
Abstract
The non-noble transition metals (Sc, Ti, V, Cr, Mn) embedded in graphene as single-atom catalysts have been comprehensively screened for CO oxidation using density functional theory calculations. Among these options, Mn–graphene is predicted to have superior activity for CO oxidation. This conclusion is based on the binding energy between metal atom and graphene substrate, diffusion barrier of metal atom on graphene, and reaction barrier based on the transition state analysis. On the other hand, Sc–, Ti–, V–, and Cr–graphene bind O2 too strongly. This will lead to catalyst poisoning by O for these systems. We expect that Mn–graphene should be straight forward to fabricate experimentally, and predict that it will be a novel, stable, and efficient single-atom catalyst. For Mn–graphene, the Langmuir–Hinshelwood (LH) and Eley–Rideal (ER) mechanisms of CO oxidation have been investigated. However, the CO molecules cannot interact with surface activated O2 on graphene to form carbonate-like CO3 complexes or other intermediates. This demonstrates that CO oxidation will not proceed via the ER mechanism. The reaction mechanism for catalysis of CO oxidation occurs in two steps: The LH mechanism CO + O2→ OCOO → CO2 + O followed by the ER mechanism CO + O → CO2. The energy barriers are 0.57–0.69 eV and 0.08 eV, respectively. These barriers are comparable to or smaller than those for Ni and Mo, indicating high activity. Brief molecular dynamics simulations were also performed on this system. We predict that Mn–graphene can be used as a single-atom catalyst for CO oxidation over a broad range of temperatures. The present work should inspire experimental work on synthesis of novel single-atom catalysts.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
CO oxidation is a technologically and scientifically important reaction. From the viewpoint of basic science research, this is often regarded as a prototypical model reaction in heterogeneous catalysis, or to test the reactivity of new catalysts. In practical application, it is a fundamental process related to both environmental problems and electrode poisoning in fuel cells. Catalytic conversion by CO oxidation is crucial in reducing CO emissions from automobiles and industry, and to avoid CO poisoning in the oxygen reduction reaction. Substantial effort has been devoted to exploring effective catalysts for CO oxidation. Typically, noble metals like Au [1,2,3], Pt [4,5,6,7], Rh [4, 7], Ir [7], Pd [7, 8], Ag [9], or noble metals supported with metal oxides (such as Au/TiO2 [10]) or their alloys (for example Au-Cu [11]) are used as effective catalysts for CO oxidation. However, noble metals are scarce and expensive. Therefore, the development of cheap and efficient non-noble metal catalysts is important for large-scale practical application.
Reducing the size of metal nanoparticles and increasing the dispersion of metal atoms are important ways to lower the consumption of catalysts. This produces a high density of active sites, and also maintains and potentially increases the catalytic performance. The extreme case is the single-atom catalyst (SAC) with 100% atomic utilization of metal atoms. The first single-atom catalyst Pt1/FeOx was fabricated and used for CO oxidation in 2011 [12]. Since then, there have been numerous studies and experiments on single-atom catalysts, which have become a new frontier in heterogeneous catalysis [13]. Single-atom catalysts may unlock new possibilities for efficient CO oxidation by catalytic conversion. The substrate is a key component of the SAC and directly affects the catalytic performance. Due to the high stability, large surface area, high aspect ratio, unique topological structure, and exotic electronic structure, graphene is a unique and ideal substrate. Recently, there have been several computational efforts to design graphene-based SACs for catalytic CO oxidation [14,15,16,17,18,19,20,21,22,23,24]. There are three primary indices which characterize the catalytic properties of SACs: the binding energy between metal atom and substrate, the diffusion barrier of metal atom on the substrate, and the reaction barrier (which is the rate-determining step of the entire reaction path). Larger binding energy and diffusion barriers produce superior performance. On the other hand, the reaction barrier needs to be sufficiently small.
