
Abstract
The detailed reaction mechanism of 3-chloro-2-methyl-1-propene (3CMP) with OH radical was investigated by employing highly accurate electronic structure calculations and kinetic modelling. Due to the unsaturated structure of 3CMP, it is highly reactive in the troposphere with OH radical. The fate of the so-formed alkyl radical intermediates and intermediate adducts in the favourable pathways is determined by its reaction with other atmospheric oxidants, such as HO2, NO and NO2 radicals. The rate constants computed within the temperature range of 200–1000 K for the favourable hydrogen atom abstraction and OH radical addition reactions are in reasonable agreement with the available experimental values. The oxidation of 3CMP results in the formation of stable chlorinated products, such as chloroacetone and formyl chloride, which are identified experimentally. These products may be transported to the stratosphere which will affect the ozone layer.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The release of volatile organic compounds (VOC) into the atmosphere is witnessed for the past few decades from anthropogenic and biogenic sources [1, 2]. The concentration of VOC in the atmosphere gone up at an alarming rate imparting a lot of environmental issues. Halogenated hydrocarbons (HHC) are a subclass of VOC, which are omnipresent in the environment due to their extensive use in the industrial, agricultural and household applications. Since the beginning of the twentieth century [3, 4], the substantial increase in concentration of HHC in the environment is a threat to human health [5, 6]. The major fate of HHC is their reaction with oxidants present in the atmosphere [7, 8].
3-Chloro-2-methyl-1-propene (3CMP) is one amongst the HHCs used as organic solvents in various industrial applications [9]. In addition, 3CMP is used as an intermediate for the synthesis of organic compounds, pesticides and as an additive in the textile industry [10,11,12]. The widespread use of 3CMP in various applications inevitably leads to its emission in the atmosphere causing environmental pollution. Due to its unsaturated structure, 3CMP is highly reactive in the troposphere. A well-established removal process for 3CMP in the atmosphere is the direct photolysis and reactions with the tropospheric oxidants, such as OH·, O3 and NO·3. The oxidation of 3CMP results in the formation of stable chlorinated products which can be transported to the stratosphere, causing additional loading of chlorine compounds with the impact on the ozone layer [12, 13]. Amongst all of the oxidizing species present in the atmosphere, OH radical is abundantly present in the troposphere and acts as a sink for organic compounds.
The oxidation of 3CMP by OH radical was experimentally studied by Rivela et al. [11] and Zhang et al. [12] by GC-FID technique at 298 K and 1 atm pressure. The room temperature rate constant for the oxidation of 3CMP by OH radical was reported as (3.23 ± 0.35) × 10−11 cm3 molecule−1 s−1 by Rivela et al. [11] and (3.83 ± 1.3) × 10−11 cm3 molecule−1 s−1 by Zhang et al. [12]. Begum et al. [14] theoretically studied the H-atom abstraction and OH-addition reactions of 3CMP with OH radical and reported the total rate constant for H-atom abstraction reaction as 1.12 × 10−13 cm3 molecule−1 s−1 and for OH-addition reaction as 2.5 × 10−11 cm3 molecule−1 s−1 at M06-2X/6-31+G(d,p) level of theory. The present investigation aims to explore the secondary reaction mechanism of the promising intermediates formed by the oxidation of 3CMP by OH radical. The favourable pathways for the formation of products were identified by comparing the energy barriers (∆E) and reaction heats (∆H) calculated for all the secondary reaction pathways. For completeness and comparison purpose, the reaction pathways and the rate constants for the primary reaction pathways were also studied.
2 Computational details
The geometry optimization and harmonic vibrational frequency calculations of all the stationary points involved in the reaction of 3CMP with OH radical were performed using the density functional theory methods, M06-2X [15,16,17,18] and ωB97XD [19], in conjunction with 6-311++G(d,p) basis set. Previous study by Zhao and Truhlar [16] has shown that the M06-2X functional of the density functional theory (DFT) has the best performance for predicting the barrier height and provides reliable results for thermochemistry and kinetics, without increasing the computational cost. All minima along the potential energy surface of the studied reactions were identified with zero imaginary frequency, and each transition state was identified with one imaginary frequency. Intrinsic reaction coordinate (IRC) [20, 21] calculations were performed to verify that the calculated transition state connects the respective reactant and product at all the above-mentioned levels of theories. In addition, to ascertain the accuracy of the DFT results, we have also performed the single-point energy calculations at the CCSD(T) level with the 6-311++G(d,p) basis set on the basis of the geometries optimized at the M06-2X/6-311++G(d,p) level of theory. CCSD(T) stands for a couple cluster single- and double-substitution method with a perturbative treatment of triple excitations [22]. The CCSD(T) method is the most popular ab initio method for the application to atmospheric chemistry problems [23]. The enthalpy of the reaction and Gibbs free energy values were calculated by including thermodynamic corrections to the energy at 298.15 K and 1 atm pressure. The thermochemical parameters, like enthalpy and Gibbs free energy for all the initial reaction pathways, were also calculated using highly accurate composite method, ROCBS-QB3 [24, 25]. All of the electronic structure calculations reported in this article were performed using Gaussian 09 [26] program.
The kinetic calculations for the titled reactions were performed on the basis of potential energy surface information obtained from M06-2X/6-311++G(d,p) level of theory. The rate constant for the favourable reactions of 3CMP with OH radical was calculated based on canonical variational transition state theory [27,28,29], including quantum tunnelling effect [28, 29]. The kinetic calculations were performed by using the GAUSSRATE 2010A [30] program which is an interface program between the Gaussian 09 and POLYRATE 2010A [31] programs.
3 Results and discussion
In the present investigation, the reactivity of 3CMP with OH radical was studied by considering all possible H-atom abstraction and OH radical addition pathways. The possible addition and abstraction pathways for the reactions of 3CMP with OH radical are shown in Scheme 1. Similar to the oxidation mechanism of volatile organic compounds initiated by OH radical [7, 32], the reaction between 3CMP and OH radical proceeds through an indirect mechanism in which pre- and post-reactive complexes was formed. Hydrogen bonding interaction, C–H…O and Cl…H–O, between 3CMP and OH radical results in the formation of reactant and product complexes.

