
Abstract
The quantum mechanics-based test for overall free-radical scavenging activity was applied for the investigation of antioxidative capacity of chlorogenic acid (5-O-caffeoylquinic acid, 5CQA) relative to trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid, Tx) as a reference compound. Hydrogen atom transfer (HAT), radical adduct formation (RAF), electron transfer (ET), and proton loss (PL) reactions of 5CQA and Tx with HO• and CH3OO• radicals in benzene and pentyl ethanoate were examined. For this purpose, two theoretical models, M06-2X/6-311++G(d,p) in combination with the CPCM solvation model, and M05-2X/6-311++G(d,p) in combination with the SMD solvation model, were employed. It was found that M05-2X/6-311++G(d,p)—SMD failed to evaluate the influence of pentyl ethanoate, whereas M06-2X—CPCM in benzene and pentyl ethanoate and M05-2X—SMD in benzene proved to be operative and showed similar trends. Both compounds can react with HO• via HAT and RAF mechanisms, whereas HAT is the only reaction pathway with CH3OO•. 5CQA is more efficient scavenger of HO• than Tx, but less efficient scavenger of CH3OO•. Less reactive free radicals are more suitable for the determination of antioxidative activity of a compound relative to Tx. Highly reactive free radicals need to be included in the investigation of all potential reaction pathways of the compound, in which case the results from different approaches can be inconsistent.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
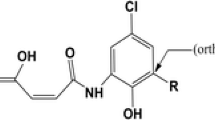
Highly reactive free radicals, such as hydroxyl and peroxyl radicals, are produced in the organism during normal metabolic processes. Imbalance between formation and consumption of free radicals leads to phenomenon known as oxidative stress [1]. These reactive species contribute to oxidative damage to proteins, DNA and lipids and are associated with some diseases [2, 3]. Thus, the compounds able to scavenge free radicals are in the focus of contemporary investigations. Numerous epidemiological studies have shown that dietary antioxidants can reduce development and progression of some chronic diseases [4,5,6,7]. One of the most abundant phenolic acid antioxidants present in fruits and vegetables is chlorogenic acid (5-O-caffeoylquinic acid, 5CQA) (Fig. 1) [8].

Possible antioxidative reaction pathways of 5CQA in nonpolar solvents. The atoms labeling scheme and product names are indicated
A wide range of experimental assays are used to evaluate antioxidative capacity [9,10,11,12]. Roughly, there are two types of these assays: chemical and biological [13]. In biological assays the damage in biomolecules caused by radical processes is measured. Chemical assays are used in studies which are devoted to evaluation of the rate constants for the reactions between free radicals and antioxidants. In these assays the antioxidative capacity of a compound is compared to that of some reference compound. The most commonly used antioxidant for this purpose is trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid, Tx) (Fig. 2).

Possible antioxidative reaction pathways of Tx in nonpolar solvents. The atoms labeling scheme and product names are indicated
Recently, a computational protocol known as the quantum mechanics-based test for overall free-radical scavenging activity (QMORSA) has been proposed for estimating antioxidative capacity of compound [14]. This approach is based on thermodynamic and kinetic investigation of all possible reactions included in antioxidative processes for examined compound. Namely, this methodology requires calculation of rate constants for all probable mechanistic pathways of an antioxidant in media of different polarity considering the influence of pH. Like experimental studies, this approach also offers possibility for determination of the relative antioxidative activity using Tx as a reference antioxidant. Thus, the ratio between overall rate constants of examined antioxidant and Tx can indicate larger or smaller activity of the antioxidant in comparison to Tx.
There are several theoretical studies devoted to explanation of antioxidative action of 5CQA [15,16,17,18,19] including thermodynamic and mechanistic approaches to the hydrogen atom transfer (HAT), radical adduct formation (RAF), sequential proton loss–electron transfer (SPLET), and single electron transfer–proton transfer (SET-PT) mechanisms. In our recent paper [17], kinetic investigations of the four mechanisms of 5CQA with HO• have revealed that in neutral and acidic media HAT and RAF mechanisms are competitive, whereas in basis media (e.g., at physiological pH) SPLET mechanism is predominant.