Many computationally designed SACs (e.g., Fe [15], Cu [19], Si [23], Al [18, 22], Ni [18, 25]) should be promising experimental candidates based on the predicted reaction barriers and other indices. However, some of the calculations are incomplete, or predict candidates which meet only one or two of the criteria. For Cu, although the reaction barrier of 0.54 eV is low enough, the diffusion barrier of 2.34 eV is too small compared to 6.78 eV for Fe or 6.82 eV for Si. Therefore, Cu may suffer from aggregation problems. Regarding Al, there are differing reports from two different groups. One group [18] calculated a binding energy of 5.65 eV and a reaction barrier of 0.32 eV, but no diffusion barrier was calculated. A second group [22] found a diffusion barrier of 2.95 eV and a reaction barrier of 0.9 eV, but without calculating the binding energy. For Ni, there is some scatter in the calculated results. One group [18] reported the binding energy and reaction barrier are 6.98 and 0.59 eV, but there is no data for the diffusion barrier. A second group [25] reported the binding energy, diffusion barrier, and reaction barrier are 7.57, 3.41, and 0.63 eV, respectively. These results suggest that Ni may be a good candidate. Some systems will have less efficient catalytic performance due to relatively high reaction barriers (such as Mo [26]); some studies involve noble metals (Au [14], Pt [16], Pd [17]) which are scarce and expensive. In Table 1, we summarize the binding energies, diffusion barriers, and reaction barriers of the rate-determining step from 13 different graphene-based SAC catalyst systems. One can see that there are few proposed SACs which simultaneously satisfy the three indices and are not noble metals. Thus, it is highly desirable to find novel, more stable, and efficient graphene-based SACs.
In this work, we have carried out first-principles density functional theory (DFT) calculations to study the catalytic reaction of CO oxidation using transition metal atoms embedded in graphene sheets. The goal is to find efficient SACs and to gain insight into the catalytic performance for the whole series. We chose the early 3d transition metal series from Sc to Mn. Among these, Mn–graphene exhibited remarkable catalytic performance for CO oxidation without catalyst poisoning. We found that Mn–graphene catalyzes CO oxidation with an energy barrier of only 0.57–0.69 eV. The reaction of CO and O2 on Mn–graphene proceeds via two steps: first the Langmuir–Hinshelwood (LH) mechanism, followed by the Eley–Rideal (ER) mechanism. We also include detailed analyses of the electronic structure of this system during the LH reaction to elucidate the superior catalytic activity exhibited by the Mn–graphene system.
2 Computational methods
Spin-polarized DFT calculations were carried out using the Dmol3 package [29]. The generalized gradient approximation with Perdew–Burke–Ernzerhof exchange-correlation functionals was used [30]. DFT semi-core pseudopotential [31] core treatment was implemented for relativistic effects, with C and O calculated in the all-electron method. (This uses a single effective potential to replace core electrons for the TM atoms.) The double numerical plus polarization (DNP) [29] basis set was used with a “fine” orbital cutoff. Energy, force, and displacement convergence were 1 × 10−5 Hartree, 0.002 Hartree/Å and 0.005Å, respectively. All self-consistent field calculations were performed with convergence criterion of 1 × 10−6 Hartree with smearing. Charge transfer was calculated using Hirshfeld charge analysis. The transition states (TSs) were confirmed by a complete linear synchronous transit (LST)/quadratic synchronous transit (QST) protocol. This involves LST maximization followed by repeated conjugated gradient (CG) refinements, and then QST maximizations with repeated CG minimizations until a transition state is located [32]. TS optimizations were followed to confirm that the located transition states have only one imaginary frequency (with the correct vibration direction). The nudged elastic band method [33] was used to confirm the minimum energy paths (MEPs) for elementary reaction steps.
A 4 × 4 × 1 supercell containing 32 atoms was chosen to model a system with one carbon atom substituted by a single transition metal atom. The minimum distance between the graphene sheet and its mirror images is set at 20 Å to avoid interactions. Unit cell was 8.5 × 9.9 × 20 Å. For the geometric optimization and for the search for transition states, the Brillouin zone was sampled with a 3 × 3 × 1 k-point mesh. For the electronic structure calculations, a Monkhorst–Pack scheme with 20 × 20 × 1 k-point grid meshes was employed.
The adsorption energy of an adsorbate is defined as:
where Eadsorbate+TM–graphene is the total energy of the TM–graphene with the adsorbed molecule, ETM–graphene is the energy of the isolated TM–graphene, and Eadsorbate is the energy of the isolated gas molecule.
Born–Oppenheimer Molecular dynamics calculations were carried out using CASTEP [34] in Materials Studio 2016 and 2018. DFT-D and the Grimme method for DFT-D correction were used with a step size of 1.5 fs. In CASTEP the Grimme method is specified for the DFT-D correction as opposed to the LS method as this was substantially more stable and reliable for SCF conversion. Other computational details were similar to our previous work (NVT, PBE, GGA) [35]. Unit cell was 9.9 × 9.9 × 12 Å, smearing = 0.15 eV, SCF tolerance approximately 8 × 10−6, 420 eV cutoff for MD). These reliable CASTEP methods were also applied to the calculation of the transition state energy TS1 going from IS to MS (SCF tolerance 2 × 10−6, 12 QST/LST steps, 570 eV cutoff, unit cell 8.5 × 9.9 × 20 Å).