Proposed reaction scheme for the hydrogen atom abstraction and OH-addition pathways of 3CMP + hydroxyl radical reaction
The structure of the reactant complexes, transition states and product complexes obtained from the M06-2X and ωB97XD methods is found to be quite similar. For example, average root-mean-square deviation (rmsd) between the internal coordinates obtained from M06-2X and ωB97XD methods for the reactant complex, transition state and intermediate complex of the favourable hydrogen atom abstraction pathway, R1, is 0.1 Å, 0.06 Å and 0.09 Å, and the rmsd for the OH radical addition pathway, R4, is 0.1 Å, 0.08 Å and 0.03 Å, and for pathway, R5, the rmsd is 0.1 Å, 0.06 Å and 0.01 Å, respectively. The similar rmsd values were found for the reactive species involved in other reactions. Note that the above-mentioned rmsd values include both bonded and non-bonded interacting atoms.
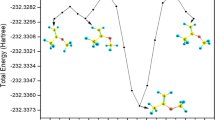
Table 1 lists the calculated energetical parameters of all the species involved in the primary reaction mechanism of 3CMP + OH using DFT, couple cluster and composite method. As given in Table 1, the energetics calculated from M06-2X and ωB97XD methods are comparable within the maximum deviation of 2 kcal/mol. The thermochemical parameters calculated from M06-2X method differ by 2–3 kcal/mol with respect to ROCBS-QB3 method which is a reasonable difference expected between DFT and composite methods. While comparing the results obtained with the methods mentioned above, it is observed that the most favourable reaction and the nature of the reaction mechanism are similar with respect to the methods of calculation. The optimized structure of all the TS and other species involved in the reaction mechanism using M06-2X/6-311++G(d,p) level of theory is shown in Fig. 2. In the present investigation, the geometry and the energetics obtained using M06-2X functional with 6-311++G(d,p) basis set are discussed in detail and are used in further kinetic calculations. Figure 1 depicts the potential energy surface of 3CMP + OH· reactions obtained at M06-2X/6-311++G(d,p) level of theory.

