulliken charge distributions in neutral molecules of isomers 270 (D3) and 271 (D5h) and their dianions, the main changes in the electron density distribution are observed on atoms of pentalene substructures for both studied isomers (Table S4) in order to coordinate endohedral metal cation(s). This is in good agreement with literature data regarding LUMO density localization [38] and suggesting that incorporation of endohedral metal can release pentalene strains due to partial charge transfer to pentalene substructures [31]. This may be useful for determining a position of metal atom(s) inside the cage. In accordance with this symmetrical pattern of electron density distribution and mostly electrostatic interaction of metal cation and fullerene anion, the position of metal is supposed to be close to pentalene substructures.
The similar results were obtained using the quantum theory of atoms in molecules developed by Bader [46, 47] that greatly complements the general charge density techniques. Quantum-topological analysis of electron density at bond critical points within Atoms in Molecules theory shows that bonds distribution performed according to our semiempirical approach coincides with observed topological parameters at bond critical points (Table S5): double bonds have a relatively high value of ρ and a negative value of ∇2ρ, and vice versa, single bonds have a relatively low value of ρ and a negative value of ∇2ρ.
In order to find exactly some structural parameters that determine the instability of studied isomers of C50, it makes sense to compare their structural parameters with those of experimental studied stable IPR isomers (e.g., most stable C60 and C70 fullerenes) [48, 49]. As a matter of fact, the corresponding bond lengths for single, double, and delocalized bonds and bond angles are all nearly equal (Table 2). The distortions of pentagons or hexagons in studied isomers of fullerene C50 (Fig. S2) are also comparable with calculated and experimentally measured values for stable fullerenes [4].
But non-IPR fullerenes that have pentalene fragments (here we do not touch any other pentagon combinations) are a special case. It is well known by the calculations as well as experimentally that the pair of fused pentagons of pentalene C8H6 or its derivatives are planar due to their sp2-hybridized atoms; the same is right concerning the molecule with linear chain of four fused pentagons [50, 51]; using the folding angle (F) as parameter (Fig. 5) F = 0° for those molecules. Now let’s take a look at the folding angles for pentalene fragments in the molecules in question (Table 3).

Folding angle F of pentalene substructure
The hypothetical molecules of the pristine isomers 271 (D5h) and 270 (D3) of C50 fullerene show folding angles near 44° (Table 3); it means that they experience high overstrain and therefore they cannot exist. However, the radical addition of chlorine atoms stabilizes the fullerene molecule as a whole due to changing the carbon hybridization from sp2 to sp3 and thus relieves the strain [9, 10]. This conclusion is consistent with ring strain relaxation in D5h–C50H10 derivatives from 529.4 to 225.1 kcal/mol (B3LYP/6-31G* level) [17]. So, the folding angles of pentagons in five pentalene substructures of the equatorial belt of isomer 271 (D5h) become equal to 68° (measured from X-ray data [10]). As to endohedral derivatives of these isomers, it would be useful to compare them with anion pentalene complexes with single metal cations [52], which have folding angles up to 400. It means that endohedral derivatives of our smaller fullerenes would have reasonable lacking of overstrain, and we may expect to obtain them.
Thus, instability of isomers 270 (D3) and 271 (D5h) of fullerene C50 is related to the high folding angle value of pentagons in pentalene fragments and, respectively, to local overstrains of the molecule. For fullerenes with substantially small sphere size, the influence of local strains on the molecule stability will obviously play a much larger role. A key role in stabilization of such fullerenes can be played by a decrease in strain caused by coordinating the endohedral metal atom with the pentalene substructures in endohedral derivatives of small fullerenes or by exohedral additions.
4 Conclusions
The distributions of single, double and delocalized π-bonds in both isomer molecules are presented for the first time, given their detailed structural formulas. It is found that the chains of π-bonds are passing through some cycles like in the previously studied higher non-IPR fullerenes C66 and C68. It is shown that chlorine atoms in the exohedral derivative C50Cl10 of isomer 271 (D5h) are attached to the indacene-like substructures confirming our recent conclusion about preference of addition to π-delocalized hexagons in radical reactions. The main factor of instability of them is high folding angle between pentagons of pentalene fragments, i.e., it is the reason of big local overstrains of the molecule. The study shows that our approach previously developed for higher fullerenes is also applicable for smaller fullerenes. The discovered features in the structures of non-IPR fullerene molecules (seems no matter smaller or higher ones) can be predictive of the ability to be synthesized as derivatives and will assist in determination positions of their reactivity. A huge number of non-IPR isomers of fullerene molecules from C20 up to C100 would open unlimited possibilities of tuning their useful properties, but unfortunately at present there are no reasonable ways to produce them.