3 Results and discussion
3.1 Searching for the best candidate
In order to ensure the long-term activity of the catalyst, we investigated the adsorption energies of CO and O2 on TM–graphene (TM = Sc, Ti, V, Cr, Mn). The variation of Ead(CO) and Ead(O2) in different TM–graphene (TM = Sc, Ti, V, Cr, Mn) frameworks is illustrated in Fig. 1. We observe that Ead(O2) is greater than Ead(CO) for all five TM. There is a large difference between Ead(O2) and Ead(CO) from Sc to Cr. However, of these five metals, only Mn–graphene has two favorable energies. The binding energies of O2 and CO with TM–graphene from recent studies are included as a comparison and to demonstrate the viability of this screening method for CO oxidation [15, 18, 19, 25, 36, 37].

We now focus on an important question which affects the feasibility of these prospective CO oxidation catalysts, namely O2 poisoning. From the data presented in Fig. 1, we see that for the four cases of TM–graphene (TM = Sc, Ti, V, Cr) the O2 adsorption energy is too strong. Therefore, these single-atom TM catalyst sheets will be dominantly covered by O2 if a 1:1 O2/CO mixture is injected with a constant gas flow rate. In this case, our results show that CO2 cannot be produced using TM–graphene (TM = Sc, Ti, V, Cr). However, the single-atom Mn–graphene sheet has suitable adsorption energies and therefore CO2 can potentially be produced. The Mn–graphene catalyst will be much more suitable to catalyze the oxidation of CO. In general, it is assumed that the reaction cannot proceed if the intermediate species are too strongly bound to the active site of the catalyst to be desorbed during the reaction. This is called “catalyst poisoning,” and is also seen in other chemical species. We use this rule to screen the potential candidates for efficient CO oxidation.
3.2 Properties of Mn–graphene
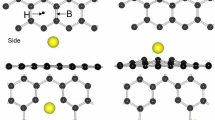
We first investigated the details of the adsorption of one Mn atom on top of pristine graphene. Representative sites of adsorption include the hollow site at the center of the hexagon, the bridging site over a C–C bond, and the top site right above a carbon atom. Stable structures and adsorption energies of these adsorption sites are shown in Fig. 2. Among these three representative adsorption sites, the maximum adsorption energy is for the hollow site at 0.69 eV. Because the adsorption energy is so small, the metal atoms can diffuse easily on pristine graphene and will form small metal clusters.

Three stable configurations of Mn adsorbed on the pristine graphene are shown in top (up panel) and side (down panel) views, respectively
The intrinsic stability of TM–embedded graphene as a catalytic substrate is based on the binding energy between metal atom and substrate, and the diffusion barrier of metal atoms on the substrate. Comparing our DFT calculations to previous results, we find that the binding energy of Mn–graphene (6.55 eV) is much higher than that for Zn–graphene (1.85 eV) [18] and Al–graphene (5.6 eV) [18], and comparable to that of other transition metal atoms (e.g., Pt–graphene (7.1 eV) [16] and Mo–graphene (7.25 eV)) [26]. The high binding energy for Mn prohibits diffusion and clumping of metal atoms, producing the high stability of the Mn–graphene catalyst.
We embedded the Mn atom directly into the top layer of the graphene surface. In other words, we add a single Mn atom into a missing atom defect on the graphene surface. Numerous experiments have previously been carried out on various embedded metal atoms (not Mn) into defective graphene, which show the stability and catalytic activity of these systems [14,15,16,17,18,19,20,21,22, 25, 26, 36, 37]. We calculate the geometry and electronic characteristics of the Mn–graphene system, as shown in Fig. 3a. The Mn atom replaces a C atom from the original graphene, but sits above the plane. The distance between the Mn atom and the adjacent C atom is 1.82 Å (this is close to the previous computational result of 1.81 Å [38]). The vertical distance from the Mn atom to the graphene layer is 1.44 Å. There is a charge transfer of 0.31 e from Mn to the graphene sheet according to a Hirshfeld charge analysis. The local density of states (LDOS) diagram reveals the bonding between the Mn atom and the adjacent C atoms. To gain deeper insight into the electronic structure, the spin-polarized LDOS is projected onto the Mn-3d and Mn-4s orbitals, and the neighboring C-2p orbital is plotted in Fig. 3b. Some of the electrons on the Mn atom are transferred to neighboring C atoms, so the Mn-4s, Mn-3d, and C-2p orbitals are partially filled. As a result, a high density of spin-polarized states is localized around the Fermi level. Furthermore, the localized Mn-3d state is important to activate reactants and to lower the reaction barriers.