Potential energy profile for the hydrogen atom abstraction and OH-addition pathways of 3CMP with hydroxyl radical calculated at M06-2X/6-311++G(d,p) level of theory
3.1 Initial reactions of 3CMP + OH·
3.1.1 H-atom abstraction reaction pathways
The proposed initial reaction scheme consists of three potential hydrogen atom abstraction pathways, namely H-atom abstraction at methylene (=CH2), methyl (–CH3) and chloromethyl (–CH2Cl) sites. Due to the presence of equivalent hydrogen atoms present in 3CMP, we consider only three hydrogen atom abstraction pathways, R1–R3, and is shown in Scheme 1. RC1 and RC2 are the reactant complexes formed to initiate the reaction pathways, R1, R2 and R3. These reactant complexes proceed towards the formation of intermediate complexes, IC1, IC2 and IC3, through the transition states, TS1, TS2 and TS3. Thus, formed intermediate complexes were identified as a true minimum on the potential energy surface, which further breaks into water molecule and C-centred radicals, I1, I2 and I3. The energy barrier calculated for the hydrogen atom abstraction from C1, C4 and C3 carbon atoms at M06-2X/6-311++G(d,p) level of theory is 12.0, 8.2 and 7.2 kcal/mol, respectively. At ωB97XD/6-311++G(d,p) level of theory, the energy barrier associated with TS1, TS2 and TS3 is 10.4, 7.3 and 5.3 kcal/mol, and at CCSD(T)/6-311++G(d,p)//M06-2X/6-311++G(d,p) level of theory, the energy barrier is 12.2, 7.6 and 6.1 kcal/mol, respectively. The calculated energy barriers at various levels of theory indicate that the H-atom abstraction from C3 and C4 carbon atoms (pathways R2 and R3) is the feasible reactions.
While analysing the geometry of the transition states, TS1, TS2 and TS3, it is observed that the breaking C-H bond is elongated by 0.1 Å with respect to the respective reactant complex, and the distance between the cleaved H-atom and O-atom of the OH radical is shorter by around 0.5 Å with respect to that of the corresponding reactant. The hydrogen atom abstraction reaction from methylene site (C1) of 3CMP is slightly exothermic and exoergic with ∆H298 = −5.83 kcal/mol and ∆G298 = −6.78 kcal/mol. For the removal of hydrogen atom from methyl (C4) and chloromethyl (C3) sites of 3CMP, the ∆H298 and ∆G298 values are found to be − 26.53 and − 27.67 kcal/mol and − 30.56 and − 31.23 kcal/mol. That is, these reactions are highly exothermic in nature. Begum et al. [14] studied the reaction of 3CMP with OH radical at M06-2X/6-31+G(d,p) level of theory and reported the enthalpy of the reactions, R1–R3, as − 4.81, − 26.37 and − 30.01 kcal/mol, which are in agreement with the present results. On comparing the energy barriers and thermochemical parameters calculated for the initial hydrogen atom abstraction reactions, it is found that the hydrogen atom abstraction from C4 (pathway R2) and C3 (pathway R3) carbon atoms is found to be the favourable reaction pathways.
3.1.2 OH radical addition reaction pathways
As shown in Scheme 1, the next significant pathway involved is the electrophilic addition of OH radical to 3CMP. Since the two carbon atoms, C1 and C2, on the double bond in 3CMP are in different environment, two OH radical addition pathways, R4 and R5, were considered. The addition of OH radical at the C1 and C2 positions of 3CMP leads to the formation of 3CMP–OH adducts, I4 and I5, through the transition states TS4 and TS5, with an energy barrier of 1.53 and 1.9 kcal mol. As shown in Fig. 2, in TS4 and TS5 the distance between the O-atom of the OH radical and the carbon atoms, C1 and C2 of 3CMP molecule, is 2.15 Å (O1–C1) and 2.13 Å (O1–C2) which is shorter by 0.6 Å with respect to that of the respective reactant complex. The 3CMP-OH adducts, I4 and I5, are formed in an exothermic reaction with an enthalpy of − 32.29 and − 30.65 kcal/mol and exoergic with ∆G298 of − 22.83 and − 19.81 kcal/mol. The enthalpy of formation of adducts, I4 and I5 reported by earlier study at M06-2X/6-31+G(d,p) level of theory is − 31.48 and − 34.53 kcal/mol. Upon comparing the energy barrier and relative enthalpy values of the studied addition pathways, it is observed that both the studied OH radical addition pathways, R4 and R5, are the most favourable reaction pathways.

Optimized structure of all the reactive species involved in the initial reaction of 3-chloro-2-methyl-1-propene with hydroxyl radical
The rate constants for the favourable H-atom abstraction and OH-addition reactions, R2, R3, R4 and R5, are computed using canonical variational transition state theory (CVT) with small curvature tunnelling correction, SCT (CVT/SCT), over the temperature range from 200 to 1000 K with zero point corrected energies, gradients and Hessians calculated at M06-2X/6-311++G(d,p) level of theory. The rate constant calculated using the transition state theory (TST), CVT, TST/SCT and CVT/SCT methods is summarized in Table S1. At 298 K, the rate constant calculated for the formation of I2, I3, I4 and I5 is 1.00 × 10−14, 2.10 × 10−14, 2.54 × 10−11 and 1.42 × 10−11 cm3 molecule−1 s−1. The rate constants calculated for pathways R2, R3 and R5 agree well with the previously reported rate constant value, while for pathway R4, the calculated rate constant is higher by two orders of magnitude than that reported by Begum et al. [14]. The transmission coefficient for the pathways R2 and R3 was ~ 1 showing a negligible tunnelling effect on the rate constants, whereas an appreciable variational effect is noticed for the studied reactions over all the studied temperature range. As observed from Table S1, for the OH radical addition reactions (pathways R4 and R5), the ratio between CVT(SCT)/CVT and CVT/TST rate constant is ~ 1 showing a negligible tunnelling and variational effect over the studied temperature range. Arrhenius plot for the H-atom abstraction and OH radical addition reaction rate constant is shown in Fig. 3.