This work is an extension of our efforts aimed at elucidation of antioxidative properties of 5CQA. The goal of the work is to determine relative antioxidative capacity of 5CQA using Tx as a reference (rT) in nonpolar solvents. For this purpose, all antioxidative pathways of 5CQA and Tx with two free radicals, HO• (extremely reactive) and CH3OO• (moderately reactive), were investigated by means of two theoretical models. An additional goal is to examine the performances of the applied theoretical models in evaluation of the rT values of 5CQA.
2 Computational details
Within this work all calculations were performed using the Gaussian 09 program package [20]. Two levels of theory were applied: M06-2X/6-311++G(d,p) [21, 22] in combination with the CPCM polarizable continuum solvation model [23] and M05-2X/6-311++G(d,p) [24] in combination with the SMD continuum solvation model [25]. The former theoretical model has recently demonstrated robustness and very good overall performance in investigations of the related problems [15, 17, 21, 26], where it was applied in a manner that the influence of solvent to the structure and energy was estimated by full geometry optimizations, including frequency calculations. The latter density functional and solvation model have often been employed [27,28,29,30,31]. Sometimes, their application was based on the gas-phase calculations [27, 29], and the solvent cage effects were included according to the corrections proposed by Okuno [32] taking into account the free volume theory [33].
Following our strategy (full optimizations with frequency calculations included) we performed the calculations taking into account the effects of two solvents of similar polarity: benzene (dielectric constant ε = 2.2706) and pentyl ethanoate (ε = 4.7297). The restricted and unrestricted calculation schemes were applied for the closed-shell and open-shell structures. The nature of the revealed stationary points was determined by analyzing the results of the frequency calculations: no imaginary vibrations for equilibrium geometries and exactly one imaginary vibration for transition states. The intrinsic reaction coordinate (IRC) calculations were carried out to confirm that each transition state connects two corresponding equilibrium geometries.
The following reactions were considered:
In Eqs. (1)–(4) Phenolic stands for 5CQA and Tx, whereas R• denotes the HO• and CH3OO• radicals. Accordingly, Phenolic•, (Phenolic-R)•, Phenolic−, and Phenolic+• represent the free radical, radical adduct, anion, and radical cation issued from the corresponding phenolic compound, respectively. Apparently, RH is the parent compound of R•, and R− is the corresponding anion. The HO• radical is produced during Fenton and Haber–Weiss reactions in organism and represents one of the most potent radicals of biological importance. CH3OO• radical is selected to imitate behavior of larger lipid peroxide radicals. Equations (1) and (2) represent the HAT and RAF mechanisms. Equations (3) and (4) characterize the first steps of the SPLET (proton loss) and SET-PT (electron transfer) mechanisms. The free energies of the reactions at T = 298.15 K in the two solvents were determined. The free energy values of the solvated proton [Eq. (3)] in benzene (− 901.8 kJ mol−1) and pentyl ethanoate (− 1015.5 kJ mol−1) were taken from the literature [34].
The rate constants were calculated for 1 M standard state using the Eckart method, also known as ZCT-0 [35]:
For this purpose TheRate program was utilized [36]. In Eq. (5) kB and h stand for the Boltzman and Planck constants, whereas \(\Delta G_{a}^{\ddag }\), σ, and γ(T) are the free activation energy, reaction path degeneracy, and transmission coefficient, respectively [37]. The energy values and partition functions were taken from the quantum mechanical calculations.
Each koverall value was obtained as a sum of the corresponding kZCT-0 values. The rT (antioxidative activity of 5CQA relative to Tx) value was calculated by dividing koverall for 5CQA with koverall for Tx at three different levels of theory. The branching ratio, i.e., the percent contribution of an antioxidative pathway i to the overall reaction of 5CQA, was calculated by dividing kZCT-0(i) with koverall and multiplying with 100.
3 Results and discussion
The first part of our investigation was devoted to thermodynamic consideration of the reactions (1)–(4). The aim of this examination was to select exergonic reaction pathways for further analysis. Bearing in mind that the rate constants for homolytic cleavage of the C–H bonds and corresponding branching ratios are much smaller in comparison to those for homolytic cleavage of the O–H bonds [27, 29, 38,39,40], examination of the HAT mechanism was focused to abstraction of hydrogen atoms from phenolic groups. As for the RAF mechanism, addition of the two free radicals to the sp2 hybridized carbon atoms was simulated. The results are presented in Table 1 and illustrated with Figs. 1 and 2. It is apparent that the ∆ r G values refereeing to the two solvents are in very good agreement. As expected for nonpolar solvents, both compounds show pronouncedly positive ∆ r G values for the loss of carboxylic proton [Eq. (3)] and for the transfer of electron to either HO• or CH3OO• [Eq. (4)]. In accordance with previous findings [15,16,17,18, 38], these results show that neither 5CQA nor Tx undergo the SPLET and SET-PT mechanisms in nonpolar media. For this reason the further steps of these antioxidative pathways were not examined, and they were excluded from the text that follows.