a Top and side views of Mn–graphene. A single Mn atom is embedded into the surface (at the location of a C vacancy). The unit cell is shown in white outline. b Spin-polarized LDOS projected on Mn-3d (red), Mn-4s (blue), and C-2p (black for neighboring carbon atoms) orbitals. The Fermi level EF is at zero. c Atomic configurations of the initial state (IS), transition state (TS), and final state (FS) for the migration of a Mn atom to an adjacent site (in eV)
We have also calculated the diffusion barrier for embedded Mn atoms in a graphene layer from one site to a neighboring site. Figure 3c shows the process of Mn diffusion from carbon vacancies to adjacent carbon vacancies, including the most stable solid structures of reactants, transition states, and products. Two C–Mn bonds are formed at the same time that the two C–C bonds break, and two pentagons form during the migration process. Effectively, a C atom and a Mn atom switch position. We find a transport barrier of 4.87 eV. This is much larger than the diffusion process of individual Mn atoms on pristine graphene (discussed above). As a comparison, the diffusion barrier of Mn atom from one carbon vacancy to another is higher than transition metals Ni (3.41 eV) [25] and Cu (2.34 eV) [19], and main-group metals Al (2.95 eV) [22] and Ge (3.03 eV) [22]. In general, with a larger diffusion barrier, less clustering will occur, and the catalyst system will be more stable. Therefore, we conclude that Mn atoms embedded into the graphene surface will be stable up to high temperatures. These Mn–graphene sites should make good and stable active sites for catalysis.
3.3 The adsorption of O2, CO, O, and CO2 on Mn–graphene
In order to study CO oxidation on Mn–graphene, the adsorption of CO, O2, O, and CO2 was investigated. The charge transfer, bond length, and adsorption energies of the adsorption structures are summarized in Table 2 and Fig. 4.

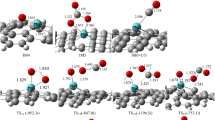
Optimized configurations (top and side views) and corresponding DOS curves for CO, O2, O, and CO2 adsorption on Mn–graphene
Figure 4 shows the most stable configuration for CO attached onto the Mn–graphene at the top Mn site with Ead(CO) = − 1.34 eV. In this configuration, CO interacts with the Mn atom with the C attached to the Mn, with the CO perpendicular to the basal plane of the graphene. The C–Mn bond length is 1.92 Å, which falls in the range of typical C–Mn chemical bonding. The covalent nature of this interaction is supported by the charge accumulation region between the C and Mn atoms. In this configuration, 0.05e are transferred from the Mn–graphene system to the CO. This charge transfer goes into the 2π* orbital of the CO and extends the bond length from 1.13 to 1.16 Å. This charge transfer and bond length extension activates the CO molecule. This activation is visualized by the charge accumulation region between C and Mn atoms. The strong hybridization between the 3d states of Mn and the 2p states of CO can be clearly seen from the computed density of states (DOS). Due to the interaction with the Mn–graphene, all the DOS peaks corresponding to CO states are down-shifted, and the peak of the 2π* orbital is split into parts and even shifted below EF.
We now consider O2 adsorption on Mn–graphene. The lowest energy state has O2 parallel to the plane of the graphene, forming two Mn–O bonds. The bond length data are shown in Table 2. The adsorption energy of O2 on Mn–graphene is Ead(O2) = − 1.87 eV (note that Co, Ni, Cu, and Zn also show a low O2 adsorption energy). This is larger than the adsorption energy of CO. In this configuration, 0.15 e are transferred from the Mn–graphene system to the O2. This transfer into the 2π* orbital of the O2 extends the bond length substantially from 1.21 to 1.39 Å. This charge transfer indicates the charge depletion region is on the embedded Mn atom, and the charge accumulation region on the O2 molecule. In addition, due to charge transfer from Mn-3d to O2-2p, the energy levels and intensities of the O2-2p state are significantly changed. This change is embodied in the elongation of the O2 bond.
We also investigated atomic O adsorption onto Mn–graphene. The adsorption energy of the lowest energy state of a single isolated O atom onto Mn–graphene is − 5.58 eV. This is much larger than Ead(O2). The Mn–O bond length is 1.61 Å, which is lower than the bond length for O2 adsorbed on Mn–graphene, due to the strong interaction energy. The adsorbed O atom gains charge from the Mn–graphene. The charge transfer is 0.30 e. In the DOS diagram, the peaks of the O-2p state around the EF are colocated with peaks at the Mn-3d state, showing strong hybridization between them. Comparing the peaks of adsorbed O2 near the Fermi level, the O-2p orbital has a higher energy level, which is a sign of potential high oxidation activity.