Availability of data and materials
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
References
Kroto HW (1987) The stability of the fullerenes Cn, with n = 24, 28, 32, 36, 50, 60 and 70. Nature 329:529–531
Schmalz TG, Seitz WA, Klein DJ, Hite GE (1998) Elemental carbon cages. J Am Chem Soc 110:1113–1127
Kovalenko VI, Khamatgalimov AR (2006) Regularities in the molecular structure of stable fullerenes. Russ Chem Rev 75:981–988
Khamatgalimov AR, Kovalenko VI (2016) Structures of unstable isolated-pentagon-rule fullerenes C72–C86 molecules. Russ Chem Rev 85:836–853
Khamatgalimov AR, Kovalenko VI (2018) Radical IPR fullerenes C74 (D3h) and C76 (Td): dimer, trimer, etc. experiments and theory. J Phys Chem C 122:3146–3151
Khamatgalimov AR, Kovalenko VI (2008) The structure of fullerene C66, which does not obey the rule of isolated pentagons, and endohedral metallofullerene Sc2@C66: quantum-chemical calculations. Russ J Phys Chem A 82:1164–1169
Khamatgalimov AR, Korolev SS, Arkhipov AA et al (2008) Stability of the non-IPR isomers 6140 (D3) and 6275 (D3) of fullerene C68. Fullerenes Nanotubes Carbon Nanostruct 16:16542–16545
Khamatgalimov AR (2016) Structure and stability of higher fullerenes in a row C60–C86. Dr. Sc. Thesis, Arbuzov Institute of Organic and Physical Chemistry, FRC Kazan Scientific Center, Russian Academy of Sciences, Kazan, Russia (in Russ.)
Xie SY, Gao F, Lu X et al (2004) Capturing the labile fullerene[50] as C50Cl10. Science 304:699
Han X, Zhou SJ, Tan YZ et al (2008) Crystal structures of saturn-like C50Cl10 and pineapple-shaped C64Cl4: geometric implications of double- and triple-pentagon-fused chlorofullerenes. Angew Chem Int Ed 47:5340–5343
Zhang BL, Wang CZ, Ho KM et al (1992) The geometry of small fullerene cages: C20 to C70. J Chem Phys 97:5007–5011
Slanina Z, Adamowicz L, Bakowies D et al (1992) Fullerene C50 isomers: temperature-induced interchange of relative stabilities. Thermochim Acta 202:249–254
Bakowies D, Thiel W (1991) MNDO study of large carbon clusters. J Am Chem Soc 113:3704–3714
Xu WG, Wang Y, Li QS (2000) Theoretical study of fullerene C50 and its derivatives. J Mol Struct Theochem 531:119–125
Chen ZF, Jiao HJ, Buhl M et al (2001) Theoretical investigation into structures and magnetic properties of smaller fullerenes and their heteroanalogues. Theor Chem Acc 106:352–363
Aihara J, Oe S (2002) Local aromaticities in fullerenes as estimated by the bond resonance energy model. Internet Electron J Mol Des 1:443–449
Lu X, Chen ZF (2005) Curved Pi-conjugation, aromaticity, and the related chemistry of small fullerenes (< C60) and single-walled carbon nanotubes. Chem Rev 105:3643–3696
Zhao X (2005) On the structure and relative stability of C50 fullerenes. J Phys Chem B 109:5267–5272
Díaz-Tendero S, Alcamí M, Martín F (2005) Fullerene C50: sphericity takes over, not strain. Chem Phys Lett 407:153–158
Sun G, Nicklaus MC, Xie RH (2005) Structure, stability, and NMR properties of lower fullerenes C38–C50 and azafullerene C44N6. J Phys Chem A 109:4617–4622
Wang DL, Shen HT, Gu HM et al (2006) Ab initio studies on the molecular electrostatic potential of C50. J Mol Struct Theochem 776:47–51
Shao N, Gao Y, Zeng XC (2007) Search for lowest-energy fullerenes 2: C38 to C80 and C112 to C120. J Phys Chem C 111:17671–17677
Xu Z, Han J, Zhu Z et al (2007) Valence of D5h, C50 fullerene. J Phys Chem A 111:656–665
Xu X, Shang Z, Li R et al (2009) From the molecular behaviors of fullerene derivatives C50X2 (X = H, F, Cl, Br, OH) to the general parallels among isostructural derivatives of fullerenes and carbon nanotubes. Phys Chem Chem Phys 11:8560–8569
Tang L, Sai L, Zhao J et al (2011) Nonclassical Cn (n = 30–40, 50) fullerenes containing five-, six-, seven-member rings. Comp Theor Chem 969:35–43
Zhang ZQ, Chen SF, Gao CL et al (2016) Regioselective oxidation of fused-pentagon chlorofullerenes. Inorg Chem 55:543–545
Miralrio A, Muñoz-Castro A, King RB et al (2019) M@C50 as higher intermediates towards large endohedral metallofullerenes: theoretical characterization, aromatic and bonding properties from relativistic DFT calculations. J Phys Chem C 123:1429–1443