On the other hand, HAT is thermodynamically favorable mechanism of 5CQA and Tx with both free radicals, where the reactions with HO• are noticeably more exergonic. In addition, the ∆ r G values for the RAF pathways with HO• are in most cases negative, except for the positions 9’ of 5CQA and 1 of Tx (Table 1). Certainly, these positions are not attractive for highly electrophilic HO• because of the large partial positive charge on C9′ of 5CQA and C1 of Tx. All RAF pathways of both phenolics with CH3OO• are endergonic, which can mainly be attributed to significantly lower reactivity of this free radical, and, to some extent, to its voluminosity. The products of all exergonic HAT and RAF pathways are presented in Figs. 1 (those related to 5CQA) and 2 (those related to Tx).
All these exergonic reaction pathways were subjected to kinetic investigation, aimed at revealing the transition states and calculating the corresponding rate constants and rT values. The located transition states for the antioxidative pathways of 5CQA and Tx are depicted in Figs. 3 and 4, whereas the activation energies, as well as the kZCT-0 and rT values related to both solvents, are collected in Table 2. Two examples of the IRC calculations for 5CQA with HO• in benzene are presented in Figs S1 and S2. Also, representative examples, including appropriate discussion, for dependence of rate constants on temperature for both 5CQA and Tx are depicted in Figs S3 and S4, respectively.

Optimized geometries of transition states figuring in the reactions of 5CQA with HO• and CH3OO• in benzene. The key bond distances are given in Å. The calculations were performed at the M06-2X/6-311++G(d,p) level of theory in combination with the CPCM solvation model

Optimized geometries of transition states figuring in the reactions of Tx with HO• and CH3OO• in benzene. The key bond distances are given in Å. The calculations were performed at the M06-2X/6-311++G(d,p) level of theory in combination with the CPCM solvation model
It is well known that HAT and RAF only slightly depend on solvent polarity [15,16,17, 26, 31, 41]. For example, it can be easily calculated from the results presented in Ref. [17] that koverall for the reaction of 5CQA with HO• in polar solvents, ethanol, and water amounts to 9.46 × 108 and 9.60 × 108 (the same order of magnitude as koverall for the reaction in benzene). Since benzene and pentyl ethanoate are of comparable polarity, it is reasonable to expect that the koverall and rT values for a certain free radical will be very similar. Table 2 shows that the rT values for CH3OO• are almost identical. To our surprise, rT for HO• is much larger in pentyl ethanoate than in benzene. To clarify this unexpected finding, we performed additional investigation. Actually, we repeated all calculations related to the kinetics of the HAT and RAF pathways of 5CQA and Tx using the M05-2X/6-311++G(d,p) level of theory in combination with the SMD solvation model. It should be pointed out that all necessary structures and corresponding free energies were obtained by full optimizations and frequency calculations. The results obtained are presented in Table 3.
It is worth emphasizing that M05-2X/6-311++G(d,p) in conjunction with SMD was not able to locate the majority of transition states for the HAT pathways of 5CQA in pentyl ethanoate. In these cases geometry convergence could not be achieved, which is illustrated with Fig S5. Thus, this level of theory could not produce either the koverall values of 5CQA in pentyl ethanoate, or the corresponding rT values for both free radicals. On the other hand, all reaction pathways in benzene were revealed, and the corresponding koverall and rT values were calculated. Thus, we can compare three sets of results in Tables 2 and 3 and calculate three sets of branching ratios (Table 4).