The most stable configuration of CO2 adsorption on Mn–graphene is shown in Fig. 5. At this point, the CO2 molecule is physisorbed on Mn–graphene with an adsorption energy of only − 0.23 eV. Since the interaction between the CO2 molecule and the base material is so weak, the CO2 to Mn–O bond length is very long at 2.86 Å. Unlike the case of CO, the adsorbed CO2 molecule donates a small amount of charge (0.06 e) to the Mn–graphene. Thus, the intersection of the DOS peaks of the CO2 states with the Mn-3d state is rarely near EF. Clearly, CO2 has weak adsorption on Mn–graphene and can easily desorb from the reaction site.

The frontier orbitals HOMO-2, HOMO-1, HOMO, and lowest unoccupied molecular orbital (LUMO) of CO, O2, O, CO2 on Mn–graphene at the Γ-point
We now discuss the frontier orbitals of CO, O2, O, and CO2 on Mn–graphene as shown in Fig. 5. There is a significant hybridization between the 2π* orbital of CO and the d orbital of Mn, the 2π* orbital of O2 and the d orbital of Mn, the p orbital of O and the d orbital of Mn in the Highest Occupied Molecular Orbital (HOMO) and HOMO-1. This reveals the adsorption mode for CO, O2, and O on Mn–graphene via HOMO and HOMO-1 interactions. The adsorption will ensure maximum overlap of the orbitals. By contrast for CO2, there is almost no orbital interaction, and the charge density is distributed between CO2 and Mn–graphene. This is consistent with the fact that the CO2 will be desorbed from Mn–graphene and then will subsequently be used for further catalytic cycles.
3.4 Reaction mechanism of CO oxidation
The surface catalytic reaction often passes through a number of basic adsorption and desorption steps, and its reaction energy affects the catalytic activity. The CO oxidation reaction has two dominant reaction mechanisms: the Langmuir–Hinshelwood (LH) mechanism and the Eley–Rideal (ER) mechanism. The LH mechanism begins with the interaction of co-adsorbed CO and O2 molecules, forming peroxide-like O–C–O–O intermediates. By contrast, the CO oxidation following the ER mechanism starts with independent CO molecules reacting directly with surface activated O2 or with atomic O generated through a surface driven O2 dissociation. Finally, a carbonate-like CO3 intermediate product is formed. Since O2 is strongly adsorbed on Mn–graphene and the O–O bond length is elongated upon its adsorption on Mn–graphene, one would expect that the ER mechanism would be dominant. However, unexpectedly, after geometry optimization and full TS search, we found that CO molecules cannot interact with surface activated O2 on graphene to form carbonate-like CO3 complexes or other intermediate formation processes. This demonstrates that the CO oxidation of this system is not via a pure ER mechanism. Therefore, the LH mechanism CO + O2 → OCOO → CO2 + O followed by the ER mechanism CO + O → CO2 is found to be the correct mechanism by which Mn–graphene catalyzes the CO oxidation reaction.
As a comparison, the energy barrier of CO oxidation catalyzed by Mn–graphene is comparable to or even lower than that of Mo-graphene (0.75 eV) [26] and Ni-graphene (0.63 eV) [25]. As a result, we predict that Mn–graphene will have higher efficiency and activity for CO oxidation than Mo-graphene.
Figure 6 shows energy profiles of the MEPs via the LH mechanism, and also views of the local optimized configurations of the adsorbates along the MEPs. When CO and O2 are co-adsorbed on Mn–graphene, one oxygen atom of O2 and one C atom of a CO can be close to each other and then form the first transition state (TS1). There is an energy barrier of 0.57–0.69 eV along this reaction pathway. Following the formation of TS1, a peroxide type intermediate state (MS) is formed, which is a mild endothermic process with a reaction energy of 0.19 eV for CO + O2 → OOCO. The length of the O–O bond of the peroxide type intermediate is continuously extended, and then the second transition state is formed with a potential barrier as low as 0.08 eV. The O–O bond then breaks when the final state (FS) is formed, releasing 3.52 eV of energy. Since the interaction between CO2 and graphene is weak, the formation of CO2 molecules leads quickly to desorption, leaving an atomic O strongly interacting with the Mn atom. The remaining atomic O adsorbed on Mn–graphene can easily react with another CO molecule over a barrier of 0.36 eV to form the final state (as shown in Fig. 6). We decided to double check the important energy barrier from IS to TS1 (which dominates the reaction rate) using accurate CASTEP calculations. This involved a complete transition state search entirely using CASTEP optimized geometries. The original calculation using Dmol3 provided a result of 0.69 eV. The CASTEP value is 0.57 eV for this barrier. Both values are included throughout the paper. The highest energy barrier of this catalytic cycle is therefore only 0.57–0.69 eV.