Arrhenius plot for the rate constant obtained for the formation of the intermediates I2, I3, I4 and I5 at a higher (550–1000 K) and b lower (200–500 K) temperature range
From Arrhenius plot, it is observed that the rate constant for the H-atom abstractions pathways R2 and R3 shows a positive temperature dependence while the rate constant for OH radical addition pathways R4 and R5 shows a negative temperature dependence in the range of 200–1000 K. The plot shows that the contribution of OH radical addition pathways, R4 and R5, is larger than that of the H-atom abstraction pathways over all the studied temperature range. The overall rate constant, k = 3.95 × 10−11 cm3 molecule−1 s−1, is in agreement with the reported experimental rate constant value [12,13,14]. The tropospheric lifetime of 3CMP can be calculated by the formula \( \tau = \frac{1}{{k_{ } \left[ {OH} \right] }} \), where [OH] is the average OH radical concentration in the troposphere. The lifetime of the 3CMP in the troposphere at 298 K and 1 atm is found to be 4 h while taking the 12-h daytime average OH radical concentration as 2 × 106 molecule cm−3 [33,34,35]. Due to the short lifetime of 3CMP, in the order of few hours, it could be degraded quickly in the troposphere and generates intermediates and products that could be the second pollutants. Hence, the atmospheric transformation of the radical intermediates I2, I3, I4 and I5 was studied by reacting them with O2, NO, HO2 radicals and through the decomposition reactions.
3.2 Atmospheric transformation of radical intermediate, I2
The scheme for the secondary reactions of the radical intermediate, I2, is shown in Scheme 2. The optimized structure of the transition states and other reactive species involved in the secondary reactions of the alkyl radical intermediates, I2, is shown in Fig. 4 and S1. The relative energy, enthalpy and Gibbs free energy are summarized in Table S2 (Supplementary material), and the profile of the potential energy surface is depicted in Fig. 5. The radical intermediate, I2, can readily react with O2 to form a peroxy radical intermediate, I6. The profile of the potential energy surface shows that the reaction of I2 with O2 has to overcome a barrier of 4.89 kcal/mol and is exothermic by − 19.18 kcal/mol. In the structure of the transition state, TS6, the C4–O2 bond distance is 2.097 Å which is 0.6 Å shorter than that of the intermediate, I2. In the atmosphere, I6 is further oxidized by NO· to form alkoxy radical intermediate, I7, and NO2. The formation of I7 is a barrierless reaction with an exothermicity of − 16.34 kcal/mol, and the reaction is exoergic with ∆G298 of − 15.57 kcal/mol. The exit pathway for alkoxy radical is its reaction with HO2 radical to form the product 2-chloromethyl-prop-2-en-1-ol (P1) along with O2 through the transition state, TS7, with an energy barrier of 9.35 kcal/mol. In TS7, the newly forming O–H bond length is 1.562 Å which is shorter by 0.6 Å than that of the intermediate complex I7 + HO2. The formation of the product 2-chloromethyl-prop-2-en-1-ol (P1) is exothermic with an enthalpy of − 22.95 kcal/mol. As shown in Scheme 2, alkoxy radical, I7, can directly decompose into methanol (P2) along with an intermediate, I8, through the transition state, TS8, with a potential barrier of 26.53 kcal/mol. In TS8, C2–C4 bond length is 2.187 Å which is 0.7 Å greater than that of the alkoxy radical, I7. However, this methanol formation reaction is highly endothermic and endoergic process with ∆H298 = 20.09 and ∆G298 = 15.66 kcal/mol, that is, the reaction is unfeasible in the atmosphere.

Possible secondary reactions from alkyl radical intermediate, I2. The relative energy (∆E) is in kcal/mol

Optimized structure of the transition states involved in the secondary reactions of the alkyl radical intermediates, I2

Relative energy profile for the subsequent reactions of radical, I2. The relative energy (kcal/mol) calculated at M06-2X/6-311++G(d,p) level of theory is given for the reactive species
3.3 Atmospheric transformation of radical intermediate, I3
Similar to I2, the initially formed alkyl radical, I3, can react with O2 and further undergo secondary reaction with HO2 and NO radicals in the atmosphere. The scheme for the secondary reactions of I3 is shown in Scheme 3, and the relative energy profile for the corresponding reactions is shown in Fig. 6. The optimized structure of the transition states and other reactive species is shown in Fig. 7 and S2. The relative energy, enthalpy and Gibbs free energy are summarized in Table S3 (Supplementary material).