Analysis of the results presented in Tables 2, 3 and 4 shows that, despite numerical differences, all three operative computational methodologies, M06-2X—CPCM in benzene and pentyl ethanoate, and M05-2X—SMD in benzene (with the same basis set), exhibit similar trends. In nonpolar solvents the two antioxidants undergo both HAT and RAF pathways with HO•, and only HAT mechanism with CH3OO•. As for the reactions of both phenolics with HO•, the RAF pathways are generally faster. The fastest pathways of 5CQA and Tx are RAFs in the positions 4′ and 6, respectively. A common feature of TS4′ and TS6 (Figs. 3, 4) is that both transition states are stabilized with hydrogen bonds between approaching HO• and phenolic groups and, thus, require low activation energies. On the other hand, attractive interactions in the HAT reaction paths [hydrogen bond between the oxygen of the hydroxyl moiety and proximate phenolic group in TS3′(HO•) and TS4′(HO•) and π-interactions between the hydrogen of the hydroxyl moiety and aromatic ring in TS(HO•)] slow down the formation of hydrogen peroxide and its leaving the reaction system. Thus, despite the finding that addition of HO• to the double bond of 5CQA is thermodynamically most favorable (Table 1), the greatest Γ value for the reaction of 5CQA with HO• comes from the RAF pathway in the 4′ position. As for the HAT pathways with CH3OO•, the Γ values for the 4′ position are larger, and in case of the SMD solvation model the difference between the branching ratios is distinct.
5CQA is more reactive toward HO• than Tx (rT > 1), but less reactive toward CH3OO• (rT < 1). Larger reactivity of 5CQA toward HO• is mainly a consequence of very fast RAF pathways. In comparison to Tx, 5CQA has more suitable positions for addition of HO•. Furthermore, the RAF pathway in the 4′ position of 5CQA is the fastest reaction path according to all three operative methodologies. The rT values for CH3OO• are mutually very similar, while those for HO• differ, where rT in pentyl ethanoate particularly deviates (Tables 2 and 3). Such behavior of rT for HO• is probably a consequence of extreme reactivity of this radical that is challenging to be numerically evaluated, whatever computational methodology is used. Thus, it is more practical to use less reactive free radicals in tasks devoted to determination of antioxidative activity of some compound relative to Tx [14, 42, 43]. However, if one’s goal is to examine all possible antioxidative pathways of the compound, they need to use highly reactive free radicals.
The reactions of 5CQA with two simple free radicals in pentyl ethanoate indicate disadvantage of the combination of the M05-2X functional and SMD solvation model to accomplish some routine computational tasks, such as finding energy maxima. On the other hand, the koverall values related to the reaction of Tx with both free radicals were obtained in this way and are mutually comparable (Tables 2, 3). Now we can compare the rate constants and koverall related to the reactions with HO• obtained in this work (Table 3) to those presented in Ref. [38]. General impression is that the values presented here are by two orders of magnitude smaller than those suggested by Alberto et al. [38]. The most noteworthy difference is that we revealed a reaction path for homolytic cleavage of the O–H bond of the phenolic group and calculated the rate constant of 5.54 × 106 M−1 s−1, whereas Alberto et al. found that this process is diffusion controlled and assigned it the rate constant of 1.91 × 109 M−1 s−1. Inconsistency between the two sets of results is, certainly, a consequence of already explained difference in applied computational methodology. We approve the approach based on geometry optimization and frequency calculation in conjunction with some continuum solvation model, as such tasks are nowadays routine.
A logical next step of our investigation is to determine rT of 5CQA in water. In slightly basic aqueous solution 5CQA exists in the form of monoanion and dianion. Extremely complex behavior of these species toward free radicals is under our intense scrutiny.
4 Summary
The quantum mechanics-based test for overall free-radical scavenging activity (QMORSA) is a very useful computational protocol for estimating rT of a compound, i.e., its antioxidative activity relative to some reference compound, usually Tx [14]. It is practical that QMORSA calculations are conducted by using a unique approach, so that the results of different investigations can be mutually compared. In this work we evaluated the rT values of 5CQA for the HO• and CH3OO• radicals in two nonpolar solvents by applying two different methodologies. It turned out that repeatedly suggested M05-2X functional in conjunction with the SMD solvation model failed to estimate the influence of pentyl ethanoate, selected to mimic lipid environment, because of unusual convergence failures. This finding is quite unexpected since SMD employs a set of parameters optimized over the M05-2X/MIDI!6D, M05-2X/6-31G(d), M05-2X/6-31+G(d,p), M05-2X/cc-pVTZ, B3LYP/6-31G(d), and HF/6-31G(d) theoretical methods [25]. On the other hand, other three methodologies, M06-2X—CPCM in benzene and pentyl ethanoate and M05-2X—SMD in benzene, proved to be operative and showed similar trends. Namely, 5CQA and Tx undergo both HAT and RAF paths with HO•, and only HAT path with CH3OO•, where 5CQA is more reactive toward HO• than Tx, but less reactive toward CH3OO•. Discrepancy of the results related to HO•, particularly in pentyl ethanoate, can be attributed to general inability of computational methodologies to numerically evaluate extreme reactivity of this radical.