Schematic energy profile and the corresponding local configurations along the Langmuir–Hinshelwood (upper panel) and Eley–Rideal (lower panel) steps using DMol3. Both top and side views are illustrated. Initial state (IS), transition state 1 (TS1), metastable state (MS), transition state 2 (TS2), and final state (FS) are shown. For the LH-step, all energies in eV are relative to the reference energy of the co-adsorbed initial state for CO and O2 on Mn–graphene. For the ER-step, all energies (in eV) are listed with respect to the reference energy of the co-adsorbed state for CO and O on Mn–graphene
3.5 Temperature effect and kinetics
The Gibbs free energy change \(\Delta G = \Delta H - T\Delta S\). In the above formula, ΔH is the change of enthalpy and ΔS is the change of entropy at temperature T. ΔH consists of two parts, namely ΔU and PΔV (change in internal energy, pressure, and change in volume). In our system, ΔS is defined by \(\Delta S = \Delta S_{vib} + \Delta S_{rot} + \Delta S_{trans}\) (change in vibrational, rotational, and translational entropy), and \(\Delta U\) is given by \(\Delta U = \Delta E_{\text{tot}} + \Delta E_{\text{vib}} + \Delta E_{\text{rot}} + \Delta E_{\text{trans}}\). \(\Delta E_{\text{tot}}\), which can be obtained from DFT calculation, is the total electronic energy change at 0 K, and \(\Delta E_{\text{vib}}\), \(\Delta E_{\text{rot}}\) and \(\Delta E_{\text{trans}}\) are the energy components from vibration, translation and rotation, respectively. The corrected free energy changes for the whole CO oxidation on the Mn–graphene sheet for temperatures from 275 to 575 K are listed in Table 3 (using the 0.69 eV barrier).
Let us consider the effect of temperature on the LH mechanism. The Gibbs free energy change increases as T increases indicating the process is thermodynamically more viable. For the ER mechanism, ΔG decreases slightly as T increases, and is thus thermodynamically less viable. The corrected energy barrier at 298 K for IS → TS in the ER mechanism is 0.40 eV. The corrected energy barrier at 298 K in the LH mechanism is 0.72 eV for IS → TS1, and 0.05 eV for MS → TS2.
Although raising the temperature can make the LH reaction thermodynamically slightly less viable and the ER a bit more viable, the Gibbs free energy change and activation barrier alter only slightly in the temperature range between 275 and 575 K. The favorable values predict stability and catalytic feasibility at relatively lower temperatures as well as higher temperatures.
On the basis of the above free energy changes (Table 3), one can get the corrected energy barrier for temperatures from 275 to 575 K. If we assume room temperature (298.15 K), one can estimate the reaction time for each elementary step along the LH pathway of CO oxidation on Mn–graphene at 298.15 K using the Arrhenius relationship (Eq. 2).
In Eq. 2, Ea is the activation energy of reaction, \(\nu\) is the attempt frequency (on the order of 1012 Hz). Note that \(\nu\) is the same as previously used value in the literature [25]. \(\kappa_{B}\) is the Boltzmann constant (8.617 × 10−5 eV/K). The estimated reaction times at three different representative temperatures (i.e., 298.15, 400, 500 K) are listed in Table 3 (using the 0.69 eV barrier). At 298.15 K, it is estimated to be τ = 1.5, 7.0 × 10−12, 5.8 × 10−6s with respect to IS → MS, MS → FS (LH), and IS → FS (ER), respectively. Alternatively, using the CASTEP barrier of 0.57 eV for IS → MS, τ = 0.01 s at 300 K.
Thus, CO oxidation on Mn–graphene can occur at relatively low temperatures. From Table 4, we can see that the order of kinetics are IS-MS < IS-FS < MS-FS with MS-FS the fastest step and IS-MS the rate-determining step. Furthermore, the IS-MS and IS-FS steps become much faster when temperature increases, whereas MS-FS step essentially stays constant. Thus, this reaction will become much faster as the temperature increases.