Fowler PW, Manolopoulos DE (2007) An atlas of fullerenes. Dover, Mineola
Lu X, Chen Z, Thiel W et al (2004) Properties of fullerene[50] and D5h decachlorofullerene[50]: a computational study. J Am Chem Soc 126:14871–14878
Mulet-Gas M, Abella L, Dunk PW et al (2015) Small endohedral metallofullerenes: exploration of the structure and growth mechanism in the Ti@C2n (2n = 26–50) family. Chem Sci 6:675–686
Tan YZ, Xie SY, Huang RB et al (2009) The stabilization of fused-pentagon fullerene molecules. Nat Chem 1:450–460
Khamatgalimov AR, Melle-Franco M, Gaynullina AA et al (2019) Ythrene: from the real radical fullerene substructure to hypothetical (yet?) radical molecules. J Phys Chem C 123:1954–1959
Becke AD (1993) Density-Functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Zhao Y, Truhlar DG (2006) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Acc 120(1–3):215–241
Frisch MJ, Trucks GW, Schlegel HB et al (2009) Gaussian 09, Revision A.1. Gaussian, Inc.: Wallingford, CT
AIMAll (Version 19.10.12), Todd A. Keith, TK Gristmill Software, Overland Park KS, USA, 2019
Sanz Matías A, Havenith RWA, Alcamí MD et al (2016) Is C50 a superaromat? Evidence from electronic structure and ring current calculations. Phys Chem Chem Phys 18:11653–11660
Hirsch A, Brettreich M (2005) Fullerenes. Chemistry and reactions. WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
Kovalenko VI, Tuktamysheva RA, Khamatgalimov AR (2014) Electronic structures of some of C84 fullerene isomers and the structures of their perfluoroalkyl derivatives. Russ J Phys Chem A 88:103–107
Tuktamysheva RA, Khamatgalimov AR, Kovalenko VI (2014) Electronic and geometric structures of some isomers of fullerene C90 and the structures of their chlorides and perfluoroalkyl polyadducts. Butlerov Commun 37:1–12 (in russ)
Khamatgalimov AR, Kovalenko VI (2012) 24 IPR isomers of fullerene C84: cage deformation as geometrical characteristic of local strains. Int J Quantum Chem 112:1055–1065
Tamm NB, Troyanov SI (2016) A minor isomer of C84 fullerene, D6h–C84(24), captured as a trifluoromethylated derivative, C84(CF3)12. Mendeleev Comm 26:312–313
Kovalenko VI, Khamatgalimov AR (2003) Open-shell fullerene C74: phenalenyl–radical substructures. Chem Phys Lett 377:263–268
Bühl M (1998) The relation between endohedral chemical shifts and local aromaticities in fullerenes. Chem Eur J 4(4):734–739
Bader RFW (1991) A quantum theory of molecular structure and its applications. Chem Rev 91:893–928
Bader RFW (1994) Atoms in molecules: a quantum theory. Clarendon Press, Oxford
Hedberg K, Hedberg L, Bethune DS et al (1991) Bond lengths in free molecules of buckminsterfullerene, C60, from gas-phase electron diffraction. Science 254:410–412
Hedberg K, Hedberg L, Buhl M et al (1997) Molecular structure of free molecules of the fullerene C70 from gas-phase electron diffraction. J Am Chem Soc 119:5314–5329
Havenith RWA, Engelberts JJ, Fowler PW et al (2004) Localisation and reversal of paratropic ring currents in molecules with formal anti-aromatic electron counts. Phys Chem Chem Phys 6:289–294
Garcia Cuesta I, Coriani S, Lazzeretti P et al (2006) From pentalene to dicyclopenta[b,g]naphthalene, or the change towards delocalized structures. Chem Phys Chem 7:240–244
Summerscales OT, Cloke FGN (2006) The organometallic chemistry of pentalene. Coord Chem Rev 250:1122–1140
Acknowledgements
This work was financial support from the government assignment for FRC Kazan Scientific Center of RAS and partially supported by the Russian Foundation for Basic Research under Grant No. 18-29-19110mk.
Author information
Authors and Affiliations
Contributions
The manuscript was written with contributions from all authors. All authors have approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Khamatgalimov, A.R., Yakupova, L.I. & Kovalenko, V.I. Features of molecular structure of small non-IPR fullerenes: the two isomers of C50. Theor Chem Acc 139, 159 (2020). https://doi.org/10.1007/s00214-020-02675-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-020-02675-z