Possible secondary reactions from alkyl radical intermediate, I3. The relative energy (∆E) is in kcal/mol

Relative energy profile for the reaction of intermediate adduct, I3, with O2, NO, HO2 radical

Optimized structure of the transition states involved in the secondary reactions of the alkyl radical intermediates, I3
The O2 addition at the C3 radical site of I3 results in the formation of peroxy radical intermediate, I9, through the transition state, TS9, with an energy barrier of 3.55 kcal/mol. In the transition state, TS9, the C3–O2 bond distance is 2.104 Å which is 0.7 Å shorter than that of the reactant. The reaction corresponding to the formation of I9 is exothermic and exoergic with ∆H298 = −15.43 and ∆G298 = −11.73 kcal/mol. Reaction of peroxy radical with HO2 is of central importance in the atmosphere, as it serves as sink for HO2 radical and terminates the ozone-generating chain reactions [36, 37]. Alkylperoxy radical (RO2) plays an important role in global and regional atmospheric chemistry. The reaction of I9 with hydroperoxy radical will lead to the formation of a stable product, 1-chloro-2-methyl-prop-2-en-1-yl-hydroperoxide (P3), along with O2 through the transition state, TS10, with an energy barrier of 2.45 kcal/mol. In the transition state, TS10, the bond length between H8- and O1-atom is 1.667 Å which is lower than the distance between H8 and O1 atoms of the reactant complex, I9 + HO2, by 0.6 Å. Here the H8-atom of HO2 binds with O1-atom of I9 forming 1-chloro-2-methyl-prop-2-en-1-yl-hydroperoxide along with O2. The product, P3, is formed in an exothermic reaction with a reaction enthalpy of − 31.77 kcal/mol and Gibbs free energy of − 34.88 kcal/mol. When the concentration of NO· is sufficiently high, the loss of I9 is dominated by its reaction with NO·, which leads to the formation of alkoxy radical intermediate, I10, in a barrierless reaction with an exothermicity of − 14.68 kcal/mol, and the formation is exoergic with ∆G298 = −15.92 kcal/mol. The formed alkoxy radical intermediate, I10, could further react with HO2 to form the product, 1-chloro-2-methyl-prop-2-en-1-ol (P4), and O2 through the transition state, TS11, with an energy barrier of 3.93 kcal/mol. In TS11, the newly forming O1–H8 bond length is 1.589 Å which is shorter by 0.4 Å than that of the reactant complex, I10 + HO2. The product, P4, is formed in an exothermic and exoergic process with ∆H298 = −41.58 and ∆G298 = −43.89 kcal/mol. Thus, the reaction of peroxy and alkoxy radicals with HO·2 is the exit pathway for radical intermediate (I3), resulting in the formation of a stable products, 1-chloro-2-methyl-prop-2-en-1-yl-hydroperoxide (P3) and 1-chloro-2-methyl-prop-2-en-1-ol (P4).
3.4 Atmospheric transformation of intermediate adduct, I4
The intermediate adduct, I4, formed from the reaction of 3CMP with OH radical is found to be the most promising radical intermediate, and hence, the subsequent reactions of I4 with atmospheric reactive species are more important. The reaction paths for I4 are shown in Scheme 4. The optimized geometry of the transition states and other reactive species is shown in Fig. 8 and S3, and the relative energy profile is shown in Fig. 9. The energy values are summarized in Table S4 (see supplementary material). As shown in Scheme 4, the energized intermediate adduct, I4, is expected to react rapidly with O2 to yield 2,1-hydroxy peroxyl radical, I11, in a barrierless reaction. The process is exothermic and exoergic with ∆H298 = −27.72 and ∆G298 = −24.01 kcal/mol. The dominant fate of peroxy radical in the atmosphere is its reaction with NO radical. The reaction of peroxy radicals with nitric oxide is of profound importance in the atmosphere, since it leads to the production of ozone from the photolysis of NO2 [38, 39]. As shown in Scheme 4, in the process of formation of 2,1-hydroxy alkoxy radical, I12, the nitric oxide radical directly abstracts the terminal oxygen atom of peroxy radical, I11, without the formation of a transition state. The formation of 2,1-hydroxy alkoxy radical, I12, is exothermic with ∆H298 = −9.86 kcal/mol and exoergic with ∆G298 = −8.8 kcal/mol. Further, the formed 2,1-hydroxy alkoxy radical, I12, can undergo two different decomposition reactions to form the stable products chloroacetone, methanol, 1-hydroxy-propan-2-one and formyl chloride. As shown in Fig. 8, the formation of product, chloroacetone (P5), in the first decomposition pathway needs to overcome an energy barrier of 6.24 kcal/mol. In the transition state, TS12, the distance between C1 and C2 atoms is increased by 0.5 Å with respect to that of 2,1-hydroxy alkoxy radical, I12. The product, P5, is accompanied with the hydroxyl methyl radical, I13, with an exothermicity of − 3.68 kcal/mol. The next exit pathway is the reaction of hydroxyl methyl radical, I13, with O2 which leads to the formation of methanol (P2) and HO2 through the transition state, TS13. The energy barrier, enthalpy and Gibbs free energy for the reaction between I13 and O2 are 12.07, − 23.82 and − 23.20 kcal/mol. In TS13, the distance between O1 and H3 is 1.301 Å, which is 0.3 Å longer than that in I13, and the distance between H3 and O3 is 1.133 Å which is 0.98 Å in HO·2.