Two MD simulations were carried out. The first run started with one O2 molecule close to the original Mn–Graphene surface. 12 CO molecules and 4 O2 molecules were added and the unit cell was reduced to 10 × 10 × 12 Å unit cell. The first frame is shown in the Supplemental Information (Figure S1). One of the O2 molecules was brought close so that there was an Mn–O bond of 2.225 Å (to jump-start the process). This was run at 1200 K for 0.9 ps total to try to follow the reaction. We confirm the attempt frequency of approximately 1012 Hz. We did observe that the O2 bonds on to the Mn and briefly and periodically the CO molecules also attach, achieving a state close to the IS state. This is shown in a movie in the SI. We did not observe catalytic reaction in this short time. The second simulation started with state TS1 from Fig. 6, and 12 CO molecules and 4 O2 molecules were added. For the MD simulations, the TS1 state needed to be recalculated using CASTEP transition state optimization for internal consistency. This allows us to follow the process from this point. A 216-fs-long MD movie at 1200 K is included in the supplemental information. One CO2 molecule is created (i.e., catalysis is successful from this starting point) and escapes. From these and other short MD simulations, we also confirm that no other lower energy pathways to CO2 exist.
3.6 Electronic structure analysis
To gain more insight into the origin of the high activity of the Mn–graphene system, we now investigate the electronic structure of the LH reaction steps. Figure 7 shows the local density of states of the CO and O2 molecules as well as the Mn-3d states in the initial state (IS), transition state 1 (TS1), metastable state (MS), transition state 2 (TS2), and final state (FS). As illustrated in Fig. 7, the 2π* level of O2 is partially shifted downward upon the adsorption in the IS due to electron transfer between the Mn atom and the adsorbate. By contrast, since the amount of charge transferred from Mn to CO is small, the 2π* level of CO is still empty in the IS. Owing to the resonance with the CO 2π* state, the O2 2π* level is further populated and broadened to hybridize with the Mn-3d state (increasing from IS to TS1). The LDOS diagram of TS1 shows that the CO 2p orbital is also partially occupied. In the MS, the O2 1π and 2π* states are strongly hybridized with the Mn-3d orbital near EF. During the transition from metastable intermediate state MS to transition state TS2, the O–O bond breaks due to the interaction of O2 atoms with CO. In addition, from IS to TS1 to MS to TS2, the O2 1π and 2π* orbitals are increasingly broadened and interacting with Mn-3d orbital. On the other hand, for CO on Mn–graphene, the 5σ and 2π* levels are shifted upward and strongly hybridized with the Mn-3d electronic orbital during the entire LH-step rather than the 1π level. Thus, the Mn-3d orbital plays a key role in the interaction between CO and O2 on Mn–graphene. For the ER reaction, when the CO approaches the O atom, and eventually forms a C–O bond, the Mn-3d orbital is occupied.

LDOS of CO (left panel), O2 (right panel) and Mn atoms in the IS, TS1, MS, and TS2 along the Langmuir–Hinshelwood step. Blue solid curve for the LDOS projected on CO, green solid curve for the LDOS projected on O2, black solid curve for the projected total LDOS, and red solid curve for the Mn d-state LDOS. The scale on the left corresponds to the LDOS of CO and O2, and the scale on the right corresponds to the total DOS
4 Conclusion
In summary, based on our systematic DFT calculations, we find that Mn–graphene as a novel single-atom catalyst exhibits superior activity for CO oxidation at low temperature without O2 poisoning. The oxidation reaction of CO on Mn–graphene is mainly carried out in two steps: CO initially starts with the LH reaction with energy barriers of 0.57–0.69 and 0.08 eV, followed by an ER reaction with energy barrier 0.36 eV. The high catalytic activity of Mn–graphene-catalyzed CO oxidation is mainly due to the orbital hybridization between CO, O2, and Mn atoms, in particular the Mn-3d, CO-2π*, and O-2π* orbitals. We hope that this work will inspire experimental work on novel single-atom catalysts. In conclusion, we predict that Mn–graphene can be used as an alternative catalyst for the oxidation of CO at relatively low temperature.
5 Supplemental Information
DFT molecular dynamics movies of Mn–graphene are included in the supplemental information. One movie starts with the TS1 state and shows 350 fs of simulation at 100 K as the system releases one free CO2 molecule. The second movie starts with Mn–graphene plus CO and O2 molecules and proceeds for 576 fs at 1200 K. This starts with one O2 molecule nearby and with one O atom very loosely bonded to the Mn atom. After 300 fs this proceeds to a state similar to the TS1 state with both CO and O2 bonded.