Our research confirms that if one’s goal is to determine antioxidative activity of a compound relative to Tx, it is more useful to include less reactive free radicals in investigation [14, 42, 43]. However, if one’s goal is to examine all possible antioxidative pathways of the compound, it is necessary to employ highly reactive free radicals, and in such case different approaches can lead to inconsistent results.
As a final point, which approach should be applied in investigations of antioxidative activity? We strongly suggest the approach based on geometry optimization and frequency calculation in conjunction with continuum solvation model, because energy is highly dependent on structure, and geometry of a molecule in the gas-phase differs from that of solvated molecule. M06-2X is more robust than M05-2X [14, 24]. Bearing in mind that radical and negatively charged species participate in antioxidative processes basis set needs to add diffuse functions to all atoms, including hydrogen.
References
Sayre LM, Perry G, Smith MA (2008) Oxidative stress and neurotoxicity. Chem Res Toxicol 21:172–188
Liu Z (2010) Chemical methods to evaluate antioxidant ability. Chem Rev 110:5675–5691
Hoye AT, Davoren JE, Wipf P et al (2008) Targeting mitochondria. Acc Chem Res 41:87–97
Crozier A, Jaganath IB, Clifford MN (2009) Dietary phenolics: chemistry, bioavailability and effects on health. Nat Prod Rep 26:1001
Hertog MG, Sweetnam PM, Fehily AM et al (1997) Antioxidant flavonols and ischemic heart disease in a Welsh population of men: the Caerphilly Study. Am J Clin Nutr 65:1489–1494
Rimm EB, Katan MB, Ascherio A et al (1996) Relation between intake of flavonoids and risk for coronary heart disease in male health professionals. Ann Intern Med 125:384–389
Xu JG, Hu QP, Liu Y (2012) Antioxidant and DNA-protective activities of chlorogenic acid isomers. J Agric Food Chem 60:11625–11630
Wang X, Wang J, Yang N (2007) Chemiluminescent determination of chlorogenic acid in fruits. Food Chem 102:422–426
Prior RL, Wu X, Schaich K (2005) Standardized methods for the determination of antioxidant capacity and phenolics in foods and dietary supplements. J Agric Food Chem 53:4290–4302
Frankel EN, Meyer AS (2000) The problems of using one-dimensional methods to evaluate multifunctional food and biological antioxidants. J Sci Food Agric 80:1925–1941
Antolovich M, Prenzler PD, Patsalides E et al (2002) Methods for testing antioxidant activity. Analyst 127:183–198
Huang D, Boxin OU, Prior RL (2005) The chemistry behind antioxidant capacity assays. J Agric Food Chem 53:1841–1856
Halliwell B, Aeschbach R, Löliger J, Aruoma OI (1995) The characterization of antioxidants. Food Chem Toxicol 33:601–617
Galano A, Alvarez-Idaboy JR (2013) A computational methodology for accurate predictions of rate constants in solution: application to the assessment of primary antioxidant activity. J Comput Chem 34:2430–2445
Marković S, Tošović J (2016) Comparative study of the antioxidative activities of caffeoylquinic and caffeic acids. Food Chem 210:585–592
Tošović J, Marković S (2016) Structural and antioxidative features of chlorogenic acid. Croat Chem Acta 89:535–541
Tošović J, Marković S, Dimitrić Marković JM et al (2017) Antioxidative mechanisms in chlorogenic acid. Food Chem 237:390–398
Chen Y, Xiao H, Zheng J, Liang G (2015) Structure-thermodynamics-antioxidant activity relationships of selected natural phenolic acids and derivatives: an experimental and theoretical evaluation. PLoS ONE 10:1–20
Uranga JG, Podio NS, Wunderlin DA, Santiago AN (2016) Theoretical and experimental study of the antioxidant behaviors of 5-O-caffeoylquinic, quinic and caffeic acids based on electronic and structural properties. ChemistrySelect 1:4113–4120
Frisch MJ, Trucks GW, Schlegel HB et al (2013) Gaussian 09, Revision D.01. Gaussian, Inc., Wallingford
Zhao Y, Truhlar DG (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor Chem Acc 120:215–241