Possible secondary reactions from radical intermediate adduct, I4. The relative energy (∆E) is in kcal/mol

Optimized structure of the transition states involved in the secondary reactions of the alkyl radical intermediates, I4

Relative energy profile for the reaction of intermediate adduct, I4, with O2, NO, HO2 radicals and decomposition reaction of alkoxy radical, I12
In the second decomposition pathway, the product, 1-hydroxy-propan-2-one (P6), is formed by the breaking of C2–C3 bond present in the alkoxy radical, I12, through the transition state, TS14. In the transition state, TS14, the C2-C3 bond was elongated by 0.5 Å with respect to that of alkoxy radical, I12. This reaction has an energy barrier of 16.1 kcal/mol and is exothermic by − 2.04 kcal/mol. The formed product, 1-hydroxy-propan-2-one (P6), is accompanied by chloromethyl radical (I14). Formation of chloromethyl radical in the atmosphere can load chlorine which adversely affects the stratospheric ozone profile [40]. Hence, it is important to study the atmospheric fate of chloromethyl radical, I14. As shown in Scheme 4, the chloromethyl radical, CH2Cl, can rapidly react with O2 yielding the chloromethylperoxy radical, I15. The reaction corresponding to the formation of chloromethylperoxy radical, I15, is an exothermic and exoergic process with ∆H298 = −27.63 and ∆G298 = −23.19 kcal/mol which is comparable to the value reported by Zhou et al. [41].
Further, the reaction of chloromethylperoxy radical, I15, with NO is of atmospheric interest because this reaction represents the predominant loss process of the CH2ClOO· in the troposphere. Under atmospheric condition, the chloromethylperoxy radical, I15, reacts with NO·. As shown in Fig. 8, chloromethylalkoxy radical, I16, is formed from the reaction of I15 and NO· with the reaction enthalpy and Gibbs free energy of − 15.41 and − 15.28 kcal/mol, respectively. The exit pathway for I16 is its reaction with O2 which leads to the formation of stable product, formyl chloride, P7, and HO2 in a highly exothermic reaction with the ∆H298 = −35.37 kcal/mol. Formyl chloride, P7, is a highly toxic compound which is either hydrolyzed to formic acid by taking cloud droplets or deposited to form secondary organic aerosol in the atmosphere [42].
3.5 Atmospheric transformation of intermediate adduct, I5
As shown in Scheme 5, the initially formed radical intermediate adduct, I5, can react with O2 and NO· to form alkyl peroxy and alkoxy radicals [7, 36]. The optimized structure of the transition states and other reactive species for the subsequent reactions of I5 is shown in Fig. 10 and S4. The calculated relative energy (ΔETot), enthalpy (ΔH298) and Gibbs energy (ΔG298) are summarized in Table S5 (Supplementary material), and the potential energy profile of the studied reaction is shown in Fig. 11. Similar to the secondary reactions of alkyl radical intermediate adduct, I5, O2 can bind at C1 site of I5 to form a 1,2-hydroxy peroxyl radical intermediate, I17, in a barrierless reaction. The reaction is exothermic and exoergic with ∆H298 = −31.06 and ∆G298 = −27.70 kcal/mol. As shown in Scheme 5, the 1,2-hydroxy peroxyl radical, I17, is further oxidized by NO· to form 1,2-hydroxy alkoxy radical, I18, and NO2 in a barrierless reaction. The formation of 1,2-hydroxy alkoxy radical, I18, is exothermic and exoergic with an enthalpy and free energy of − 10.93 and − 11.75 kcal/mol. As shown in Scheme 5, the exit pathway for the 1,2-hydroxy alkoxy radical, I18, is its reaction with O2 and decomposition reaction. The reaction between I18 and O2 leads to the formation of the product, 3-chloro-2-hydroxy-2-methyl-propionaldehyde (P8), along with HO2 through the transition state, TS15, with an energy barrier of 11.54 kcal/mol. The product P8 is formed in a highly exothermic and exoergic reaction with ∆H298 = −37.8 kcal/mol and ∆G298 = −35.77 kcal/mol. The second exit pathway studied for alkoxy radical, I18, is the decomposition reaction. The decomposition of 1,2-hydroxy alkoxy radical, I8, into methanol (P2) and a carbon-centred radical, I19, surmounts an energy barrier of 11.58 kcal/mol. The decomposition reaction is found to be exothermic and exoergic with reaction enthalpy of − 6.54 kcal/mol and Gibbs free energy of − 3.90 kcal/mol. Further, the carbon-centred radical intermediate, I19, can react with the molecular oxygen to form a stable product, chloroacetone (P5) and HO·2. The chloroacetone is the main product formed in the oxidation of 3CMP. The atmospheric lifetime of the parent molecule, 3CMP, is around 4 h, as reported in previous studies [43]; the lifetime of the product, chloroacetone, is 900 h. That is, the degradation of chloroacetone is very hard which will lead to adverse effect on the balanced atmosphere.