References
Kimble ML, Castleman AW, Mitrić R, Bürgel C, Bonačić-Koutecký V (2004) J Am Chem Soc 126:2526
Lopez N, Nørskov JK (2002) J Am Chem Soc 124:11262
Liu Z-P, Hu P, Alavi A (2002) J Am Chem Soc 124:14770
Oh S-H, Hoflund GB (2007) J Catal 245:35
Lee SW, Chen S, Sheng W, Yabuuchi N, Kim Y-T, Mitani T, Vescovo E, Shao-Horn Y (2009) J Am Chem Soc 131:15669
Ackermann MD, Pedersen TM, Hendriksen BLM, Robach O, Bobaru SC, Popa I, Quiros C, Kim H, Hammer B, Ferrer S, Frenken JWM (2005) Phys Rev Lett 95:255505
Gong X-Q, Liu Z-P, Raval R, Hu P (2004) J Am Chem Soc 126:8
Zhang CJ, Hu P (2001) J Am Chem Soc 123:1166
Su H-Y, Yang M-M, Bao X-H, Li W-X (2008) J Phys Chem C 112:17303
Liu Z-P, Gong X-Q, Kohanoff J, Sanchez C, Hu P (2003) Phys Rev Lett 91:266102
Gao Y, Shao N, Pei Y, Zeng XC (2010) Nano Lett 10:1055
Qiao B, Wang A, Yang X, Allard LF, Jiang Z, Cui Y, Liu J, Li J, Zhang T (2011) Nat Chem 3:634
Yang X-F, Wang A, Qiao B, Li J, Liu J, Zhang T (2013) Acc Chem Res 46:1740
Lu Y-H, Zhou M, Zhang C, Feng Y-P (2009) J Phys Chem C 113:20157
Li Y, Zhou Z, Yu G, Chen W, Chen Z (2010) J Phys Chem C 114:6252
Liu X, Sui Y, Duan T, Meng C, Han Y (2014) Phys Chem Chem Phys 16:23584
Esrafili MD, Nematollahi P, Nurazar R (2016) Superlattices Microstruct 92:60
Tang Y, Dai X, Yang Z, Liu Z, Pan L, Ma D, Lu Z (2014) Carbon 71:139
Song EH, Wen Z, Jiang Q (2011) J Phys Chem C 115:3678
Tang Y, Liu Z, Dai X, Yang Z, Chen W, Ma D, Lu Z (2014) Appl Surf Sci 308:402
Esrafili MD, Saeidi N (2015) Physica E 74:382
Esrafili MD, Nematollahi P, Abdollahpour H (2016) Appl Surf Sci 378:418
Zhao J-X, Chen Y, Fu H-G (2012) Theor Chem Acc 131:1243
Jiang QG, Ao ZM, Li S, Wen Z (2014) RSC Adv 4:20290
Xu X-Y, Li J, Xu H, Xu X, Zhao C (2016) New J Chem 40:9361
Tang Y, Pan L, Chen W, Li C, Shen Z, Dai X (2015) Appl Phys A 119:475
Tang Y, Yang Z, Dai X (2012) Phys Chem Chem Phys 14:16566
Tang Y, Ma D, Chen W, Dai X (2015) Sens Actuators B 211:227
Delley B (1990) J Chem Phys 92:508
Perdew JP, Burke K, Ernzerhof M (1996) Phys Rev Lett 77:3865
Delley B (2002) Phys Rev B 66:155125
Halgren TA, Lipscomb WN (1977) Chem Phys Lett 49:225
Henkelman G, Jónsson H (2000) J Chem Phys 113:9978
Clark SJ, Segall MD, Pickard CJ, Hasnip PJ, Probert MIJ, Refson K, Payne MC (2005) Z Kristallogr 220:567
Ganz E, Ganz AB, Yang L-M, Dornfeld M (2017) Phys Chem Chem Phys 19:3756
Chu M, Liu X, Sui Y, Luo J, Meng C (2015) Molecules 20:19540
Tang Y, Zhou J, Shen Z, Chen W, Li C, Dai X (2016) RSC Adv 6:93985
Krasheninnikov AV, Lehtinen PO, Foster AS, Pyykkö P, Nieminen RM (2009) Phys Rev Lett 102:126807
Acknowledgements
L. X. and L.-M. Y. gratefully acknowledges the support from National Natural Science Foundation of China (Grant No. 21673087), startup fund (2006013118 and 3004013105) and independent innovation research fund (0118013090) from Huazhong University of Science and Technology. We thank the Minnesota Supercomputing Institute (MSI) at the University of Minnesota for supercomputing resources. The DFT calculations were carried out on the Mesabi and Itasca supercomputers at the MSI.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xu, L., Yang, LM. & Ganz, E. Mn–graphene single-atom catalyst evaluated for CO oxidation by computational screening. Theor Chem Acc 137, 98 (2018). https://doi.org/10.1007/s00214-018-2270-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-018-2270-8