Zhao Y, Truhlar DG (2008) Density functionals with broad applicability in chemistry. Acc Chem Res 41:157–167
Cossi M, Rega N, Scalmani G, Barone V (2003) Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J Comput Chem 24:669–681
Zhao Y, Schultz NE, Truhlar DG (2006) Design of density functionals by combining the method of constraint satisfaction with parametrization for thermochemistry, thermochemical kinetics, and noncovalent interactions. J Chem Theory Comput 2:364–382
Marenich AV, Cramer CJ, Truhlar DG (2009) Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B 113:6378–6396
Redžepović I, Marković S, Tošović J (2017) Antioxidative activity of caffeic acid—mechanistic DFT study. Kragujev J Sci 39:109–122
Iuga C, Alvarez-Idaboy JR, Russo N (2012) Antioxidant activity of trans -resveratrol toward hydroxyl and hydroperoxyl radicals: a quantum chemical and computational kinetics study. J Org Chem 77:3868–3877
Markovic Z, Amic D, Milenkovic D et al (2013) Examination of the chemical behavior of the quercetin radical cation towards some bases. Phys Chem Chem Phys 15:7370–7378
Medina ME, Iuga C, Alvarez-Idaboy JR (2014) Antioxidant activity of fraxetin and its regeneration in aqueous media. A density functional theory study. RSC Adv 4:52920–52932
Amić A, Lučić B, Stepanić V et al (2017) Free radical scavenging potency of quercetin catecholic colonic metabolites: thermodynamics of 2H +/2e − processes. Food Chem 218:144–151
Milenković D, Đorović J, Petrović V et al (2017) Hydrogen atom transfer versus proton coupled electron transfer mechanism of gallic acid with different peroxy radicals. React Kinet Mech Catal 123:215–230
Okuno Y (1997) Theoretical investigation of the mechanism of the Baeyer–Villiger reaction in nonpolar solvents. Eur J Chem 3:212–218
Alberty RA (1960) The foundations of chemical kinetics (Benson, Sidney W.). J Chem Educ 37:660
Marković Z, Tošović J, Milenković D, Marković S (2016) Revisiting the solvation enthalpies and free energies of the proton and electron in various solvents. Comput Theor Chem 1077:11–17
Eckart C (1930) The penetration of a potential barrier by electrons. Phys Rev 35:1303–1309
Duncan WT, Bell RL, Truong TN (1998) TheRate: program for ab initio direct dynamics calculations of thermal and vibrational-state-selected rate constants. J Comput Chem 19:1039–1052
Fernández-Ramos A, Miller JA, Klippenstein SJ, Truhlar DG (2006) Modeling the kinetics of bimolecular reactions. Chem Rev 106:4518–4584
Alberto ME, Russo N, Grand A, Galano A (2013) A physicochemical examination of the free radical scavenging activity of Trolox: mechanism, kinetics and influence of the environment. Phys Chem Chem Phys 15:4642
Medina ME, Iuga C, Álvarez-Idaboy JR (2013) Antioxidant activity of propyl gallate in aqueous and lipid media: a theoretical study. Phys Chem Chem Phys 15:13137
Marino T, Russo N, Galano A (2016) A deeper insight on the radical scavenger activity of two simple coumarins toward OOH radical. Comput Theor Chem 1077:133–138
Dimić DS, Milenković DA, Marković JMD, Marković ZS (2017) Thermodynamic and kinetic analysis of the reaction between biological catecholamines and chlorinated methylperoxy radicals. Mol Phys 8976:1–13
Galano A, Tan DX, Reiter RJ (2011) Melatonin as a natural ally against oxidative stress: a physicochemical examination. J Pineal Res 51:1–16
Rose RC, Bode AM (1993) Biology of free radical scavengers: an evaluation of ascorbate. FASEB J 7:1135–1142
Acknowledgements
This work was supported by the Ministry of Education, Science and Technological Development of Serbia, Project No 172016. The authors are grateful to Reviewer for useful suggestions regarding this work.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tošović, J., Marković, S. Reactivity of chlorogenic acid toward hydroxyl and methyl peroxy radicals relative to trolox in nonpolar media. Theor Chem Acc 137, 76 (2018). https://doi.org/10.1007/s00214-018-2251-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-018-2251-y