Reaction scheme for subsequent reactions of radical intermediate adduct, I5

Optimized structure of the transition states involved in the secondary reactions of the alkyl radical intermediates, I5

Relative energy profile for the reaction of intermediate adduct, I5, with O2, NO·, HO2 radicals and decomposition reaction of alkoxy radical, I13
4 Conclusion
The gas-phase reactions of 3-chloro-2-methyl-1-propene (3CMP) with OH radical are studied by using electronic structure calculations based on DFT methods and variational transition state theory. Five reaction pathways, three hydrogen atom abstraction reactions and two OH radical addition reactions were identified for the initial reaction. The calculated potential energy surface and thermochemical parameters show that the studied OH-addition reactions and H-atom abstraction reaction are the most favourable reactions. The rate constant is calculated for the favourable initial H-atom abstraction reactions (formation of radical intermediates, I2 and I3) and OH radical addition reactions (formation of intermediate adducts, I5 and I6). At 298 K, the calculated rate constant for H-atom abstraction reaction is 1.00 × 10−14 and 2.10 × 10−14 cm3 molecule−1 s−1, and for OH-addition reactions, the rate constant is 2.54 × 10−11 and 1.41 × 10−11 cm3 molecule−1 s−1. That is, OH-addition reactions are found to be more favourable than the H-atom abstraction reactions. The estimated lifetime of 3CMP in the normal atmospheric OH· concentration is 4 h. Calculated lifetime of 3CMP shows that it is easily degraded in the troposphere into reactive intermediates and then to end products which could be the second pollutants. Hence, the subsequent secondary reactions were studied for the initially formed alkyl radicals I2, I3 and 3CMP-OH adducts, I5 and I6, which results in the formation of stable products. Amongst the identified chlorinated products, chloroacetone and formyl chloride are the potential source for chlorine which may alter the ozone balance in the atmosphere. The reported reaction pathways, rate constants and products identified in the present work clearly infer the role of 3CMP in the regional atmosphere.
References
Laothawornkitkul J, Taylor JE, Paul ND, Hewitt CN (2009) New Phytol 183:27–51
Koppmann R (2007) Volatile organic compounds in the atmosphere. Wiley, London
Kodavanti PRS, Senthilkumar K, Loganathan G (2008) Int Encycl Public Health 4:686–693
Adriaens P, Gruden C, McCormick M (2003) Treatise Geochem 9:612
Budnik LT, Fahrenholtz S, Kloth S, Baur X (2010) J Environ Monit 12:936–942
Kittrell JR, Quinlan CW, Eldridge JW (1991) J Air Waste Manag Assoc 41:1129–1133
Atkinson R, Arey J (2003) Chem Rev 103:4605–4638
Finlayson-Pitts BJ, Pitts JN Jr (1999) Chemistry of the upper and lower atmosphere: theory, experiments, and applications. Elsevier, Amsterdam
Appendix C (1994) Chlorinated alkenes. Regul Toxicol Pharmacol 20:S757–S818
Carcinogens (2011) U.S. Department of Health and Human Services Secretary Kathleen Sebelius released the 12th report on carcinogens
Rivela C, Gibilisco RG, Teruel MA (2015) J Phys Organ Chem 28:480–484
Zhang Q, Chen Y, Tong S, Ge M, Shenolikar J, Johnson MS, Wang Y, Tsona NT, Mellouki A, Du L (2017) Atmos Environ 170:12–21
Yujing M, Mellouki A (2001) Phys Chem Chem Phys 3:2614–2617
Begum SS, Gour NK, Baruah SD, Deka RC (2018) Mol Phys 117:1–9
Zhao Y, Truhlar DG (2008) Acc Chem Res 41:157–167
Zhao Y, Truhlar DG (2008) Theor Chem Acc 120:215–241
Karton A, Tarnopolsky A, Schatz GC, Martin JML (2008) J Phys Chem A 112:12868–12886
Xie HB, Li C, He N, Wang C, Zhang S, Chen J (2014) Environ Sci Technol 48:1700–1706
Chai JD, Head-Gordon M (2008) Phys Chem Chem Phys 10:6615–6620
Gonzalez C, Schlegel HB (1989) J Chem Phys 90:2154–2161
Gonzalez C, Schlegel HB (1990) J Phys Chem 94:5523–5527
Purvis GD, Bartlett RJ (1982) J Chem Phys 76:1910–1918
Vereecken L, Francisco JS (2012) Chem Soc Rev 41:6259–6293
Montgomery JA, Frisch MJ, Ochterski JW, Petersson GA (1999) J Chem Phys 110:2822–2827
Montgomery JA, Frisch MJ, Ochterski JW, Petersson GA (2000) J Chem Phys 112:6532–6542
Frisch MJ, Trucks GE, Schlegel HB, Scuseria GE, RobbMA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA et al. (2009) Gaussian 09
Garrett BC, Truhlar DG (1979) J Am Chem Soc 101:4534–4548
Garrett BC, Truhlar DG (1979) J Chem Phys 70(4):1593–1598
Garrett BC, Truhlar DG, Grev RS, Magnuson AW (1980) J Phys Chem 84:1730–1748
Zheng J, Zhang S, Corchado J, Chuang Y, Coitino E, Ellingson B, Truhlar D (2015) GAUSSRATE. University of Minnesota, Minneapolis
Zheng J, Zhang S, Lynch BJ, Corchado JC, Chuang YY, Fast P, Hu W, Liu YP, Lynch G, Nguyen K (2010) POLYRATE, version 2010
Bhuvaneswari R, Sandhiya L, Senthilkumar K (2017) J Phys Chem A 121:6028–6035
Jacob DJ, Potter T, Colman B, Fishman J, Hill MA (2003) The oxidizing power of the atmosphere. In: Handbook of weather, climate and water, pp 29–46
Arey J, Atkinson R, Zielinska B, McElroy PA (1989) Environ Sci Technol 23:321–327
Bunce NJ, Dryfhout HG (1992) Can J Chem 70:1966–1970
Orlando JJ, Tyndall GS (2012) Chem Soc Rev 41:6294–6317
Orlando JJ, Tyndall GS, Wallington TJ (2003) Chem Rev 103:4657–4690
Lightfoot PD, Cox RA, Crowley JN, Destriau M, Hayman GD, Jenkin ME, Moortgat GK, Zabel F (1992) Atmos Environ 26:1805–1961
Zhang J, Dransfield T, Donahue NM (2004) J Phys Chem A 108:9082–9095
Li Y, Francisco JS (2001) J Chem Phys 114:2879–2882
Zhou M, Ma R, Yuan D, Chen M (2009) J Phys Chem A 113:2826–2830
Kleijn R, Elshkaki A, De Koning A, Tukker A (2001) Literature study on degradation products of known emissions
Tanaka N, Yamagishi S, Nishikiori H (2013) Comput Theor Chem 1020:108–112
Acknowledgements
The authors are thankful to UGC and Department of Science and Technology (DST), India, for funding to establish the high-performance computing facility under the SAP and PURSE programs.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
214_2019_2518_MOESM1_ESM.doc
The TST, CVT, TST (SCT) and CVT (SCT) rate constants (cm3 molecule−1 s−1) for the initial H-abstraction reactions (R2 and R3) and OH-addition reactions (R4 and R5) of 3CMP with OH radical are given in Table S1. Relative energy, ΔETot (kcal/mol), enthalpy, ΔH298 (kcal/mol), and Gibbs free energy, ΔG298 (kcal/mol), for the reactions of radical intermediates, I2, I3, 45 and I5, calculated at M06-2X, ωB97XD and CCSD(T) level of theories are summarized in Table S2, S3, S4 and S5. (DOC 1109 kb)
Rights and permissions
About this article

Cite this article
Bhuvaneswari, R., Senthilkumar, K. A comprehensive quantum chemical study on the mechanism and kinetics of atmospheric reactions of 3-chloro-2-methyl-1-propene with OH radical. Theor Chem Acc 139, 2 (2020). https://doi.org/10.1007/s00214-019-2518-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-019-2518-y